
Alterations of the Sialylation Machinery in Brugada Syndrome
 , , , , ,
, , , , ,  , , , ,
, , , ,
Abstract
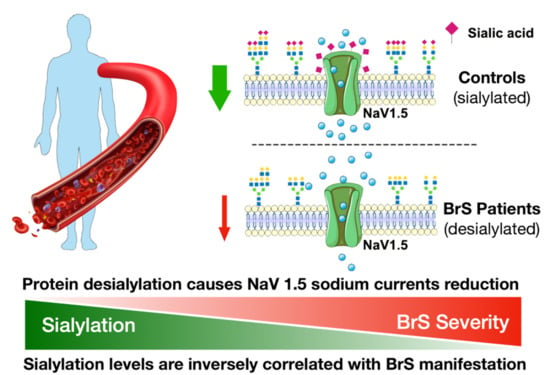
:
1. Introduction
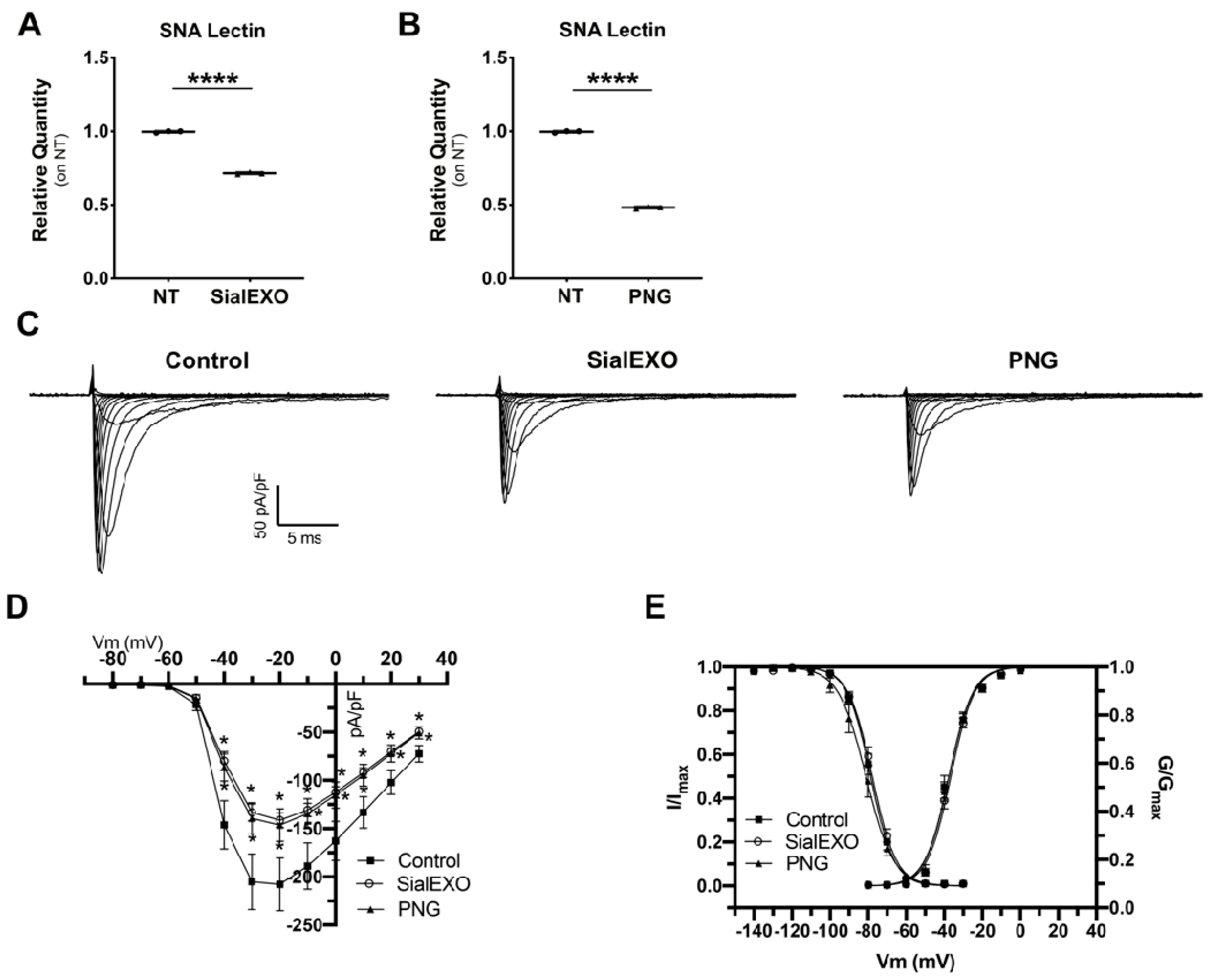
2. Results
2.1. Study Population
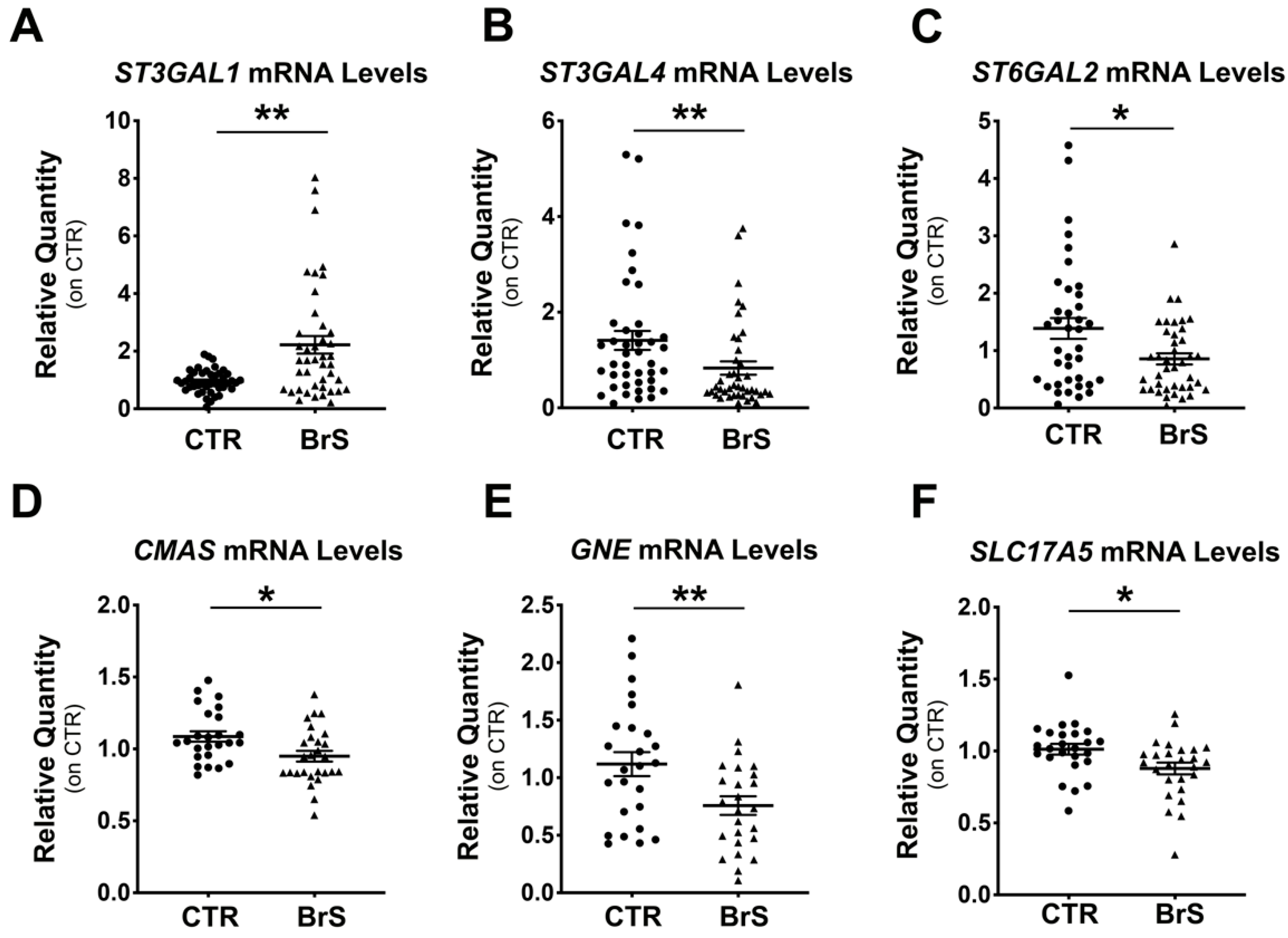
2.2. Gene Expression Analysis Shows Alteration of the Sialylation Machinery in BrS Patients
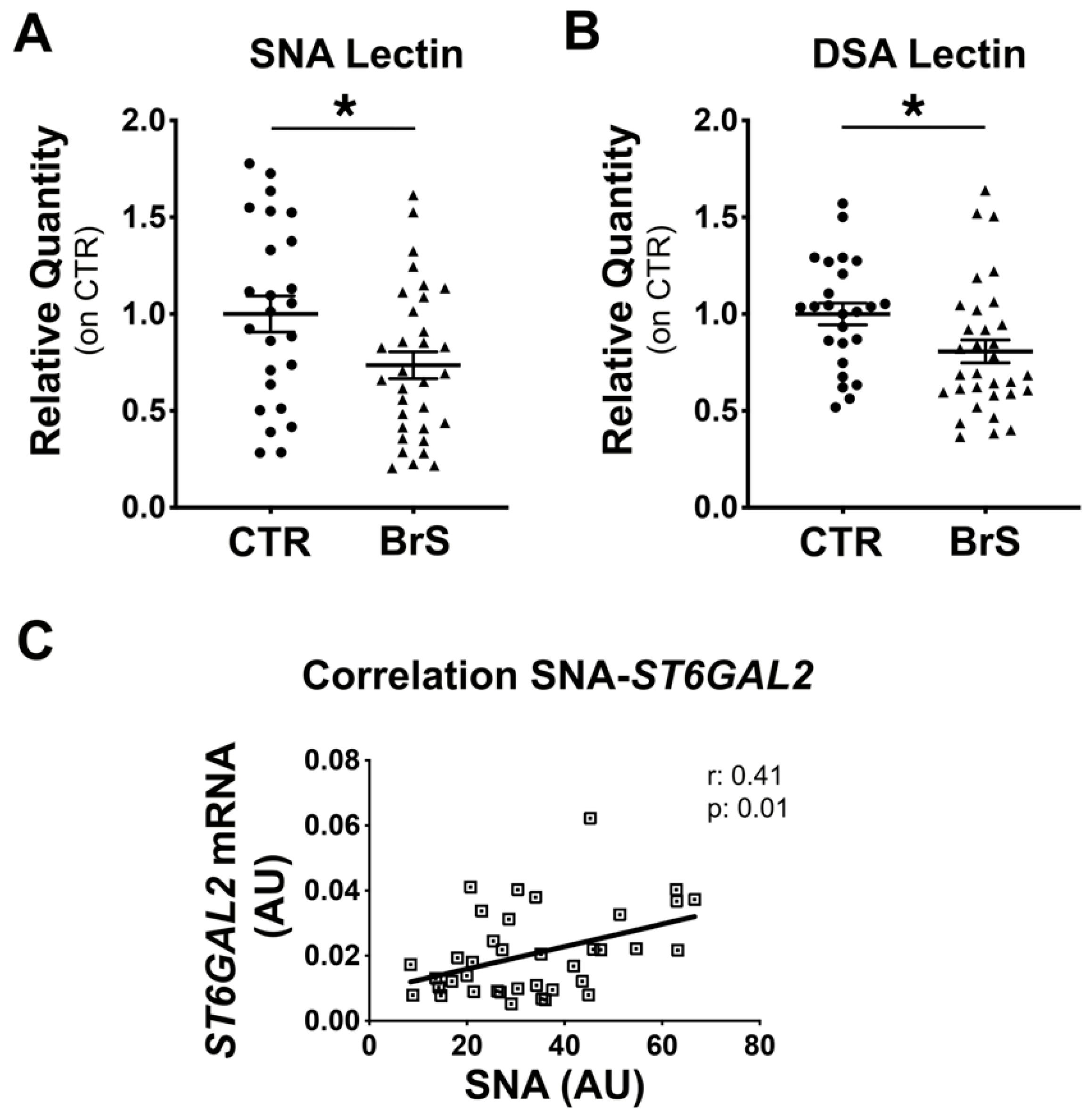
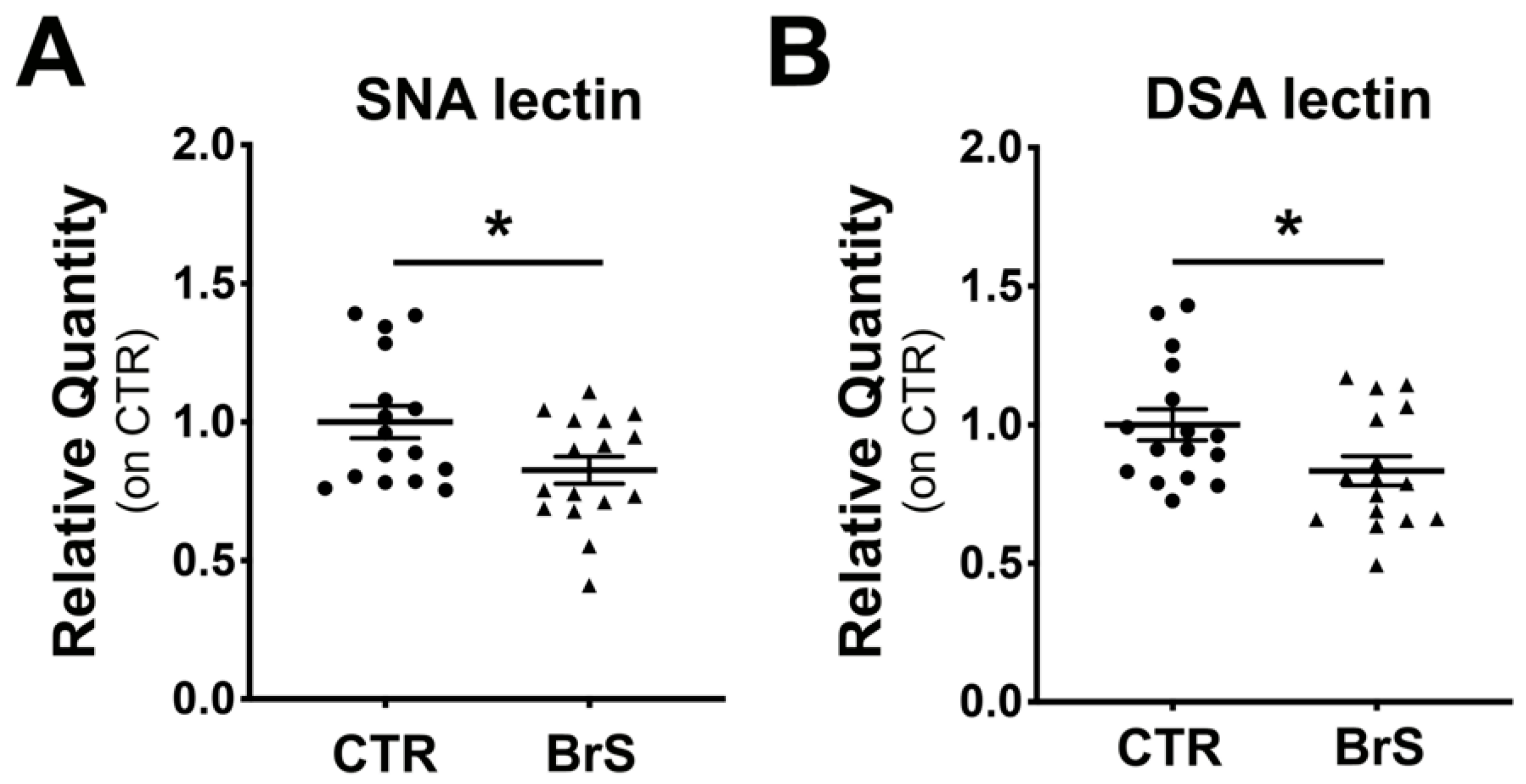
2.3. Flow Cytometric Analysis of PBMCs Reveals a Reduction in Membrane Protein Sialylation Levels
2.4. Western Blot Analysis of PBMCs Show a Decrease of Intracellular Protein Sialylation Levels in BrS Patients
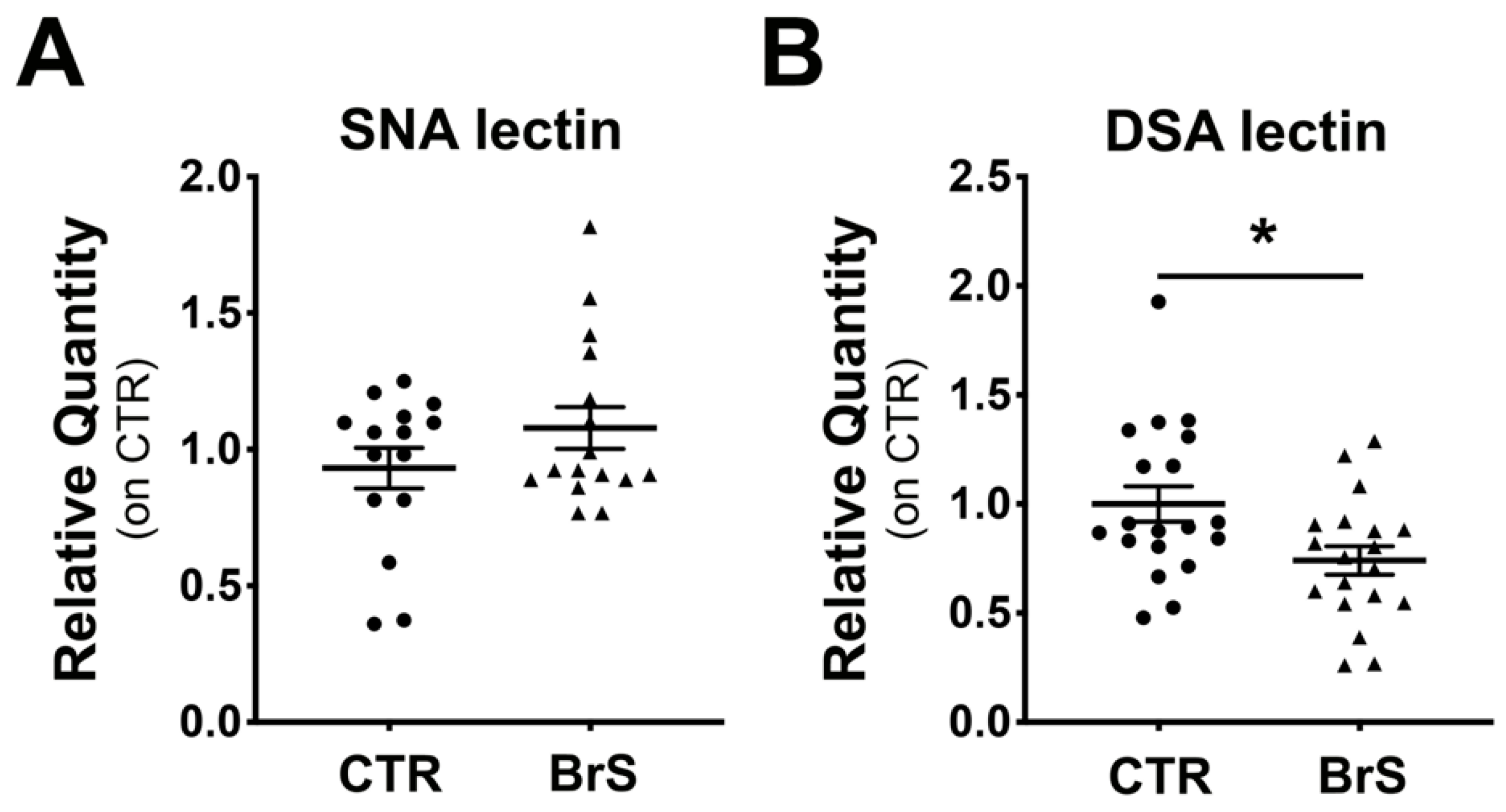
2.5. Analysis of Circulating Plasma Proteins Show Changes in Protein Sialylation Levels in BrS Patients
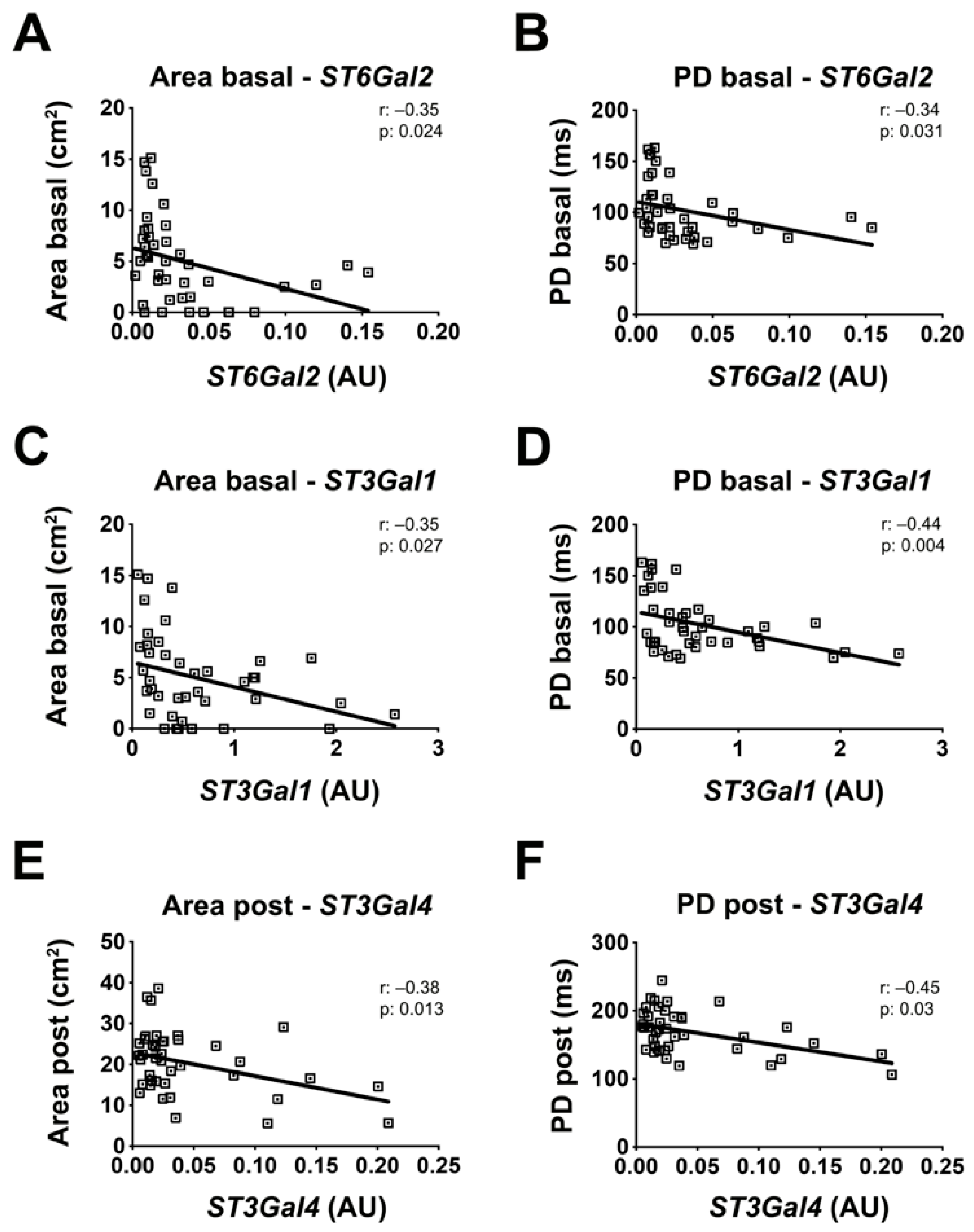
2.6. Sialyltransferases Expression Levels Correlate with the BrS Severity
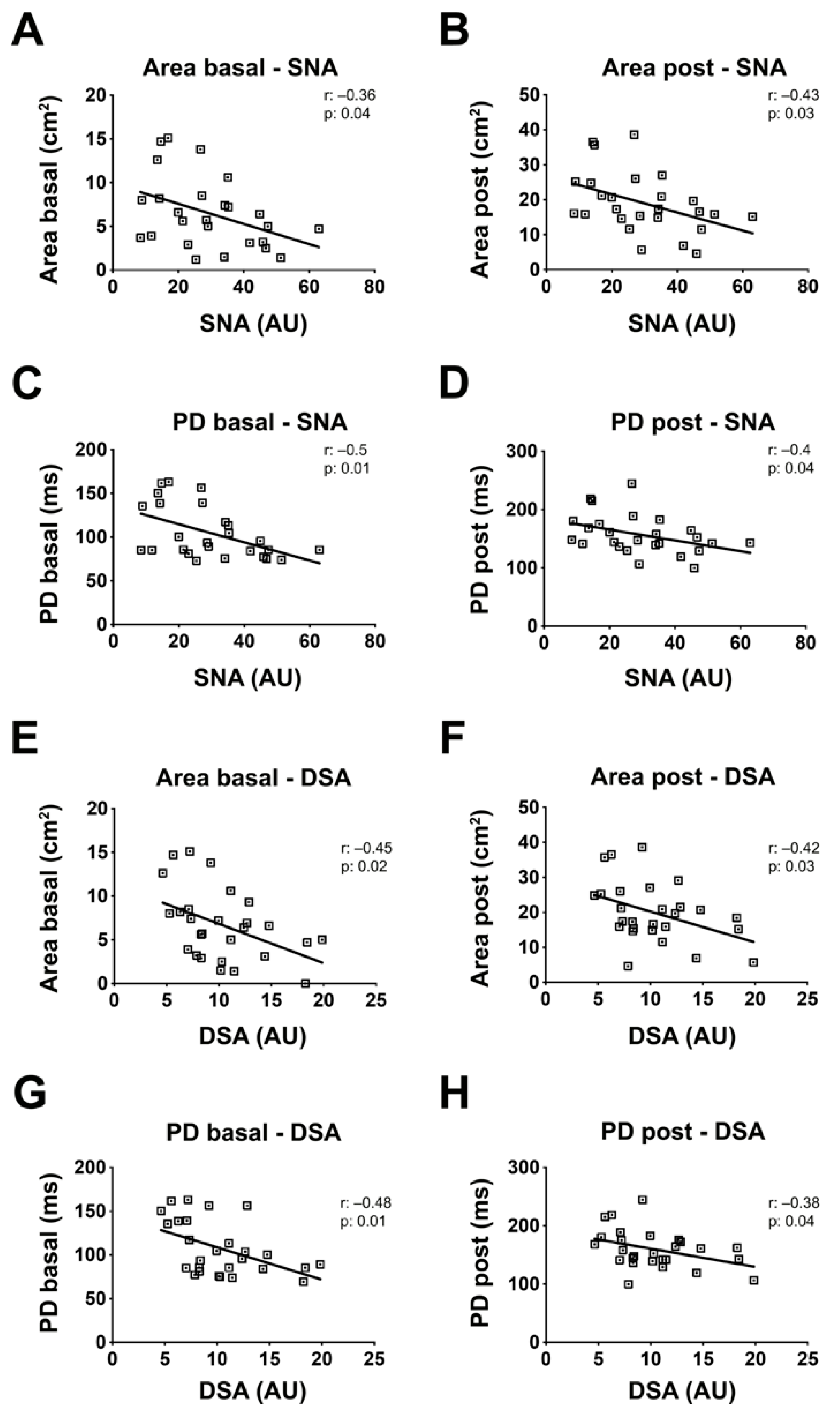
2.7. Sialylation of PBMC Proteins Are Associated with the BrS Phenotype Manifestation
2.8. Inward Sodium Current Is Reduced as a Result of Desialylation
3. Discussion
Study Limitations
4. Materials and Methods
4.1. Study Population
4.2. Electrophysiological Study and Arrhythmogenic Substrate Mapping
4.3. Plasma Isolation and Processing
4.4. Isolation and Processing of Peripheral Blood Mononuclear Cells
4.5. Flow Cytometry Analysis
4.6. Real-Time PCR
- -
- ST3Gal1 FW: 5′-CGGGAGCTGGGAGATAATGT-3′
- -
- ST3Gal1 Rev: 5′-TGATGAAGGCTGGGTGGTAG-3′
- -
- ST3Gal4 FW: 5′-TCCTGGTAGCTTTCAAGGCA-3′
- -
- ST3Gal4 Rev: 5′-CAGGCTCAGCAGTTTGTC-3′
- -
- ST6Gal2 FW: 5′-CCCCAGCCATCACTTCATTG-3′
- -
- ST6Gal2 Rev: 5′-TGGGTTTCTCTGACGATGCT-3′
- -
- Neu3 FW: 5′-TGGTCATCCCTGCGTATACC-3′
- -
- Neu3 Rev: 5′-TCACCTCTGCCACTTCACAT-3′
- -
- CMAS FW: 5′-CTGTTGTGAGACGCCATCAG-3′
- -
- CMAS Rev: 5′-CCACACTATGTTCAGCTCGC-3′
- -
- GNE FW: 5′-GTGGTACTTGGCTCTCACCT-3′
- -
- GNE Rev: 5′-ATCTGGCAGCTTCACTAGGG-3′
- -
- SLC17A5 FW: 5′-TGGAGGATATGTTGCCAGCA-3′
- -
- SLC17A5 Rev: 5′-GAGCCCAAGAAGACCACATG-3ì
- -
- HPRT FW: 5′-TATGGCGACCCGCAGCCCT-3′
- -
- HPRT Rev: 5′-CATCTCGAGCAAGACGTTCAG-3′
4.7. Western Blot
4.8. Cell Culture and Transfection
4.9. Sialic Acid Removal
4.10. Electrophysiological Measurements
4.11. Statistical Analysis
4.12. Study Approval
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Antzelevitch, C.; Brugada, P.; Borggrefe, M.; Brugada, J.; Brugada, R.; Corrado, D.; Gussak, I.; LeMarec, H.; Nademanee, K.; Riera, A.R.P.; et al. Brugada Syndrome: Report of the Second Consensus Conference: Endorsed by the Heart Rhythm Society and the European Heart Rhythm Association. Circulation 2005, 111, 659–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brugada, P.; Brugada, J. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: A distinct clinical and electrocardiographic syndrome: A multicenter report. J. Am. Coll. Cardiol. 1992, 20, 1391–1396. [Google Scholar] [CrossRef]
- Nademanee, K.; Raju, H.; de Noronha, S.V.; Papadakis, M.; Robinson, L.; Rothery, S.; Makita, N.; Kowase, S.; Boonmee, N.; Vitayakritsirikul, V.; et al. Fibrosis, Connexin-43, and Conduction Abnormalities in the Brugada Syndrome. J. Am. Coll. Cardiol. 2015, 66, 1976–1986. [Google Scholar] [CrossRef] [Green Version]
- Pappone, C.; Santinelli, V. Brugada Syndrome: Progress in Diagnosis and Management. Arrhythmia Electrophysiol. Rev. 2019, 8, 13–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quan, X.-Q.; Li, S.; Liu, R.; Zheng, K.; Wu, X.-F.; Tang, Q. A meta-analytic review of prevalence for Brugada ECG patterns and the risk for death. Medicine 2016, 95, e5643. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Kirsch, G.E.; Zhang, D.; Brugada, R.; Brugada, J.; Brugada, P.; Potenza, D.; Moya, A.; Borggrefe, M.; Breithardt, G.; et al. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature 1998, 392, 293–296. [Google Scholar] [CrossRef]
- Hofman, N.; Tan, H.L.; Alders, M.; Kolder, I.; de Haij, S.; Mannens, M.M.; Lombardi, M.P.; Deprez, R.H.L.D.; van Langen, I.; Wilde, A.A. Yield of Molecular and Clinical Testing for Arrhythmia Syndromes: Report of 15 years’ experience. Circulation 2013, 128, 1513–1521. [Google Scholar] [CrossRef] [Green Version]
- Kapplinger, J.D.; Tester, D.J.; Alders, M.; Benito, B.; Berthet, M.; Brugada, J.; Brugada, P.; Fressart, V.; Guerchicoff, A.; Harris-Kerr, C.; et al. An international compendium of mutations in the SCN5A-encoded cardiac sodium channel in patients referred for Brugada syndrome genetic testing. Heart Rhythm. 2010, 7, 33–46. [Google Scholar] [CrossRef] [Green Version]
- Herren, A.W.; Bers, D.M.; Grandi, E. Post-translational modifications of the cardiac Na channel: Contribution of CaMKII-dependent phosphorylation to acquired arrhythmias. Am. J. Physiol. Heart. Circ. Physiol. 2013, 305, H431–H445. [Google Scholar] [CrossRef] [Green Version]
- Ciconte, G.; Monasky, M.M.; Santinelli, V.; Micaglio, E.; Vicedomini, G.; Anastasia, L.; Negro, G.; Borrelli, V.; Giannelli, L.; Santini, F.; et al. Brugada syndrome genetics is associated with phenotype severity. Eur. Heart J. 2021, 42, 1082–1090. [Google Scholar] [CrossRef]
- Denham, N.C.; Pearman, C.M.; Ding, W.Y.; Waktare, J.; Gupta, D.; Snowdon, R.; Hall, M.; Cooper, R.; Modi, S.; Todd, D.; et al. Systematic re-evaluation of SCN5A variants associated with Brugada syndrome. J. Cardiovasc. Electrophysiol. 2019, 30, 118–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monasky, M.M.; Micaglio, E.; Ciconte, G.; Pappone, C. Brugada Syndrome: Oligogenic or Mendelian Disease? Int. J. Mol. Sci. 2020, 21, 1687. [Google Scholar] [CrossRef] [Green Version]
- Blok, M.; Boukens, B.J. Mechanisms of Arrhythmias in the Brugada Syndrome. Int. J. Mol. Sci. 2020, 21, 7051. [Google Scholar] [CrossRef] [PubMed]
- Behr, E.R.; Ben-Haim, Y.; Ackerman, M.J.; Krahn, A.D.; Wilde, A.A.M. Brugada syndrome and reduced right ventricular outflow tract conduction reserve: A final common pathway? Eur. Heart J. 2021, 42, 1073–1081. [Google Scholar] [CrossRef]
- Marionneau, C.; Abriel, H. Regulation of the cardiac Na+ channel NaV1.5 by post-translational modifications. J. Mol. Cell. Cardiol. 2015, 82, 36–47. [Google Scholar] [CrossRef] [PubMed]
- Reily, C.; Stewart, T.J.; Renfrow, M.B.; Novak, J. Glycosylation in health and disease. Nat. Rev. Nephrol. 2019, 15, 346–366. [Google Scholar] [CrossRef]
- Baycin-Hizal, D.; Gottschalk, A.; Jacobson, E.; Mai, S.; Wolozny, D.; Zhang, H.; Krag, S.S.; Betenbaugh, M.J. Physiologic and pathophysiologic consequences of altered sialylation and glycosylation on ion channel function. Biochem. Biophys. Res. Commun. 2014, 453, 243–253. [Google Scholar] [CrossRef] [Green Version]
- Montpetit, M.L.; Stocker, P.J.; Schwetz, T.A.; Harper, J.M.; Norring, S.A.; Schaffer, L.; North, S.J.; Jang-Lee, J.; Gilmartin, T.; Head, S.R.; et al. Regulated and aberrant glycosylation modulate cardiac electrical signaling. Proc. Natl. Acad. Sci. USA 2009, 106, 16517–16522. [Google Scholar] [CrossRef] [Green Version]
- Ednie, A.R.; Bennett, E.S. Modulation of Voltage-Gated Ion Channels by Sialylation. Compr. Physiol. 2012, 2, 1269–1301. [Google Scholar] [CrossRef]
- Bao, L.; Ding, L.; Yang, M.; Ju, H. Noninvasive imaging of sialyltransferase activity in living cells by chemoselective recognition. Sci. Rep. 2015, 5, 10947. [Google Scholar] [CrossRef]
- Ufret-Vincenty, C.A.; Baro, D.J.; Lederer, W.J.; Rockman, H.A.; Quiñones, L.E.; Santana, L. Role of Sodium Channel Deglycosylation in the Genesis of Cardiac Arrhythmias in Heart Failure. J. Biol. Chem. 2001, 276, 28197–28203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marques-Da-Silva, D.; Francisco, R.; Webster, D.; Ferreira, V.D.R.; Jaeken, J.; Pulinilkunnil, T. Cardiac complications of congenital disorders of glycosylation (CDG): A systematic review of the literature. J. Inherit. Metab. Dis. 2017, 40, 657–672. [Google Scholar] [CrossRef] [PubMed]
- Sparks, S.E.; Krasnewich, D.M. Congenital Disorders of N-Linked Glycosylation and Multiple Pathway Overview. In GeneReviews((R)); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; NCBI: Seattle, WA, USA, 1993. [Google Scholar]
- Cuda, G.; Di Domenico, M.; Scumaci, D.; Grasso, S.; Gaspari, M.; Curcio, A.; Oliva, A.; Ausania, F.; Di Nunzio, C.; Ricciardi, C.; et al. Biomarker discovery by plasma proteomics in familial Brugada Syndrome. Front. Biosci. 2013, 18, 564–571. [Google Scholar] [CrossRef] [Green Version]
- Scumaci, D.; Oliva, A.; Concolino, A.; Curcio, A.; Fiumara, C.V.; Tammè, L.; Campuzano, O.; Pascali, V.L.; Coll, M.; Iglesias, A.; et al. Integration of “Omics” Strategies for Biomarkers Discovery and for the Elucidation of Molecular Mechanisms Underlying Brugada Syndrome. Proteom. -Clin. Appl. 2018, 12, e1800065. [Google Scholar] [CrossRef] [PubMed]
- D’Imperio, S.; Monasky, M.M.; Micaglio, E.; Ciconte, G.; Anastasia, L.; Pappone, C. Brugada Syndrome: Warning of a Systemic Condition? Front. Cardiovasc. Med. 2021, 8, 771349. [Google Scholar] [CrossRef] [PubMed]
- Wallentin, L.; Eriksson, N.; Olszowka, M.; Grammer, T.B.; Hagström, E.; Held, C.; Kleber, M.E.; Koenig, W.; März, W.; Stewart, R.A.H.; et al. Plasma proteins associated with cardiovascular death in patients with chronic coronary heart disease: A retrospective study. PLoS Med. 2021, 18, e1003513. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Alroy, J.; Pereira, M.E. A neuraminidase from Trypanosoma cruzi removes sialic acid from the surface of mammalian myocardial and endothelial cells. J. Clin. Investig. 1986, 77, 127–135. [Google Scholar] [CrossRef]
- Deng, W.; Ednie, A.R.; Qi, J.; Bennett, E.S. Aberrant sialylation causes dilated cardiomyopathy and stress-induced heart failure. Basic Res. Cardiol. 2016, 111, 57. [Google Scholar] [CrossRef] [Green Version]
- Rong, J.; Han, J.; Dong, L.; Tan, Y.; Yang, H.; Feng, L.; Wang, Q.-W.; Meng, R.; Zhao, J.; Wang, S.-Q.; et al. Glycan Imaging in Intact Rat Hearts and Glycoproteomic Analysis Reveal the Upregulation of Sialylation during Cardiac Hypertrophy. J. Am. Chem. Soc. 2014, 136, 17468–17476. [Google Scholar] [CrossRef]
- Zhang, D.; Zhu, L.; Li, C.; Mu, J.; Fu, Y.; Zhu, Q.; Zhou, Z.; Liu, P.; Han, C. Sialyltransferase7A, a Klf4-responsive gene, promotes cardiomyocyte apoptosis during myocardial infarction. Basic Res. Cardiol. 2015, 110, 28. [Google Scholar] [CrossRef]
- Suzuki, O.; Kanai, T.; Nishikawa, T.; Yamamoto, Y.; Noguchi, A.; Takimoto, K.; Koura, M.; Noguchi, Y.; Uchio-Yamada, K.; Tsuji, S.; et al. Adult onset cardiac dilatation in a transgenic mouse line with Gal.BETA.1,3GalNAc. ALPHA.2,3-sialyltransferase II (ST3Gal-II) transgenes: A new model for dilated cardiomyopathy. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2011, 87, 550–562. [Google Scholar] [CrossRef] [Green Version]
- Ednie, A.R.; Horton, K.-K.; Wu, J.; Bennett, E.S. Expression of the sialyltransferase, ST3Gal4, impacts cardiac voltage-gated sodium channel activity, refractory period and ventricular conduction. J. Mol. Cell. Cardiol. 2013, 59, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Hosseini, S.M.; Kim, R.; Udupa, S.; Costain, G.; Jobling, R.; Liston, E.; Jamal, S.M.; Szybowska, M.; Morel, C.F.; Bowdin, S.; et al. Reappraisal of Reported Genes for Sudden Arrhythmic Death. Circulation 2018, 138, 1195–1205. [Google Scholar] [CrossRef] [PubMed]
- Bezzina, C.R.; Barc, J.; Mizusawa, Y.; Remme, C.A.; Gourraud, J.-B.; Simonet, F.; Verkerk, A.O.; Schwartz, P.J.; Crotti, L.; Dagradi, F.; et al. Common variants at SCN5A-SCN10A and HEY2 are associated with Brugada syndrome, a rare disease with high risk of sudden cardiac death. Nat. Genet. 2013, 45, 1044–1049. [Google Scholar] [CrossRef] [PubMed]
- Cortada, E.; Brugada, R.; Verges, M. N-Glycosylation of the voltage-gated sodium channel β2 subunit is required for efficient trafficking of NaV1.5/β2 to the plasma membrane. J. Biol. Chem. 2019, 294, 16123–16140. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.; Montpetit, M.L.; Stocker, P.J.; Bennett, E.S. The Sialic Acid Component of the β1 Subunit Modulates Voltage-gated Sodium Channel Function. J. Biol. Chem. 2004, 279, 44303–44310. [Google Scholar] [CrossRef] [Green Version]
- Sendfeld, F.; Selga, E.; Scornik, F.S.; Pérez, G.J.; Mills, N.L.; Brugada, R. Experimental Models of Brugada syndrome. Int. J. Mol. Sci. 2019, 20, 2123. [Google Scholar] [CrossRef] [Green Version]
- Detta, N.; Frisso, G.; Salvatore, F. The multi-faceted aspects of the complex cardiac Nav1.5 protein in membrane function and pathophysiology. Biochim. Biophys. Acta (BBA)—Proteins Proteom. 2015, 1854, 1502–1509. [Google Scholar] [CrossRef] [Green Version]
- Liang, P.; Sallam, K.; Wu, H.; Li, Y.; Itzhaki, I.; Garg, P.; Zhang, Y.; Termglichan, V.; Lan, F.; Gu, M.; et al. Patient-Specific and Genome-Edited Induced Pluripotent Stem Cell–Derived Cardiomyocytes Elucidate Single-Cell Phenotype of Brugada Syndrome. J. Am. Coll. Cardiol. 2016, 68, 2086–2096. [Google Scholar] [CrossRef]
- Kelly, M.I.; Albahrani, M.; Castro, C.; Poon, E.; Yan, B.; Littrell, J.; Waas, M.; Boheler, K.R.; Gundry, R.L. Importance of evaluating protein glycosylation in pluripotent stem cell-derived cardiomyocytes for research and clinical applications. Pflugers Arch. 2021, 473, 1041–1059. [Google Scholar] [CrossRef]
- Dias, T.P.; Pinto, S.N.; Santos, J.I.; Fernandes, T.G.; Fernandes, F.; Diogo, M.M.; Prieto, M.; Cabral, J.M. Biophysical study of human induced Pluripotent Stem Cell-Derived cardiomyocyte structural maturation during long-term culture. Biochem. Biophys. Res. Commun. 2018, 499, 611–617. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wang, L.; Liu, Z.; Alimohamadi, S.; Yin, C.; Liu, J.; Qian, L. Comparative Gene Expression Analyses Reveal Distinct Molecular Signatures between Differentially Reprogrammed Cardiomyocytes. Cell Rep. 2017, 20, 3014–3024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campostrini, G.; Kosmidis, G.; Ward-van Oostwaard, D.; Davis, R.P.; Yiangou, L.; Ottaviani, D.; Veerman, C.C.; Mei, H.; Orlova, V.V.; Wilde, A.A.M.; et al. Maturation of hiPSC-derived cardiomyocytes promotes adult alternative splicing of SCN5A and reveals changes in sodium current associated with cardiac arrhythmia. Cardiovasc Res 2022, cvac059. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.C.; Harmer, S.C.; Poliandri, A.; Nobles, M.; Edwards, E.C.; Ware, J.S.; Sharp, T.V.; McKay, T.R.; Dunkel, L.; Lambiase, P.D.; et al. Ajmaline blocks I Na and I Kr without eliciting differences between Brugada syndrome patient and control human pluripotent stem cell-derived cardiac clusters. Stem Cell Res. 2017, 25, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Veerman, C.C.; Mengarelli, I.; Guan, K.; Stauske, M.; Barc, J.; Tan, H.L.; Wilde, A.A.M.; Verkerk, A.O.; Bezzina, C.R. hiPSC-derived cardiomyocytes from Brugada Syndrome patients without identified mutations do not exhibit clear cellular electrophysiological abnormalities. Sci. Rep. 2016, 6, 30967. [Google Scholar] [CrossRef] [Green Version]
- Kawada, S.; Morita, H.; Antzelevitch, C.; Morimoto, Y.; Nakagawa, K.; Watanabe, A.; Nishii, N.; Nakamura, K.; Ito, H. Shanghai Score System for Diagnosis of Brugada Syndrome: Validation of the Score System and System and Reclassification of the Patients. JACC Clin. Electrophysiol. 2018, 4, 724–730. [Google Scholar] [CrossRef]
- Antzelevitch, C.; Yan, G.-X.; Ackerman, M.J.; Borggrefe, M.; Corrado, D.; Guo, J.; Gussak, I.; Hasdemir, C.; Horie, M.; Huikuri, H.; et al. J-Wave syndromes expert consensus conference report: Emerging concepts and gaps in knowledge. Europace 2017, 19, 665–694. [Google Scholar] [CrossRef]
- Monasky, M.; Micaglio, E.; Ciconte, G.; Rivolta, I.; Borrelli, V.; Ghiroldi, A.; D’Imperio, S.; Binda, A.; Melgari, D.; Benedetti, S.; et al. Novel SCN5A p.Val1667Asp Missense Variant Segregation and Characterization in a Family with Severe Brugada Syndrome and Multiple Sudden Deaths. Int. J. Mol. Sci. 2021, 22, 4700. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study Sample (n = 84) | Patient Group (n = 42) | Control Group (n = 42) | p-Value § | |
|---|---|---|---|---|
| Male, n (%) | 62 (73.8) | 35 (83.3) | 27 (64.3) | 0.287 |
| Age (years) (mean +/− SD) | 40.2 ± 15.5 | 38.6 ± 12.4 | 40.5 ± 15.3 | 0.193 |
| Spontaneous type 1 pattern, n (%) | 19 (22.6) | 19 (45.2) | 0 | <0.001 |
| Fever induced type 1 pattern, n (%) | 5 (6.0) | 5 (12.0) | 0 | 0.113 |
| SCA, n (%) | 9 (10.7) | 9 (21.4) | 0 | <0.001 |
| Spontaneous VT/VF requiring ICD therapy, n (%) | 18 (21.4) | 18 (42.8) | 0 | <0.001 |
| Nocturnal agonal respirations, n (%) | 4 (4.7) | 4 (9.4) | 0 | 0.113 |
| Suspected arrhythmic syncope, n (%) | 10 (11.9) | 7 (16.6) | 3 (7.1) | 0.708 |
| Unclear syncope, n (%) | 16 (19.0) | 9 (21.4) | 7(16.6) | 0.556 |
| AF/Flutter, n (%) | 16 (19.0) | 11 (26.2) | 5 (16.1) | 0.365 |
| Family history of BrS, n (%) | 30 (35.7) | 20 (47.6) | 10 (23.8) | <0.001 |
| Family history of SD, n (%) | 37 (44.0) | 23 (54.7) | 10 (23.8) | <0.001 |
| Shanghai Score, n (%) | ||||
| Score < 3 | 35 (41.6) | 0 | 35 (83.2) | <0.001 |
| Score < 4 | 7 (8.3) | 5 (11.2) | 2 (4.7) | 1.000 |
| Score < 5 | 8 (9.5) | 4 (9.5) | 4 (9.5) | 0.053 |
| Score < 6 | 5 (5.9) | 4 (9.5) | 1 (2.3) | 1.000 |
| Score < 7 | 6 (7.1) | 6 (14.2) | 0 | 0.001 |
| Score ≥ 7 | 23 (27.4) | 23 (54.8) | 0 | <0.001 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghiroldi, A.; Ciconte, G.; Creo, P.; Tarantino, A.; Melgari, D.; D’Imperio, S.; Piccoli, M.; Cirillo, F.; Micaglio, E.; Monasky, M.M.; et al. Alterations of the Sialylation Machinery in Brugada Syndrome. Int. J. Mol. Sci. 2022, 23, 13154. https://doi.org/10.3390/ijms232113154
Ghiroldi A, Ciconte G, Creo P, Tarantino A, Melgari D, D’Imperio S, Piccoli M, Cirillo F, Micaglio E, Monasky MM, et al. Alterations of the Sialylation Machinery in Brugada Syndrome. International Journal of Molecular Sciences. 2022; 23(21):13154. https://doi.org/10.3390/ijms232113154
Chicago/Turabian StyleGhiroldi, Andrea, Giuseppe Ciconte, Pasquale Creo, Adriana Tarantino, Dario Melgari, Sara D’Imperio, Marco Piccoli, Federica Cirillo, Emanuele Micaglio, Michelle M. Monasky, and et al. 2022. "Alterations of the Sialylation Machinery in Brugada Syndrome" International Journal of Molecular Sciences 23, no. 21: 13154. https://doi.org/10.3390/ijms232113154