
Regulation of Airway Smooth Muscle Cell Proliferation by Diacylglycerol Kinase: Relevance to Airway Remodeling in Asthma
 , , ,
, , ,
Abstract
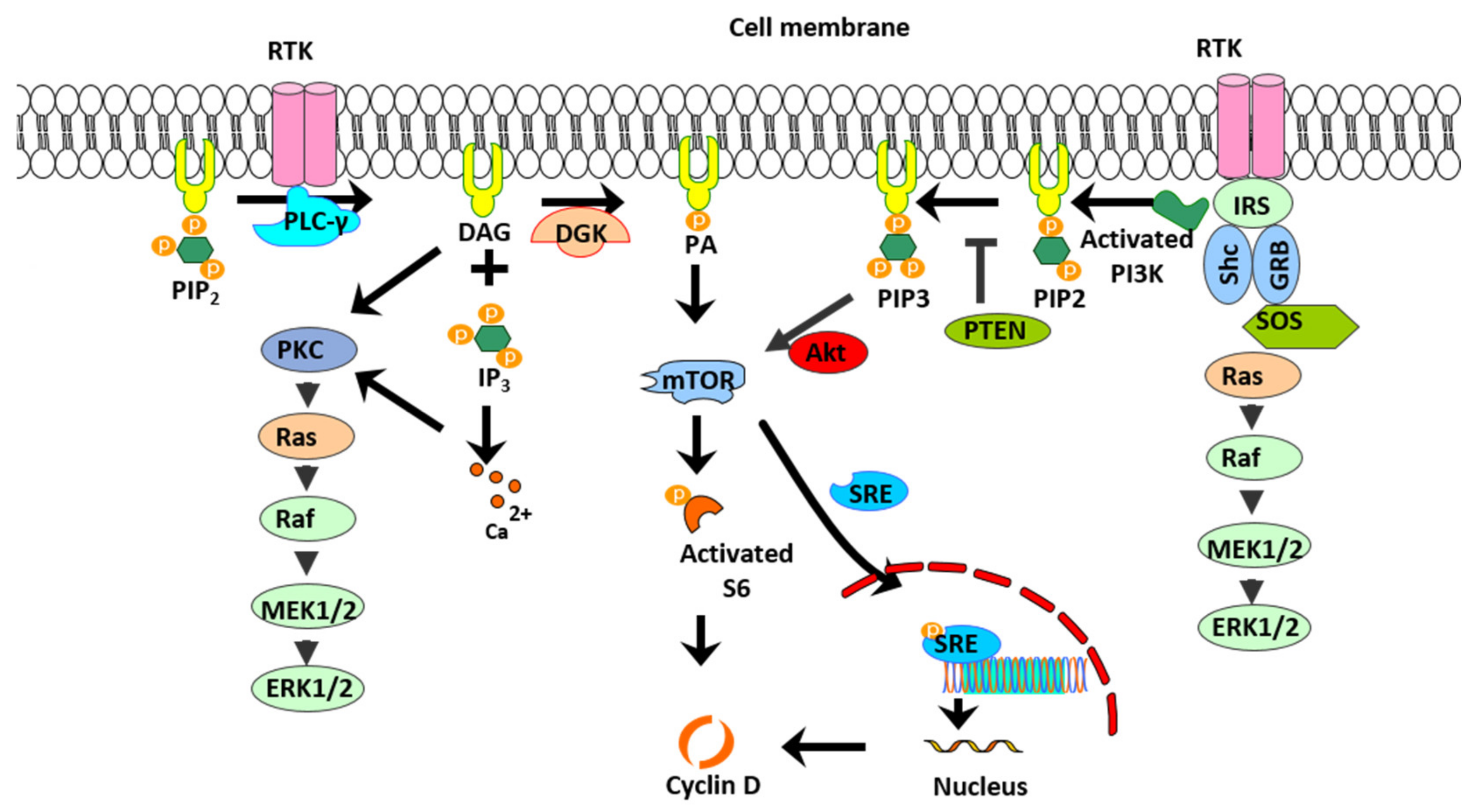
:1. Introduction
2. Results
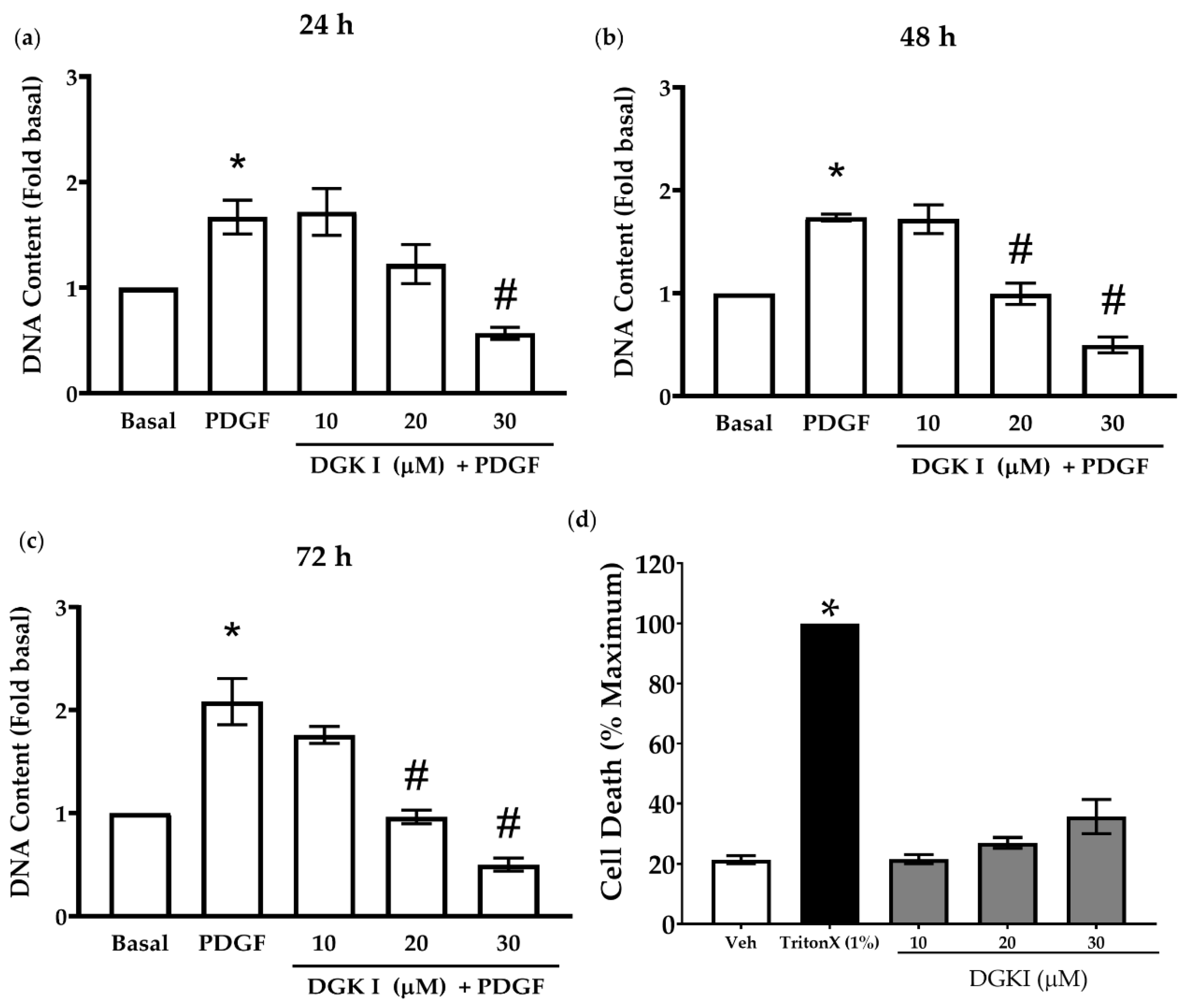
2.1. DGK Inhibition Reduces Human ASM Cell Proliferation
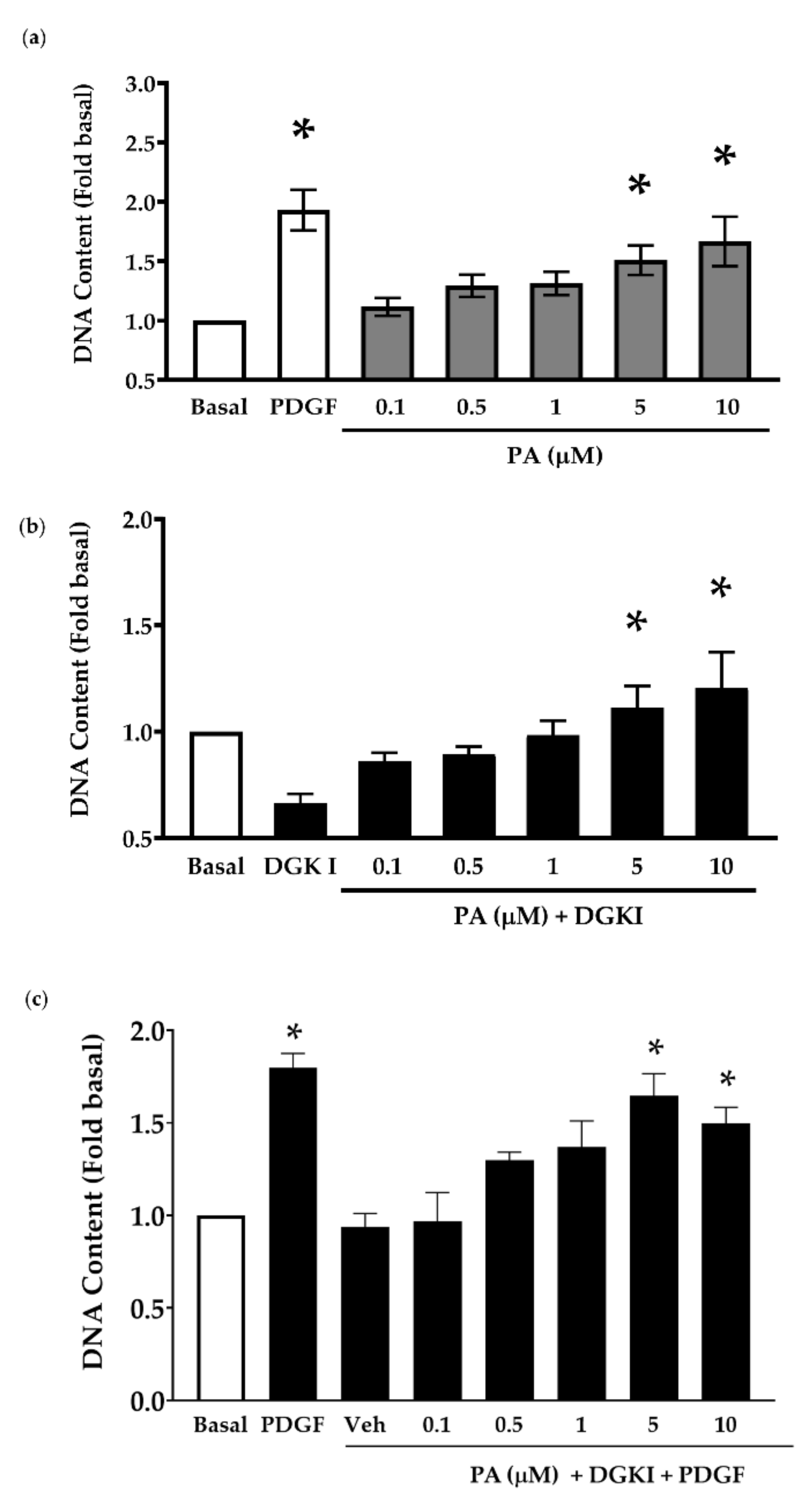
2.2. Exogenous PA Promotes Human ASM Cell Proliferation and Reverses the Anti-Mitogenic Effect of DGK Inhibition
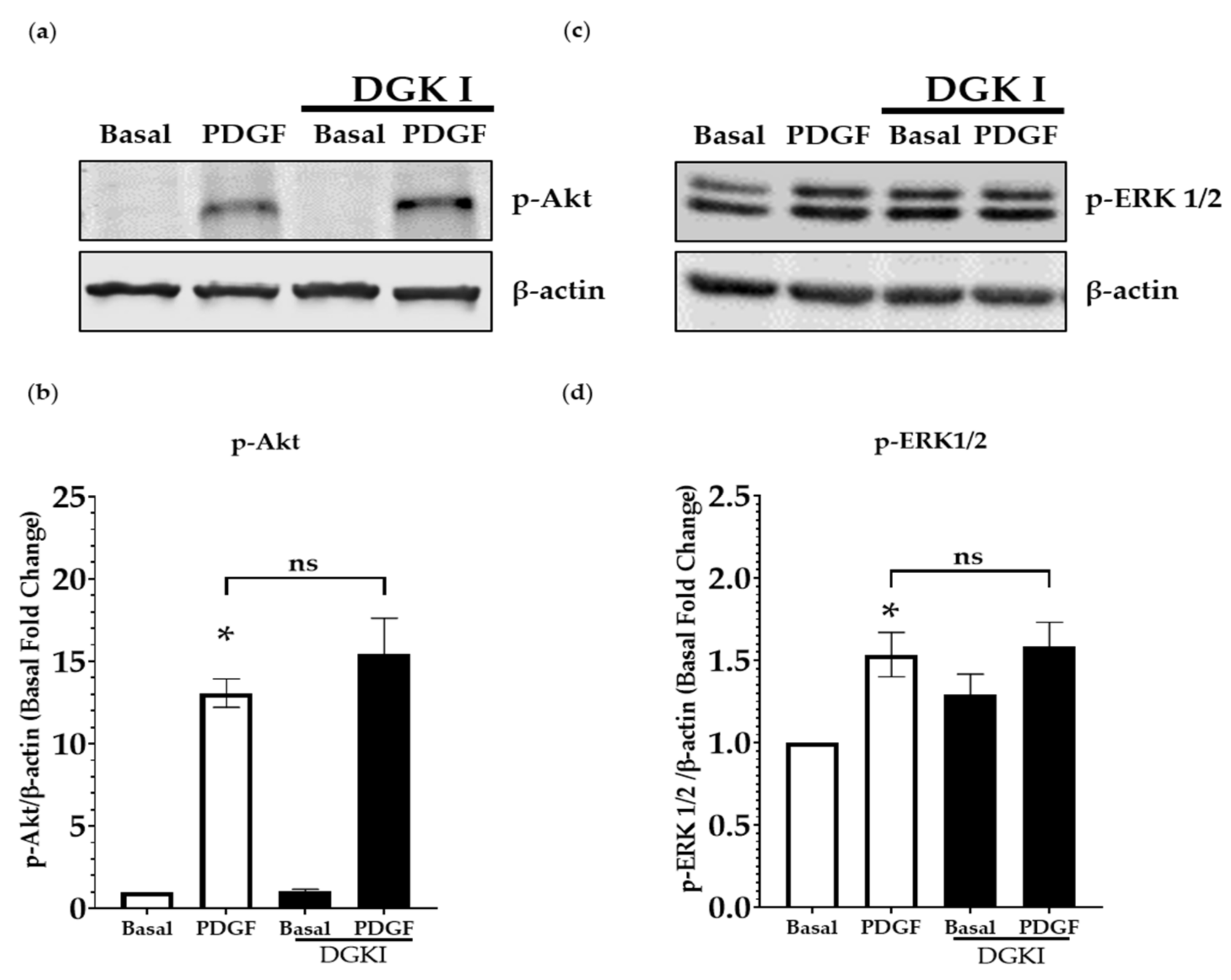
2.3. DGK Inhibition Does Not Influence PDGF-Induced MAP Kinase and PI3 Kinase Signaling
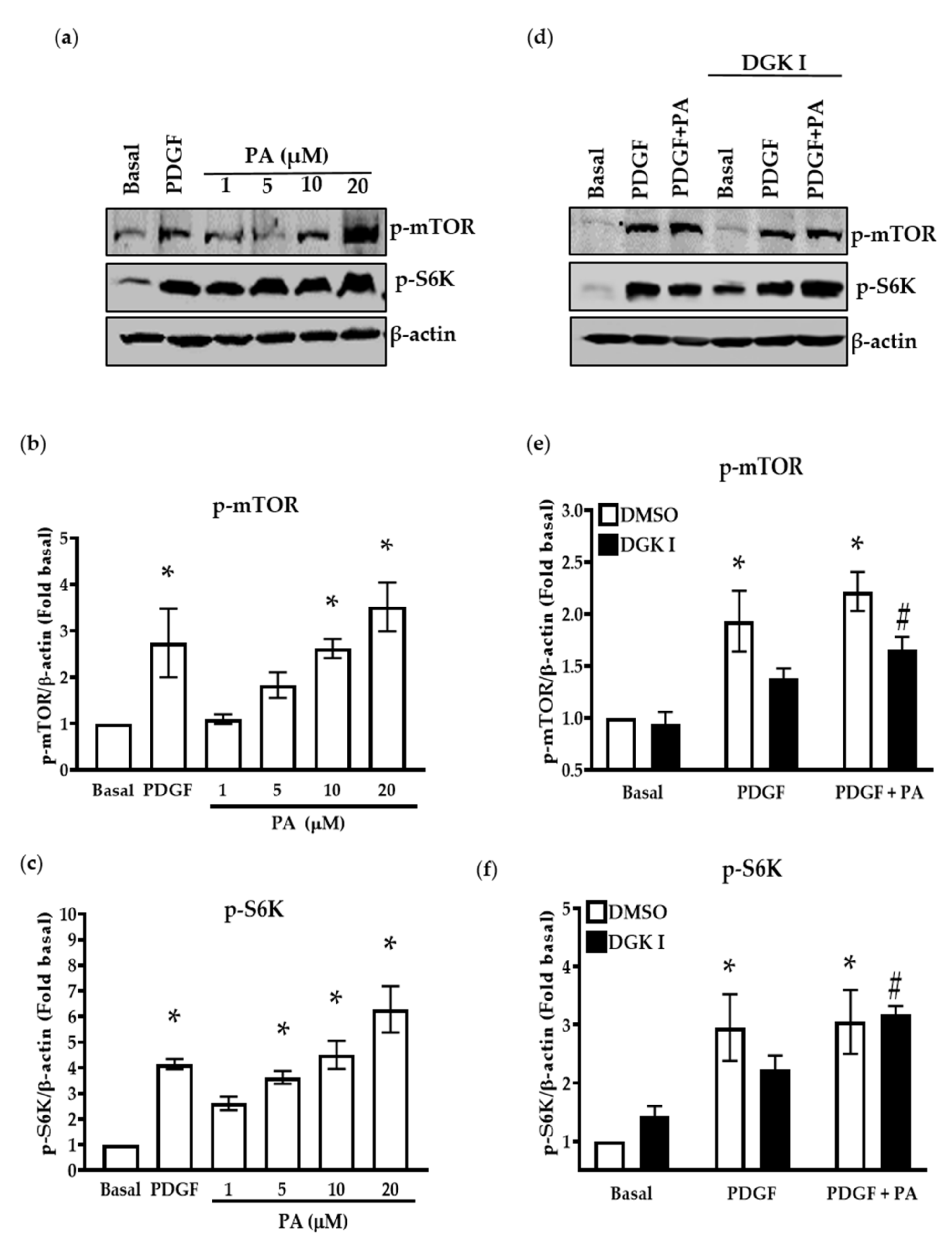
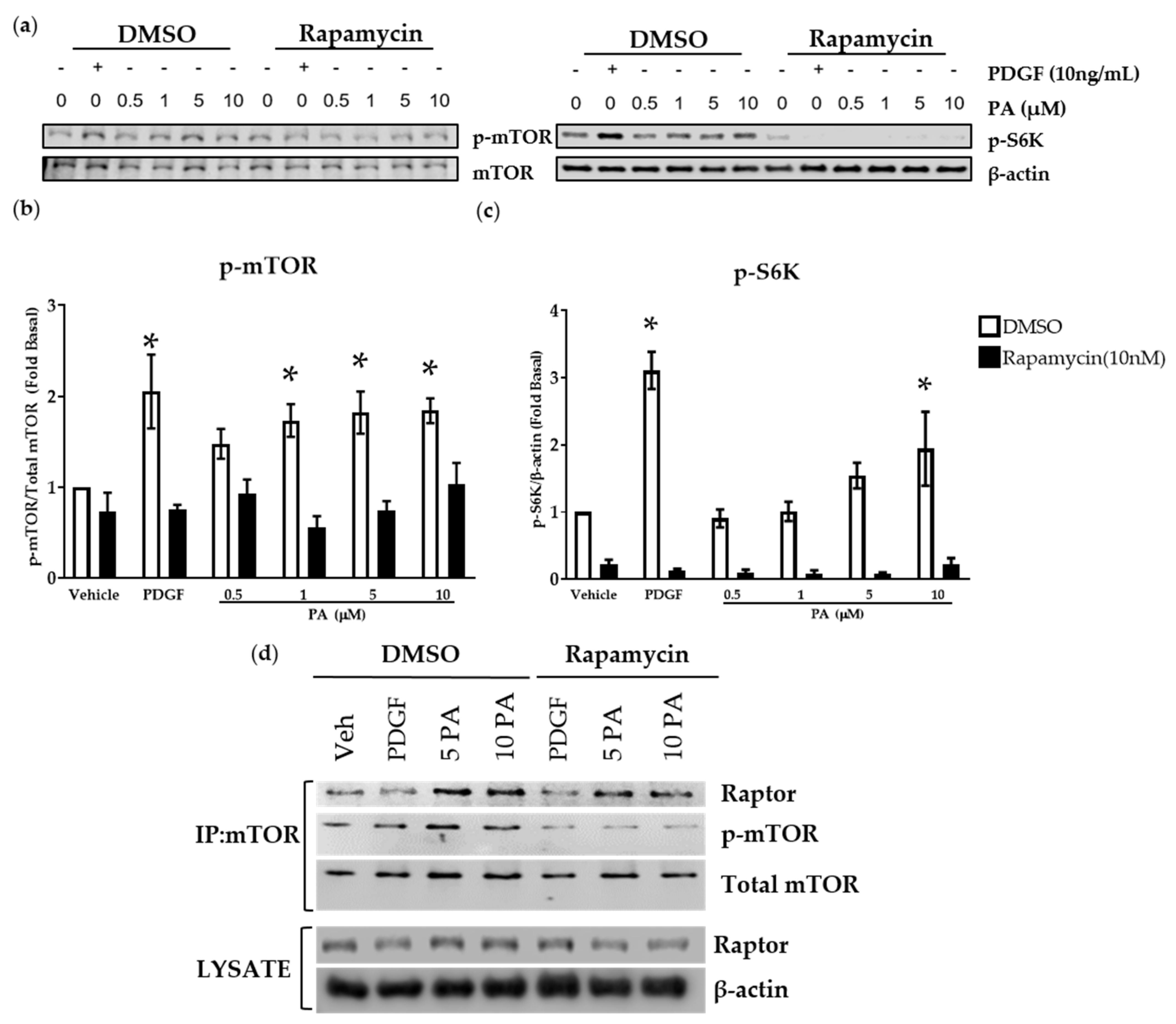
2.4. DGK Inhibition and Exogenous PA Modulate mTOR Signaling
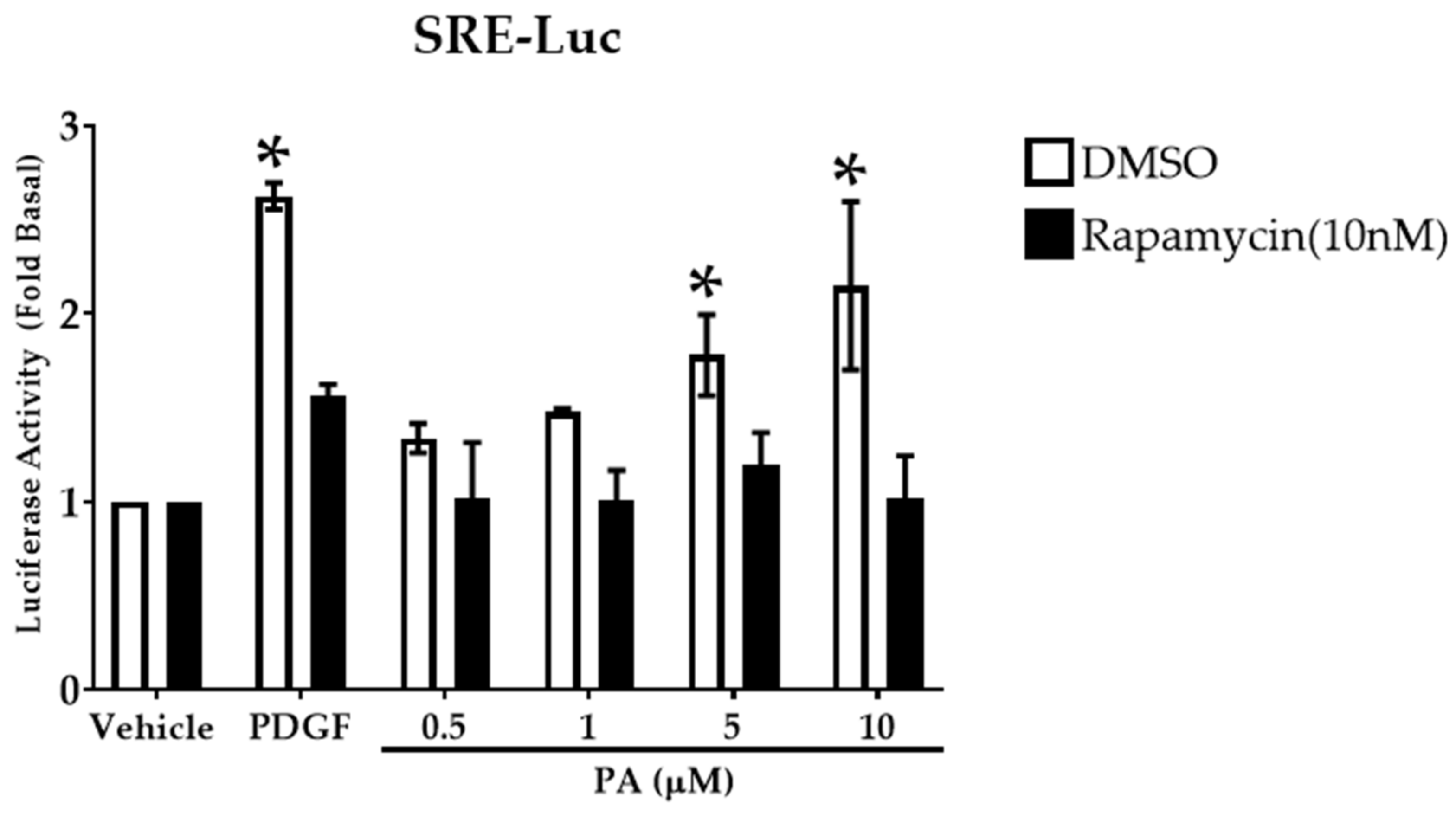
2.5. PA Activates Transcription Factor Serum Response Element
2.6. DGK Inhibition and PA Modulate the Expression of Pro-Mitogenic Genes
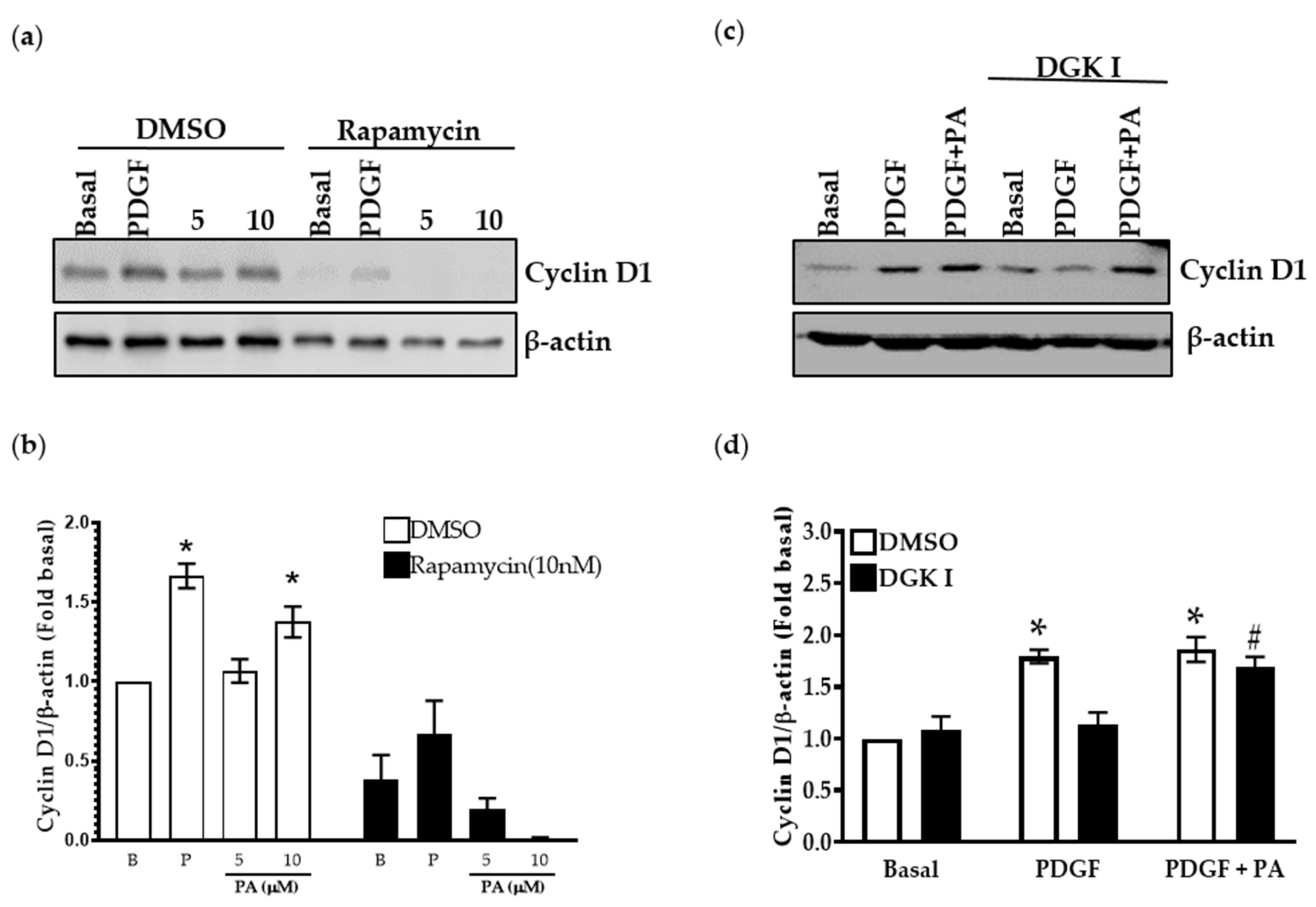
2.7. PA Increases Cyclin D1 Levels Attenuated by DGK Inhibition
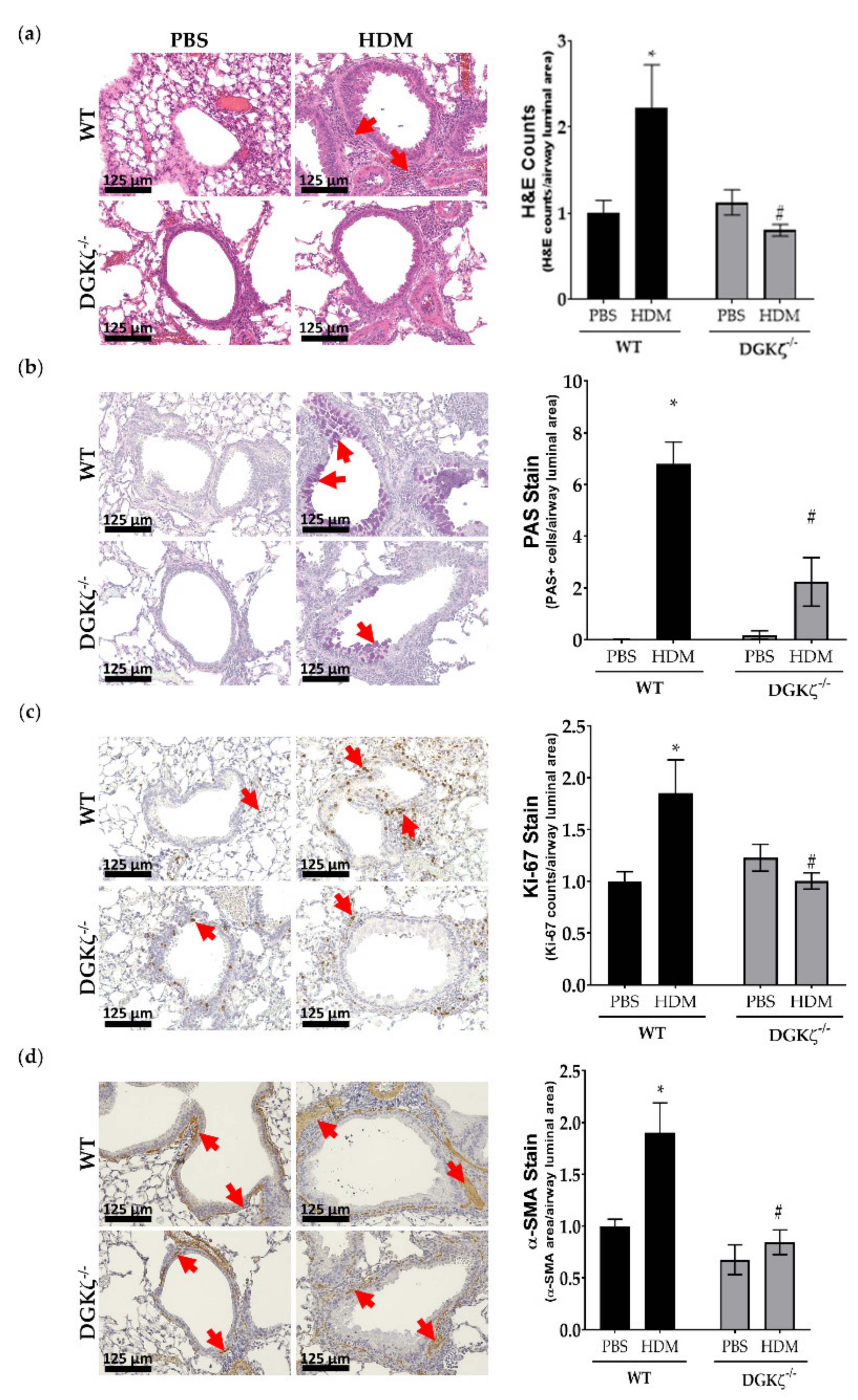
2.8. DGKζ KO Mice Exhibit Reduced Expression of Airway Remodeling Markers upon HDM Challenge
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. Viral Transduction of Human ASM Cells
4.4. Luciferase (Luc) Reporter Assay
4.5. CyQuantCell Proliferation and MTT Assay
4.6. Western Blotting
4.7. Co-Immunoprecipitation
4.8. RNA Isolation and Real-Time PCR Array
4.9. Murine Model of Allergen-Induced Asthma
4.10. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dekkers, B.G.J.; Maarsingh, H.; Meurs, H.; Gosens, R. Airway Structural Components Drive Airway Smooth Muscle Remodeling in Asthma. Proc. Am. Thorac. Soc. 2009, 6, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Prakash, Y.S. Airway smooth muscle in airway reactivity and remodeling: What have we learned? Am. J. Physiol. Cell. Mol. Physiol. 2013, 305, L912–L933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nayak, A.P.; Deshpande, D.A.; Penn, R.B. New targets for resolution of airway remodeling in obstructive lung diseases. F1000Research 2018, 7, 680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benayoun, L.; Druilhe, A.; Dombret, M.-C.; Aubier, M.; Pretolani, M. Airway Structural Alterations Selectively Associated with Severe Asthma. Am. J. Respir. Crit. Care Med. 2003, 167, 1360–1368. [Google Scholar] [CrossRef]
- Halwani, R.; Al-Muhsen, S.; Hamid, Q. Airway remodeling in asthma. Curr. Opin. Pharmacol. 2010, 10, 236–245. [Google Scholar] [CrossRef]
- Hassan, M.; Jo, T.; Risse, P.-A.; Tolloczko, B.; Lemière, C.; Olivenstein, R.; Hamid, Q.; Martin, J.G. Airway smooth muscle remodeling is a dynamic process in severe long-standing asthma. J. Allergy Clin. Immunol. 2010, 125, 1037–1045.e3. [Google Scholar] [CrossRef]
- Orsini, M.J.; Krymskaya, V.P.; Eszterhas, A.J.; Benovic, J.L.; Panettieri, R.A., Jr.; Penn, R.B. MAPK superfamily activation in human airway smooth muscle: Mitogenesis requires prolonged p42/p44 activation. Am. J. Physiol. Content 1999, 277, L479–L488. [Google Scholar] [CrossRef]
- Billington, C.K.; Kong, K.C.; Bhattacharyya, R.; Wedegaertner, P.B.; Panettieri, R.A.; Chan, T.O.; Penn, R.B. Cooperative Regulation of p70S6 Kinase by Receptor Tyrosine Kinases and G Protein-Coupled Receptors Augments Airway Smooth Muscle Growth. Biochemistry 2005, 44, 14595–14605. [Google Scholar] [CrossRef]
- Krymskaya, V.P.; Orsini, M.J.; Eszterhas, A.J.; Brodbeck, K.C.; Benovic, J.L.; Panettieri, R.A.; Penn, R.B. Mechanisms of Proliferation Synergy by Receptor Tyrosine Kinase and G Protein–Coupled Receptor Activation in Human Airway Smooth Muscle. Am. J. Respir. Cell Mol. Biol. 2000, 23, 546–554. [Google Scholar] [CrossRef]
- Johnson, P.R.A.; Roth, M.; Tamm, M.; Hughes, M.; Ge, Q.; King, G.; Burgess, J.K.; Black, J.L. Airway Smooth Muscle Cell Proliferation Is Increased in Asthma. Am. J. Respir. Crit. Care Med. 2001, 164, 474–477. [Google Scholar] [CrossRef]
- Black, J.L.; Roth, M.; Lee, J.; Carlin, S.; Johnson, P.R.A. Mechanisms of Airway Remodeling. Am. J. Respir. Crit. Care Med. 2001, 164, S63–S66. [Google Scholar] [CrossRef]
- Gosens, R.; Stelmack, G.L.; Dueck, G.; McNeill, K.D.; Yamasaki, A.; Gerthoffer, W.T.; Unruh, H.; Gounni, A.S.; Zaagsma, J.; Halayko, A.J. Role of caveolin-1 in p42/p44 MAP kinase activation and proliferation of human airway smooth muscle. Am. J. Physiol. Cell. Mol. Physiol. 2006, 291, L523–L534. [Google Scholar] [CrossRef]
- Hershenson, M.B.; Abe, M.K. Mitogen-Activated Signaling in Airway Smooth Muscle. Am. J. Respir. Cell Mol. Biol. 1999, 21, 651–654. [Google Scholar] [CrossRef]
- Hershenson, M.B.; Naureckas, E.T.; Li, J. Mitogen-activated signaling in cultured airway smooth muscle cells. Can. J. Physiol. Pharmacol. 1997, 75, 898–910. [Google Scholar] [CrossRef]
- Krymskaya, V.P.; Penn, R.B.; Orsini, M.J.; Scott, P.H.; Plevin, R.J.; Walker, T.R.; Eszterhas, A.J.; Amrani, Y.; Chilvers, E.R.; Panettieri, R.A., Jr. Phosphatidylinositol 3-kinase mediates mitogen-induced human airway smooth muscle cell proliferation. Am. J. Physiol. Cell. Mol. Physiol. 1999, 277, L65–L78. [Google Scholar] [CrossRef]
- Defnet, A.E.; Huang, W.; Polischak, S.; Yadav, S.K.; Kane, M.A.; Shapiro, P.; Deshpande, D.A. Effects of ATP-competitive and function-selective ERK inhibitors on airway smooth muscle cell proliferation. FASEB J. 2019, 33, 10833–10843. [Google Scholar] [CrossRef]
- Deshpande, D.A.; Penn, R.B. Targeting G protein-coupled receptor signaling in asthma. Cell. Signal. 2006, 18, 2105–2120. [Google Scholar] [CrossRef]
- Du, C.-L.; Xu, Y.-J.; Liu, X.-S.; Xie, J.-G.; Xie, M.; Zhang, Z.-X.; Zhang, J.; Qiao, L.-F. Up-regulation of cyclin D1 expression in asthma serum-sensitized human airway smooth muscle promotes proliferation via protein kinase Cα. Exp. Lung Res. 2010, 36, 201–210. [Google Scholar] [CrossRef]
- Shulga, Y.V.; Topham, M.K.; Epand, R.M. Regulation and Functions of Diacylglycerol Kinases. Chem. Rev. 2011, 111, 6186–6208. [Google Scholar] [CrossRef]
- Singh, B.K.; Lu, W.; Paustian, A.M.S.; Ge, M.Q.; Koziol-White, C.J.; Flayer, C.H.; Killingbeck, S.S.; Wang, N.; Dong, X.; Riese, M.J.; et al. Diacylglycerol kinase ζ promotes allergic airway inflammation and airway hyperresponsiveness through distinct mechanisms. Sci. Signal. 2019, 12, eaax3332. [Google Scholar] [CrossRef]
- Yadav, S.K.; Sharma, P.; Shah, S.D.; Panettieri, R.A., Jr.; Kambayashi, T.; Penn, R.B.; Deshpande, D.A. Autocrine regulation of airway smooth muscle contraction by diacylglycerol kinase. J. Cell. Physiol. 2021, 237, 603–616. [Google Scholar] [CrossRef]
- Sharma, P.; Yadav, S.K.; Shah, S.D.; Javed, E.; Lim, J.M.; Pan, S.; Nayak, A.P.; Panettieri, J.R.A.; Penn, R.B.; Kambayashi, T.; et al. Diacylglycerol Kinase Inhibition Reduces Airway Contraction by Negative Feedback Regulation of Gq-Signaling. Am. J. Respir. Cell Mol. Biol. 2021, 65, 658–671. [Google Scholar] [CrossRef]
- Baldanzi, G.; Bettio, V.; Malacarne, V.; Graziani, A. Diacylglycerol Kinases: Shaping Diacylglycerol and Phosphatidic Acid Gradients to Control Cell Polarity. Front. Cell Dev. Biol. 2016, 4, 140. [Google Scholar] [CrossRef] [Green Version]
- Reeves, H.L.; Thompson, M.G.; Dack, C.L.; Burt, A.D.; Day, C.P. The role of phosphatidic acid in platelet-derived growth factor-induced proliferation of rat hepatic stellate cells. Hepatology 2000, 31, 95–100. [Google Scholar] [CrossRef]
- Ha, K.S.; Exton, J.H. Activation of actin polymerization by phosphatidic acid derived from phosphatidylcholine in IIC9 fibroblasts. J. Cell Biol. 1993, 123, 1789–1796. [Google Scholar] [CrossRef]
- Tanguy, E.; Wang, Q.; Moine, H.; Vitale, N. Phosphatidic Acid: From Pleiotropic Functions to Neuronal Pathology. Front. Cell. Neurosci. 2019, 13, 2. [Google Scholar] [CrossRef] [Green Version]
- Fang, Y.; Vilella-Bach, M.; Bachmann, R.; Flanigan, A.; Chen, J. Phosphatidic Acid-Mediated Mitogenic Activation of mTOR Signaling. Science 2001, 294, 1942–1945. [Google Scholar] [CrossRef] [PubMed]
- Yoon, M.-S.; Sun, Y.; Arauz, E.; Jiang, Y.; Chen, J. Phosphatidic Acid Activates Mammalian Target of Rapamycin Complex 1 (mTORC1) Kinase by Displacing FK506 Binding Protein 38 (FKBP38) and Exerting an Allosteric Effect. J. Biol. Chem. 2011, 286, 29568–29574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ávila-Flores, A.; Santos, T.; Rincón, E.; Mérida, I. Modulation of the Mammalian Target of Rapamycin Pathway by Diacylglycerol Kinase-produced Phosphatidic Acid. J. Biol. Chem. 2005, 280, 10091–10099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakai, H.; Murakami, C.; Usuki, T.; Lu, Q.; Matsumoto, K.-I.; Urano, T.; Sakane, F. Diacylglycerol kinase η regulates C2C12 myoblast proliferation through the mTOR signaling pathway. Biochimie 2020, 177, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Joshi, R.P.; Schmidt, A.M.; Das, J.; Pytel, D.; Riese, M.J.; Lester, M.; Diehl, J.A.; Behrens, E.M.; Kambayashi, T.; Koretzky, G.A. The ζ Isoform of Diacylglycerol Kinase Plays a Predominant Role in Regulatory T Cell Development and TCR-Mediated Ras Signaling. Sci. Signal. 2013, 6, ra102. [Google Scholar] [CrossRef] [Green Version]
- Zhong, X.-P.; Hainey, E.A.; Olenchock, B.A.; Zhao, H.; Topham, M.K.; Koretzky, G.A. Regulation of T Cell Receptor-induced Activation of the Ras-ERK Pathway by Diacylglycerol Kinase ζ. J. Biol. Chem. 2002, 277, 31089–31098. [Google Scholar] [CrossRef] [Green Version]
- Fingar, D.C.; Richardson, C.J.; Tee, A.R.; Cheatham, L.; Tsou, C.; Blenis, J. mTOR Controls Cell Cycle Progression through Its Cell Growth Effectors S6K1 and 4E-BP1/Eukaryotic Translation Initiation Factor 4E. Mol. Cell. Biol. 2004, 24, 200–216. [Google Scholar] [CrossRef] [Green Version]
- Toschi, A.; Lee, E.; Xu, L.; Garcia, A.; Gadir, N.; Foster, D.A. Regulation of mTORC1 and mTORC2 Complex Assembly by Phosphatidic Acid: Competition with Rapamycin. Mol. Cell. Biol. 2009, 29, 1411–1420. [Google Scholar] [CrossRef] [Green Version]
- Sharma, P.; Panebra, A.; Pera, T.; Tiegs, B.C.; Hershfeld, A.; Kenyon, L.C.; Deshpande, D.A. Antimitogenic effect of bitter taste receptor agonists on airway smooth muscle cells. Am. J. Physiol. Cell. Mol. Physiol. 2016, 310, L365–L376. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, Y.; Fukami, K. Regulation and physiological functions of mammalian phospholipase C. J. Biochem. 2017, 161, 315–321. [Google Scholar] [CrossRef] [Green Version]
- Mallows, R.S.; Bolton, T.B. Relationship between stimulated phosphatidic acid production and inositol lipid hydrolysis in intestinal longitudinal smooth muscle from guinea pig. Biochem. J. 1987, 244, 763–768. [Google Scholar] [CrossRef] [Green Version]
- Tolan, D.G.; Pyne, N.; Pyne, S. Phosphatidic acid phosphohydrolase in guinea-pig airway smooth muscle. Biochem. Soc. Trans. 1995, 23, 198S. [Google Scholar] [CrossRef]
- Ohanian, J.; Ollerenshaw, J.; Collins, P.; Heagerty, A. Agonist-induced production of 1,2-diacylglycerol and phosphatidic acid in intact resistance arteries. Evidence that accumulation of diacylglycerol is not a prerequisite for contraction. J. Biol. Chem. 1990, 265, 8921–8928. (In English). Available online: https://www.jbc.org/content/265/15/8921.full.pdf (accessed on 2 October 2022). [CrossRef]
- Lassègue, B.; Alexander, R.W.; Clark, M.; Akers, M.; Griendling, K. Phosphatidylcholine is a major source of phosphatidic acid and diacylglycerol in angiotensin II-stimulated vascular smooth-muscle cells. Biochem. J. 1993, 292, 509–517. [Google Scholar] [CrossRef]
- Ha, K.S.; Yeo, E.J.; Exton, J.H. Lysophosphatidic acid activation of phosphatidylcholine-hydrolysing phospholipase D and actin polymerization by a pertussis toxin-sensitive mechanism. Biochem. J. 1994, 303, 55–59. [Google Scholar] [CrossRef]
- Carpio, L.; Dziak, R. Phosphatidic acid effects on cytosolic calcium and proliferation in osteoblastic cells. Prostaglandins, Leukot. Essent. Fat. Acids 1998, 59, 101–109. [Google Scholar] [CrossRef]
- Hornberger, T.A.; Chu, W.K.; Mak, Y.W.; Hsiung, J.W.; Huang, S.A.; Chien, S. The role of phospholipase D and phosphatidic acid in the mechanical activation of mTOR signaling in skeletal muscle. Proc. Natl. Acad. Sci. USA 2006, 103, 4741–4746. [Google Scholar] [CrossRef] [Green Version]
- Foster, D.A. Regulation of mTOR by Phosphatidic Acid? Cancer Res. 2007, 67, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Jhanwar-Uniyal, M.; Amin, A.G.; Cooper, J.B.; Das, K.; Schmidt, M.H.; Murali, R. Discrete signaling mechanisms of mTORC1 and mTORC2: Connected yet apart in cellular and molecular aspects. Adv. Biol. Regul. 2017, 64, 39–48. [Google Scholar] [CrossRef]
- Flores, I.; Casaseca, T.; Martinez-A, C.; Kanoh, H.; Merida, I. Phosphatidic Acid Generation through Interleukin 2 (IL-2)-induced α-Diacylglycerol Kinase Activation Is an Essential Step in IL-2-mediated Lymphocyte Proliferation. J. Biol. Chem. 1996, 271, 10334–10340. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, S.; Trice, J.; Shinde, P.; Willis, R.E.; Pressley, T.A.; Perez-Zoghbi, J.F. Ca2+ oscillations, Ca2+ sensitization, and contraction activated by protein kinase C in small airway smooth muscle. J. Gen. Physiol. 2013, 141, 165–178. [Google Scholar] [CrossRef] [Green Version]
- Ross, E.M.; Mateu, D.; Gomes, A.V.; Arana, C.; Tran, T.; Litosch, I. Structural Determinants for Phosphatidic Acid Regulation of Phospholipase C-β1. J. Biol. Chem. 2006, 281, 33087–33094. [Google Scholar] [CrossRef] [Green Version]
- You, J.-S.; Lincoln, H.C.; Kim, C.-R.; Frey, J.W.; Goodman, C.; Zhong, X.-P.; Hornberger, T. The Role of Diacylglycerol Kinase ζ and Phosphatidic Acid in the Mechanical Activation of Mammalian Target of Rapamycin (mTOR) Signaling and Skeletal Muscle Hypertrophy. J. Biol. Chem. 2014, 289, 1551–1563. [Google Scholar] [CrossRef] [Green Version]
- Michael, J.V.; Gavrila, A.; Nayak, A.P.; Pera, T.; Liberato, J.R.; Polischak, S.R.; Shah, S.D.; Deshpande, D.A.; Penn, R.B. Cooperativity of E-prostanoid receptor subtypes in regulating signaling and growth inhibition in human airway smooth muscle. FASEB J. 2019, 33, 4780–4789. [Google Scholar] [CrossRef]
- Deshpande, D.A.; Wang, W.C.H.; McIlmoyle, E.L.; Robinett, K.S.; Schillinger, R.M.; An, S.S.; Sham, J.S.K.; Liggett, S.B. Bitter taste receptors on airway smooth muscle bronchodilate by localized calcium signaling and reverse obstruction. Nat. Med. 2010, 16, 1299–1304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pera, T.; Deshpande, D.A.; Ippolito, M.; Wang, B.; Gavrila, A.; Michael, J.V.; Nayak, A.P.; Tompkins, E.; Farrell, E.; Kroeze, W.K.; et al. Biased signaling of the proton-sensing receptor OGR1 by benzodiazepines. FASEB J. 2018, 32, 862–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saxena, H.; Deshpande, D.; Tiegs, B.; Yan, H.; Battafarano, R.; Burrows, W.; Damera, G.; Panettieri, R.; Dubose, T.D., Jr.; An, S.; et al. The GPCR OGR1 (GPR68) mediates diverse signalling and contraction of airway smooth muscle in response to small reductions in extracellular pH. J. Cereb. Blood Flow Metab. 2011, 166, 981–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Misior, A.M.; Deshpande, D.A.; Loza, M.J.; Pascual, R.M.; Hipp, J.D.; Penn, R.B. Glucocorticoid- and Protein Kinase A–Dependent Transcriptome Regulation in Airway Smooth Muscle. Am. J. Respir. Cell Mol. Biol. 2009, 41, 24–39. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.-H.; Machado, F.S.; Guo, R.; Nichols, K.E.; Burks, A.W.; Aliberti, J.C.; Zhong, X.-P. Diacylglycerol kinase ζ regulates microbial recognition and host resistance to Toxoplasma gondii. J. Exp. Med. 2007, 204, 781–792. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, A.M.; Zou, T.; Joshi, R.P.; Leichner, T.M.; Pimentel, M.A.; Sommers, C.L.; Kambayashi, T. Diacylglycerol Kinase ζ Limits the Generation of Natural Regulatory T Cells. Sci. Signal. 2013, 6, ra101. [Google Scholar] [CrossRef] [Green Version]
- Sharma, P.; Yi, R.; Nayak, A.P.; Wang, N.; Tang, F.; Knight, M.J.; Pan, S.; Oliver, B.; Deshpande, D.A. Bitter Taste Receptor Agonists Mitigate Features of Allergic Asthma in Mice. Sci. Rep. 2017, 7, srep46166. [Google Scholar] [CrossRef] [Green Version]
- McAlinden, K.D.; Deshpande, D.A.; Ghavami, S.; Xenaki, D.; Sohal, S.S.; Oliver, B.G.; Haghi, M.; Sharma, P. Autophagy Activation in Asthma Airways Remodeling. Am. J. Respir. Cell Mol. Biol. 2019, 60, 541–553. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phase of Cell Cycle | Protein | Up Regulated by PDGF | Down Regulated by DGK | Rescued by PA |
|---|---|---|---|---|
| G1 Phase & G1/S Transition | Anaphase promoting complex subunit 2 | ANAPC2 | ||
| Cyclin-dependent kinase 6 | CDK6 | CDK6 | CDK6 | |
| S Phase & DNA Replication | Minichromosome maintenance complex component 2 | MCM2 | ||
| Minichromosome maintenance complex component 3 | MCM3 | MCM3 | ||
| G2 Phase & G2/M Transition | BRCA2 and CDKN1A interacting protein | BCCIP | BCCIP | |
| Baculoviral IAP repeat containing 5 | BIRC5 | BIRC5 | ||
| Cyclin G1 | CCNG1 | |||
| Cyclin T1 | CCNT1 | CCNT1 | CCNT1 | |
| CDC28 protein kinase regulatory subunit 1B | CKS1B | |||
| CDC28 protein kinase regulatory subunit 2 | CKS2 | CKS2 | CKS2 | |
| Karyopherin alpha 2 | KPNA2 | KPNA2 | KPNA2 | |
| M Phase | Cyclin F | CCNF | CCNF | CCNF |
| Cell division cycle 16 homolog (S. cerevisiae) | CDC16 | |||
| Cell Cycle Checkpoint & Cell Cycle Arrest | Cyclin-dependent kinase inhibitor 1A (p21, Cip1) | CDKN1A | CDKN1A | CDKN1A |
| Cyclin-dependent kinase inhibitor 1B (p27, Kip1) | CDKN1B | CDKN1B | CDKN1B | |
| Cyclin-dependent kinase inhibitor 2B (p15, inhibits CDK4) | CDKN2B | CDKN2B | CDKN2B | |
| CHK1 checkpoint homolog (S. pombe) | CHEK1 | |||
| CHK2 checkpoint homolog (S. pombe) | CHEK2 | |||
| HUS1 checkpoint homolog (S. pombe) | HUS1 | HUS1 | ||
| Regulation of the Cell Cycle | Cyclin C | CCNC | CCNC | CCNC |
| Cyclin D2 | CCND2 | CCND2 | CCND2 | |
| Cyclin-dependent kinase 4 | CDK4 | CDK4 | ||
| E2F transcription factor 4, p107/p130-binding | E2F4 | |||
| Transcription factor Dp-1 | TFDP1 | TFDP1 | TFDP1 | |
| Negative Regulation of the Cell Cycle | Ataxia telangiectasia mutated | ATM |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hernandez-Lara, M.A.; Yadav, S.K.; Shah, S.D.; Okumura, M.; Yokoyama, Y.; Penn, R.B.; Kambayashi, T.; Deshpande, D.A. Regulation of Airway Smooth Muscle Cell Proliferation by Diacylglycerol Kinase: Relevance to Airway Remodeling in Asthma. Int. J. Mol. Sci. 2022, 23, 11868. https://doi.org/10.3390/ijms231911868
Hernandez-Lara MA, Yadav SK, Shah SD, Okumura M, Yokoyama Y, Penn RB, Kambayashi T, Deshpande DA. Regulation of Airway Smooth Muscle Cell Proliferation by Diacylglycerol Kinase: Relevance to Airway Remodeling in Asthma. International Journal of Molecular Sciences. 2022; 23(19):11868. https://doi.org/10.3390/ijms231911868
Chicago/Turabian StyleHernandez-Lara, Miguel Angel, Santosh K. Yadav, Sushrut D. Shah, Mariko Okumura, Yuichi Yokoyama, Raymond B. Penn, Taku Kambayashi, and Deepak A. Deshpande. 2022. "Regulation of Airway Smooth Muscle Cell Proliferation by Diacylglycerol Kinase: Relevance to Airway Remodeling in Asthma" International Journal of Molecular Sciences 23, no. 19: 11868. https://doi.org/10.3390/ijms231911868