
EMD-57033 Augments the Contractility in Porcine Myocardium by Promoting the Activation of Myosin in Thick Filaments



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
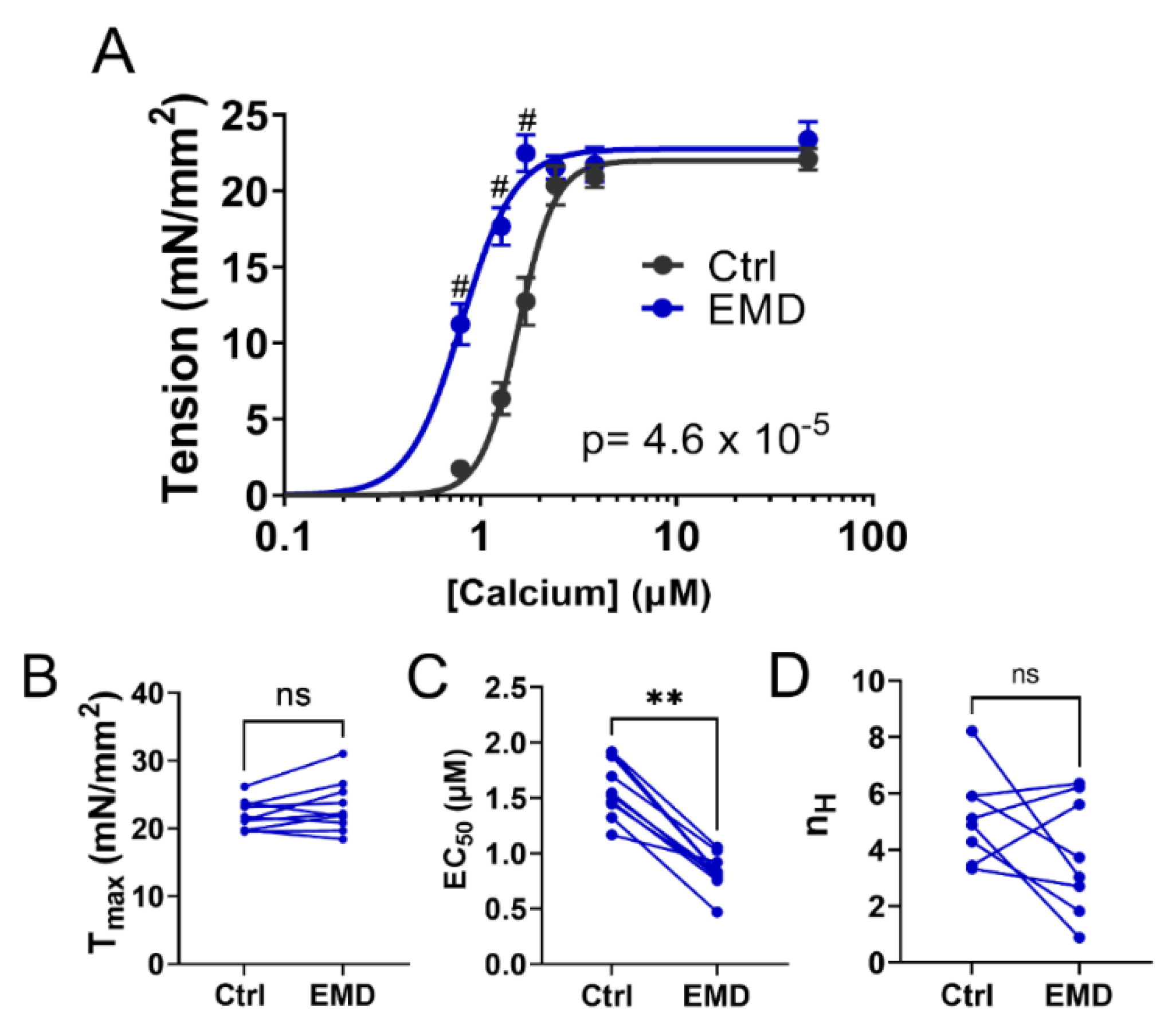
2.1. Contractility of Muscle Tissue after the EMD Treatment
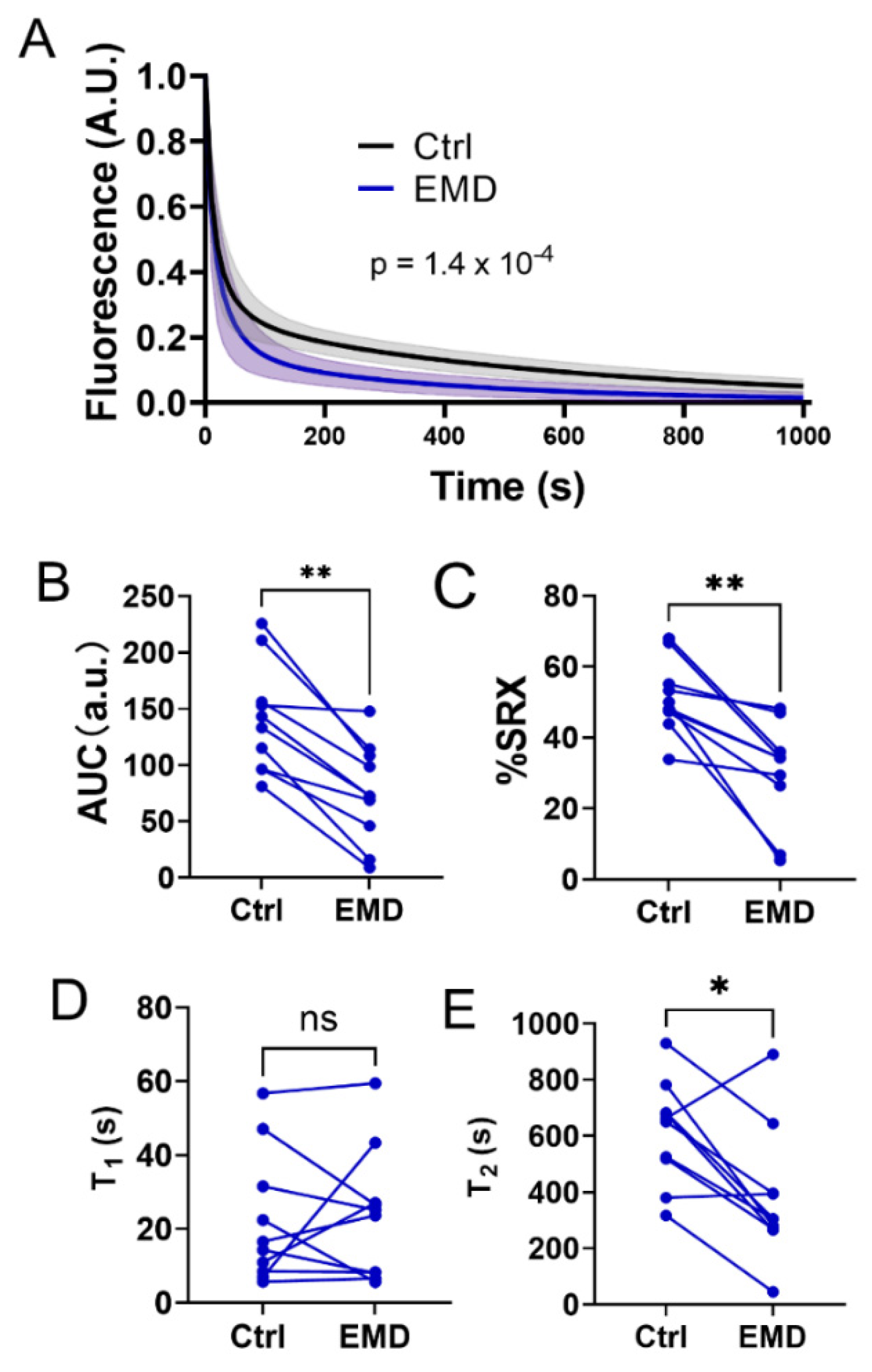
2.2. Alterations in the Biochemical States of the Myosin Heads with EMD Treatment
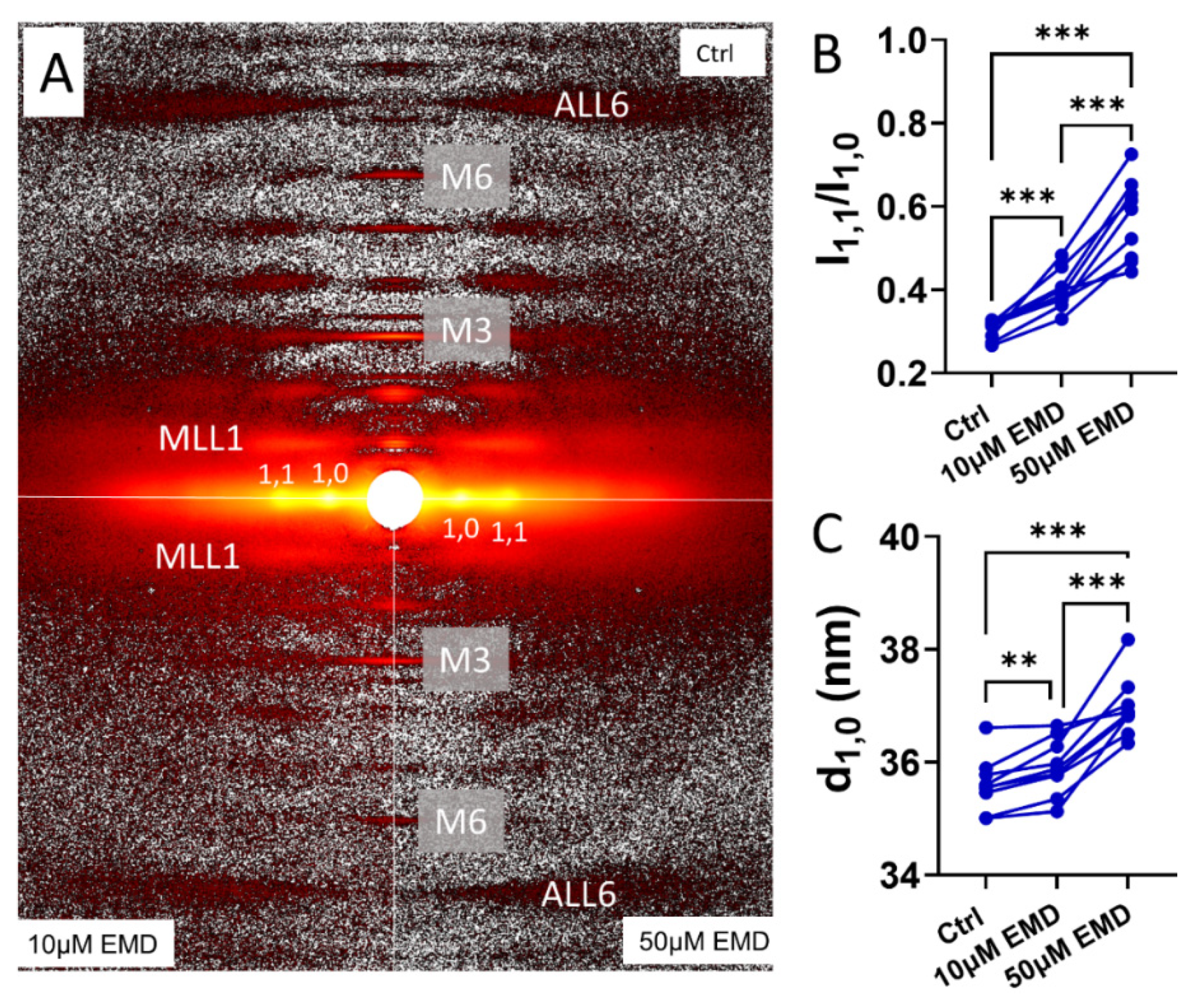
2.3. Changes in the X-ray Equatorial Diffraction Patterns with EMD
2.4. Changes in the Meridional X-ray Reflections and the Layer Lines with EMD
3. Discussion
3.1. EMD Recruits Myosin from the Biochemically-Defined SRX State
3.2. EMD Recruits Myosin from the Structurally-Defined OFF State
3.3. Sarcomeric Activators as an Approach for Rescuing the Contractility in Myocardium
4. Materials and Methods
4.1. Isometric Tension-Calcium Relationships
4.2. Myosin ATP Turnover Kinetics
4.3. Muscle Preparations for the Small-Angle X-Diffraction
4.4. Statistics
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Hanft, L.M.; Emter, C.A.; McDonald, K.S. Cardiac myofibrillar contractile properties during the progression from hypertension to decompensated heart failure. Am. J. Physiol. Heart Circ. Physiol. 2017, 313, H103–H113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirk, J.A.; Chakir, K.; Lee, K.H.; Karst, E.; Holewinski, R.J.; Pironti, G.; Tunin, R.S.; Pozios, I.; Abraham, T.P.; de Tombe, P.; et al. Pacemaker-induced transient asynchrony suppresses heart failure progression. Sci. Transl. Med. 2015, 7, 319ra207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blair, C.A.; Brundage, E.A.; Thompson, K.L.; Stromberg, A.; Guglin, M.; Biesiadecki, B.J.; Campbell, K.S. Heart Failure in Humans Reduces Contractile Force in Myocardium From Both Ventricles. JACC Basic Transl. Sci. 2020, 5, 786–798. [Google Scholar] [CrossRef]
- van der Velden, J.; Merkus, D.; Klarenbeek, B.R.; James, A.T.; Boontje, N.M.; Dekkers, D.H.; Stienen, G.J.; Lamers, J.M.; Duncker, D.J. Alterations in myofilament function contribute to left ventricular dysfunction in pigs early after myocardial infarction. Circ. Res. 2004, 95, e85–e95. [Google Scholar] [CrossRef] [Green Version]
- Aslam, M.I.; Hahn, V.S.; Jani, V.; Hsu, S.; Sharma, K.; Kass, D.A. Reduced Right Ventricular Sarcomere Contractility in Heart Failure With Preserved Ejection Fraction and Severe Obesity. Circulation 2021, 143, 965–967. [Google Scholar] [CrossRef] [PubMed]
- Hsu, S.; Kokkonen-Simon, K.M.; Kirk, J.A.; Kolb, T.M.; Damico, R.L.; Mathai, S.C.; Mukherjee, M.; Shah, A.A.; Wigley, F.M.; Margulies, K.B.; et al. Right Ventricular Myofilament Functional Differences in Humans With Systemic Sclerosis-Associated Versus Idiopathic Pulmonary Arterial Hypertension. Circulation 2018, 137, 2360–2370. [Google Scholar] [CrossRef]
- Teerlink, J.R.; Diaz, R.; Felker, G.M.; McMurray, J.J.V.; Metra, M.; Solomon, S.D.; Adams, K.F.; Anand, I.; Arias-Mendoza, A.; Biering-Sorensen, T.; et al. Cardiac Myosin Activation with Omecamtiv Mecarbil in Systolic Heart Failure. N. Engl. J. Med. 2021, 384, 105–116. [Google Scholar] [CrossRef]
- Tobacman, L.S. Thin filament-mediated regulation of cardiac contraction. Annu. Rev. Physiol. 1996, 58, 447–481. [Google Scholar] [CrossRef]
- Wakabayashi, T. Mechanism of the calcium-regulation of muscle contraction—in pursuit of its structural basis. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2015, 91, 321–350. [Google Scholar] [CrossRef] [Green Version]
- Lehman, W.; Craig, R.; Vibert, P. Ca2+ induced tropomyosin movement in Limulus thin filaments revealed by three-dimensional reconstruction. Nature 1994, 368, 65–67. [Google Scholar] [CrossRef]
- Yamada, Y.; Namba, K.; Fujii, T. Cardiac muscle thin filament structures reveal calcium regulatory mechanism. Nat. Commun. 2020, 11, 153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Risi, C.M.; Pepper, I.; Belknap, B.; Landim-Vieira, M.; White, H.D.; Dryden, K.; Pinto, J.R.; Chase, P.B.; Galkin, V.E. The structure of the native cardiac thin filament at systolic Ca(2+) levels. Proc. Natl. Acad. Sci. USA 2021, 118, e2024288118. [Google Scholar] [CrossRef]
- Fitzsimons, D.P.; Moss, R.L. Strong binding of myosin modulates length-dependent Ca2+ activation of rat ventricular myocytes. Circ. Res. 1998, 83, 602–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spudich, J.A. Three perspectives on the molecular basis of hypercontractility caused by hypertrophic cardiomyopathy mutations. Pflug. Arch. Eur. J. Physiol. 2019, 471, 701–717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooke, R. The role of the myosin ATPase activity in adaptive thermogenesis by skeletal muscle. Biophys. Rev. 2011, 3, 33–45. [Google Scholar] [CrossRef] [Green Version]
- Hooijman, P.; Stewart, M.A.; Cooke, R. A New State of Cardiac Myosin with Very Slow ATP Turnover: A Potential Cardioprotective Mechanism in the Heart. Biophys. J. 2011, 100, 1969–1976. [Google Scholar] [CrossRef] [Green Version]
- McNamara, J.W.; Li, A.; Dos Remedios, C.G.; Cooke, R. The role of super-relaxed myosin in skeletal and cardiac muscle. Biophys. Rev. 2015, 7, 5–14. [Google Scholar] [CrossRef] [Green Version]
- Nag, S.; Trivedi, D.V. To lie or not to lie: Super-relaxing with myosins. Elife 2021, 10, e63703. [Google Scholar] [CrossRef]
- Haselgrove, J.C. X-ray evidence for conformational changes in the myosin filaments of vertebrate striated muscle. J. Mol. Biol. 1975, 92, 113–143. [Google Scholar] [CrossRef]
- Huxley, H.E. Structural changes in actin- and myosin-containing filaments during contraction. Cold Spring Harbor Symp. Quant. Biol. 1973, 37, 361–376. [Google Scholar] [CrossRef]
- Ait-Mou, Y.; Hsu, K.; Farman, G.P.; Kumar, M.; Greaser, M.L.; Irving, T.C.; de Tombe, P.P. Titin strain contributes to the Frank-Starling law of the heart by structural rearrangements of both thin- and thick-filament proteins. Proc. Natl. Acad. Sci. USA 2016, 113, 2306–2311. [Google Scholar] [CrossRef] [Green Version]
- Irving, M. Regulation of Contraction by the Thick Filaments in Skeletal Muscle. Biophys. J. 2017, 113, 2579–2594. [Google Scholar] [CrossRef] [Green Version]
- Linari, M.; Brunello, E.; Reconditi, M.; Fusi, L.; Caremani, M.; Narayanan, T.; Piazzesi, G.; Lombardi, V.; Irving, M. Force generation by skeletal muscle is controlled by mechanosensing in myosin filaments. Nature 2015, 528, 276–279. [Google Scholar] [CrossRef] [Green Version]
- Woodhead, J.L.; Craig, R. Through Thick and Thin—Interfilament Communication in Muscle. Biophys. J. 2015, 109, 665–667. [Google Scholar] [CrossRef] [Green Version]
- Ma, W.; Irving, T.C. Small Angle X-ray Diffraction as a Tool for Structural Characterization of Muscle Disease. Int. J. Mol. Sci. 2022, 23, 3052. [Google Scholar] [CrossRef]
- Ma, W.; Henze, M.; Anderson, R.L.; Gong, H.; Wong, F.L.; Del Rio, C.L.; Irving, T. The Super-Relaxed State and Length Dependent Activation in Porcine Myocardium. Circ. Res. 2021, 129, 617–630. [Google Scholar] [CrossRef]
- Chu, S.; Muretta, J.M.; Thomas, D.D. Direct detection of the myosin super-relaxed state and interacting-heads motif in solution. J. Biol. Chem. 2021, 297, 101157. [Google Scholar] [CrossRef]
- Anderson, R.L.; Trivedi, D.V.; Sarkar, S.S.; Henze, M.; Ma, W.; Gong, H.; Rogers, C.S.; Gorham, J.M.; Wong, F.L.; Morck, M.M.; et al. Deciphering the super relaxed state of human beta-cardiac myosin and the mode of action of mavacamten from myosin molecules to muscle fibers. Proc. Natl. Acad. Sci. USA 2018, 115, E8143–E8152. [Google Scholar] [CrossRef] [Green Version]
- Solaro, R.J.; Gambassi, G.; Warshaw, D.M.; Keller, M.R.; Spurgeon, H.A.; Beier, N.; Lakatta, E.G. Stereoselective actions of thiadiazinones on canine cardiac myocytes and myofilaments. Circ. Res. 1993, 73, 981–990. [Google Scholar] [CrossRef] [Green Version]
- Li, M.X.; Spyracopoulos, L.; Beier, N.; Putkey, J.A.; Sykes, B.D. Interaction of cardiac troponin C with Ca(2+) sensitizer EMD 57033 and cardiac troponin I inhibitory peptide. Biochemistry 2000, 39, 8782–8790. [Google Scholar] [CrossRef]
- Stevens, C.M.; Rayani, K.; Singh, G.; Lotfalisalmasi, B.; Tieleman, D.P.; Tibbits, G.F. Changes in the dynamics of the cardiac troponin C molecule explain the effects of Ca(2+)-sensitizing mutations. J. Biol. Chem. 2017, 292, 11915–11926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radke, M.B.; Taft, M.H.; Stapel, B.; Hilfiker-Kleiner, D.; Preller, M.; Manstein, D.J. Small molecule-mediated refolding and activation of myosin motor function. Elife 2014, 3, e01603. [Google Scholar] [CrossRef] [PubMed]
- Haselgrove, J.C.; Huxley, H.E. X-ray evidence for radial cross-bridge movement and for the sliding filament model in actively contracting skeletal muscle. J. Mol. Biol. 1973, 77, 549–568. [Google Scholar] [CrossRef]
- Ma, W.; Gong, H.; Irving, T. Myosin Head Configurations in Resting and Contracting Murine Skeletal Muscle. Int. J. Mol. Sci. 2018, 19, 2643. [Google Scholar] [CrossRef] [Green Version]
- Reconditi, M. Recent Improvements in Small Angle X-ray Diffraction for the Study of Muscle Physiology. Rep. Prog. Phys. Phys. Soc. 2006, 69, 2709–2759. [Google Scholar] [CrossRef]
- Craig, R.; Padron, R. Structural basis of the super- and hyper-relaxed states of myosin II. J. Gen. Physiol. 2022, 154, e202113012. [Google Scholar] [CrossRef]
- Jani, V.; Aslam, I.; Ma, W.; Gong, H.; Cammarato, A.; Irving, C.; Kass, D.; Hsu, S. RV Sarcomeres from LV-HFrEF Patients with Low PAPi Have Abnormal RV Thick Filament Structure. Circ. Res. 2021, 129 (Suppl. S1), AP505. [Google Scholar] [CrossRef]
- Senzaki, H.; Isoda, T.; Paolocci, N.; Ekelund, U.; Hare, J.M.; Kass, D.A. Improved mechanoenergetics and cardiac rest and reserve function of in vivo failing heart by calcium sensitizer EMD-57033. Circulation 2000, 101, 1040–1048. [Google Scholar] [CrossRef] [Green Version]
- Irving, T.C.; Millman, B.M. Changes in thick filament structure during compression of the filament lattice in relaxed frog sartorius muscle. J. Muscle Res. Cell Motil. 1989, 10, 385–394. [Google Scholar] [CrossRef]
- Millman, B.M. The filament lattice of striated muscle. Physiol. Rev. 1998, 78, 359–391. [Google Scholar] [CrossRef]
- Aslam, M.I.; Jani, V.; Lin, B.L.; Dunkerly-Eyring, B.; Livingston, C.E.; Ramachandran, A.; Ranek, M.J.; Bedi, K.C.; Margulies, K.B.; Kass, D.A.; et al. Pulmonary artery pulsatility index predicts right ventricular myofilament dysfunction in advanced human heart failure. Eur. J. Heart Fail. 2021, 23, 339–341. [Google Scholar] [CrossRef] [PubMed]
- de Zeeuw, S.; Trines, S.A.; Krams, R.; Verdouw, P.D.; Duncker, D.J. Cardiovascular profile of the calcium sensitizer EMD 57033 in open-chest anaesthetized pigs with regionally stunned myocardium. Br. J. Pharmacol. 2000, 129, 1413–1422. [Google Scholar] [CrossRef] [Green Version]
- Soergel, D.G.; Georgakopoulos, D.; Stull, L.B.; Kass, D.A.; Murphy, A.M. Augmented systolic response to the calcium sensitizer EMD-57033 in a transgenic model with troponin I truncation. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H1785–H1792. [Google Scholar] [CrossRef] [Green Version]
- Chung, J.H.; Biesiadecki, B.J.; Ziolo, M.T.; Davis, J.P.; Janssen, P.M. Myofilament Calcium Sensitivity: Role in Regulation of In vivo Cardiac Contraction and Relaxation. Front. Physiol. 2016, 7, 562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Kampourakis, T.; Yan, Z.; Sevrieva, I.; Irving, M.; Sun, Y.B. Distinct contributions of the thin and thick filaments to length-dependent activation in heart muscle. Elife 2017, 6, e24081. [Google Scholar] [CrossRef] [Green Version]
- Solaro, R.J.; Rarick, H.M. Troponin and tropomyosin: Proteins that switch on and tune in the activity of cardiac myofilaments. Circ. Res. 1998, 83, 471–480. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Nag, S.; Gong, H.; Qi, L.; Irving, T. Cardiac myosin filaments are directly regulated by calcium. J. Gen. Physiol. 2022, 154, e202213213. [Google Scholar] [CrossRef] [PubMed]
- Alsulami, K.; Marston, S. Small Molecules acting on Myofilaments as Treatments for Heart and Skeletal Muscle Diseases. Int. J. Mol. Sci. 2020, 21, 9599. [Google Scholar] [CrossRef]
- Kass, D.A.; Solaro, R.J. Mechanisms and use of calcium-sensitizing agents in the failing heart. Circulation 2006, 113, 305–315. [Google Scholar] [CrossRef] [Green Version]
- Schamp, K.; Schreder, S.A.; Dressman, J. Development of an in vitro/in vivo correlation for lipid formulations of EMD 50733, a poorly soluble, lipophilic drug substance. Eur. J. Pharm. Biopharm. 2006, 62, 227–234. [Google Scholar] [CrossRef]
- Vogt, M.; Vertzoni, M.; Kunath, K.; Reppas, C.; Dressman, J.B. Cogrinding enhances the oral bioavailability of EMD 57033, a poorly water soluble drug, in dogs. Eur. J. Pharm. Biopharm. 2008, 68, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Stewart, M.A.; Franks-Skiba, K.; Chen, S.; Cooke, R. Myosin ATP turnover rate is a mechanism involved in thermogenesis in resting skeletal muscle fibers. Proc. Natl. Acad. Sci. USA 2010, 107, 430–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walklate, J.; Kao, K.; Regnier, M.; Geeves, M.A. Exploring the Super-relaxed State of Myosin in Myofibrils from Fast-twitch, Slow-twitch and Cardiac Muscle. J. Biol. Chem. 2022, 298, 101640. [Google Scholar] [CrossRef]
- Ma, W.; Gong, H.; Jani, V.; Lee, K.H.; Landim-Vieira, M.; Papadaki, M.; Pinto, J.R.; Aslam, M.I.; Cammarato, A.; Irving, T. Myofibril orientation as a metric for characterizing heart disease. Biophys. J. 2022, 121, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Fischetti, R.; Stepanov, S.; Rosenbaum, G.; Barrea, R.; Black, E.; Gore, D.; Heurich, R.; Kondrashkina, E.; Kropf, A.J.; Wang, S.; et al. The BioCAT undulator beamline 18ID: A facility for biological non-crystalline diffraction and X-ray absorption spectroscopy at the Advanced Photon Source. J. Synchrotron. Radiat. 2004, 11, 399–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiratrakanvong, J.; Shao, J.; Menendez, M.; Li, X.; Li, J.; Ma, W.; Agam, G.; Irving, T. MuscleX: Software Suite for Diffraction X-ray Imaging; V1.13.1; BioCAT: Chicago, IL, USA, 2018. [Google Scholar] [CrossRef]
- Ma, W.; Gong, H.; Kiss, B.; Lee, E.J.; Granzier, H.; Irving, T. Thick-Filament Extensibility in Intact Skeletal Muscle. Biophys. J. 2018, 115, 1580–1588. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jani, V.; Qian, W.; Yuan, S.; Irving, T.; Ma, W. EMD-57033 Augments the Contractility in Porcine Myocardium by Promoting the Activation of Myosin in Thick Filaments. Int. J. Mol. Sci. 2022, 23, 14517. https://doi.org/10.3390/ijms232314517
Jani V, Qian W, Yuan S, Irving T, Ma W. EMD-57033 Augments the Contractility in Porcine Myocardium by Promoting the Activation of Myosin in Thick Filaments. International Journal of Molecular Sciences. 2022; 23(23):14517. https://doi.org/10.3390/ijms232314517
Chicago/Turabian StyleJani, Vivek, Wenjing Qian, Shengyao Yuan, Thomas Irving, and Weikang Ma. 2022. "EMD-57033 Augments the Contractility in Porcine Myocardium by Promoting the Activation of Myosin in Thick Filaments" International Journal of Molecular Sciences 23, no. 23: 14517. https://doi.org/10.3390/ijms232314517