
Discovery and In Vivo Efficacy of Trace Amine-Associated Receptor 1 (TAAR1) Agonist 4-(2-Aminoethyl)-N-(3,5-dimethylphenyl)piperidine-1-carboxamide Hydrochloride (AP163) for the Treatment of Psychotic Disorders

 ,
,  , , , ,
, , , ,  and
and
Abstract
:1. Introduction
2. Results and Discussion
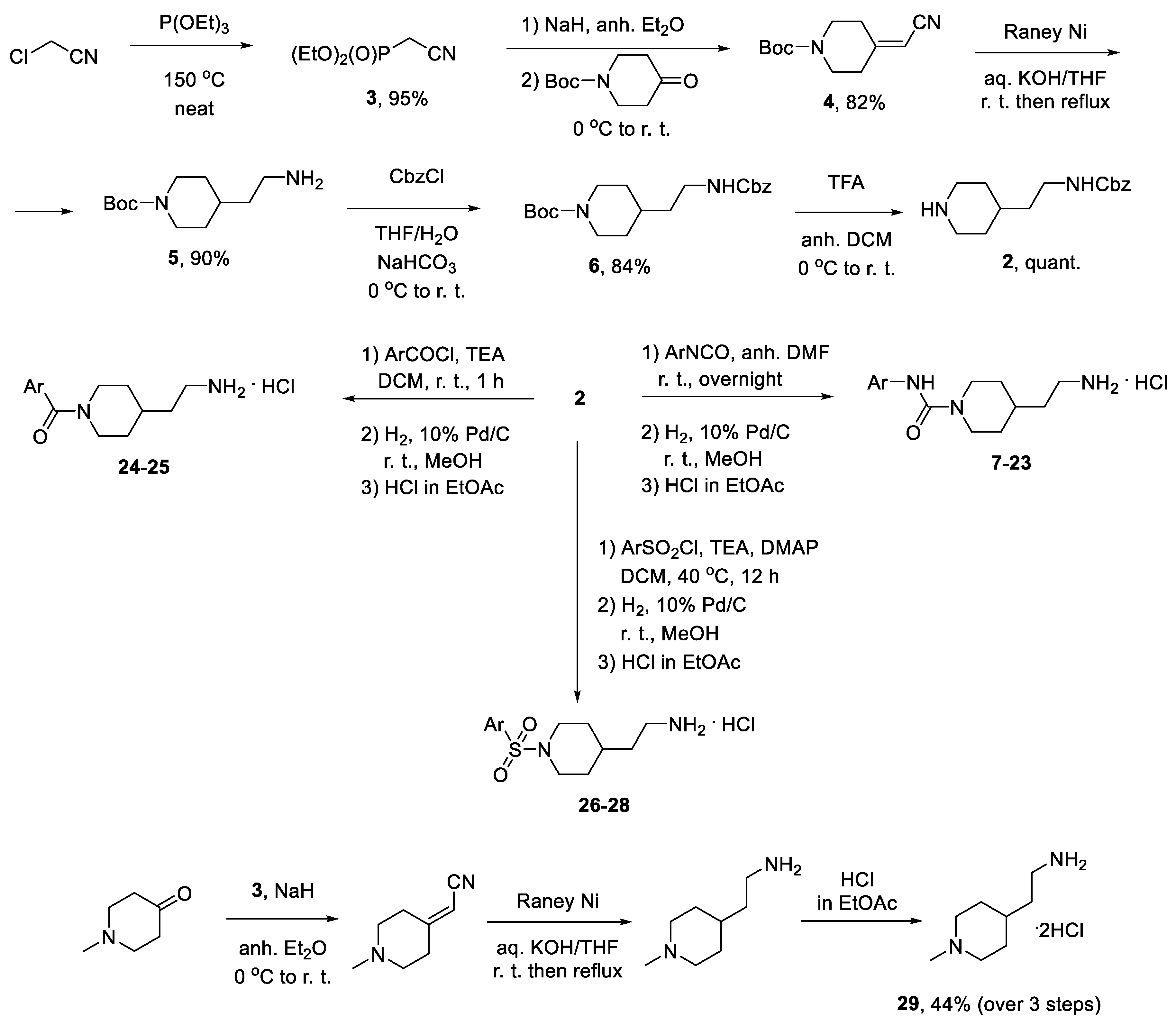
2.1. Compounds Synthesis
2.2. Agonistic Activity against TAAR1 In Vitro
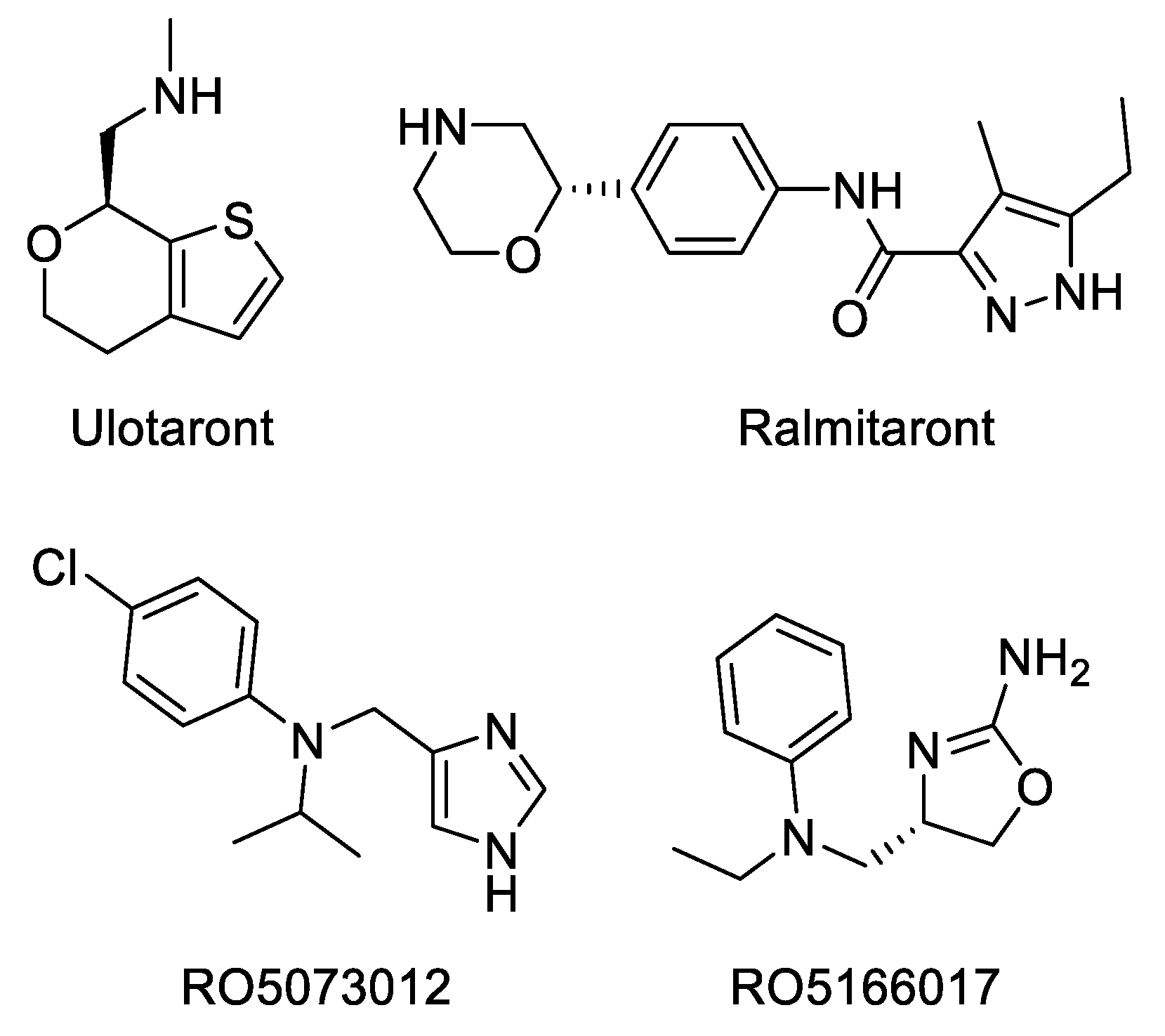
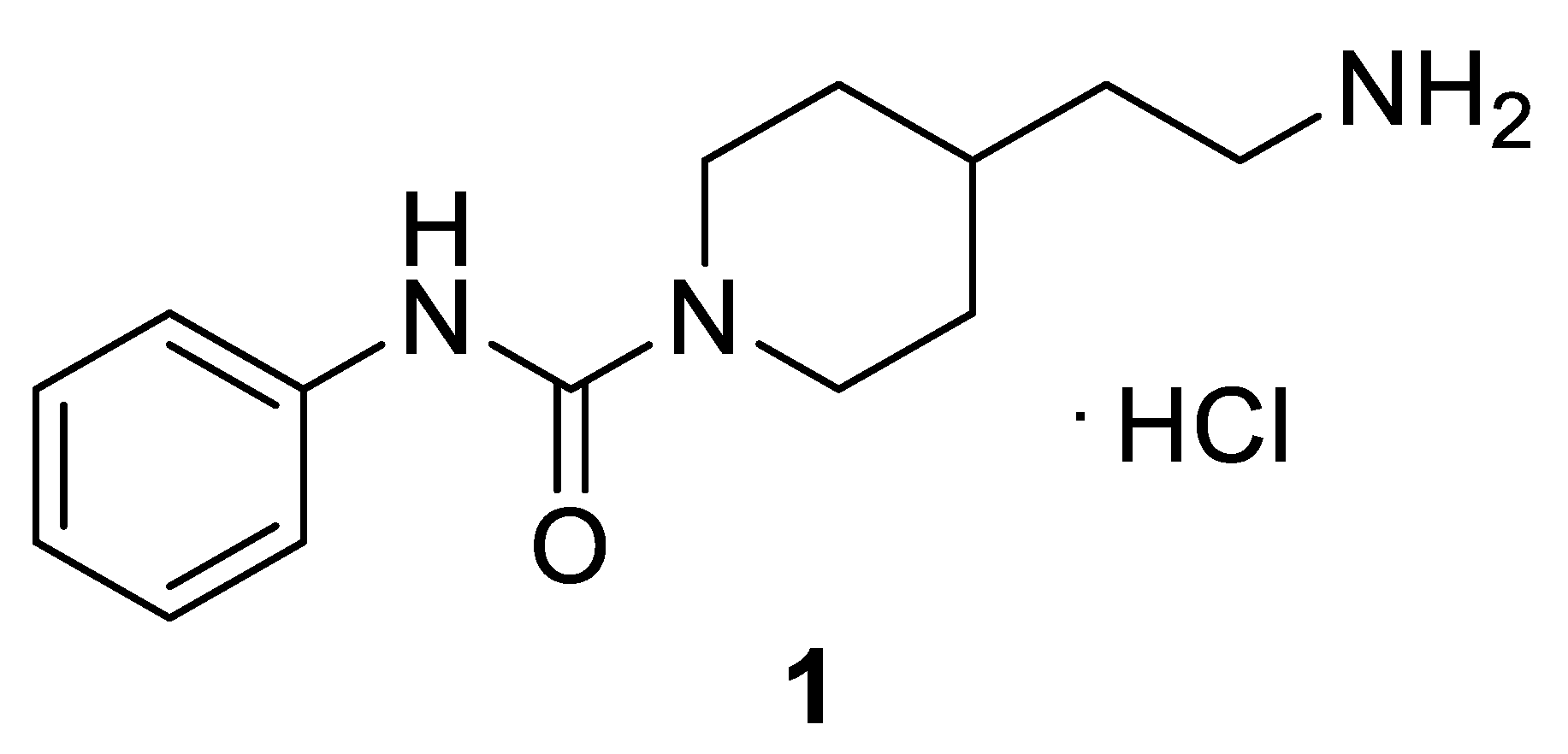
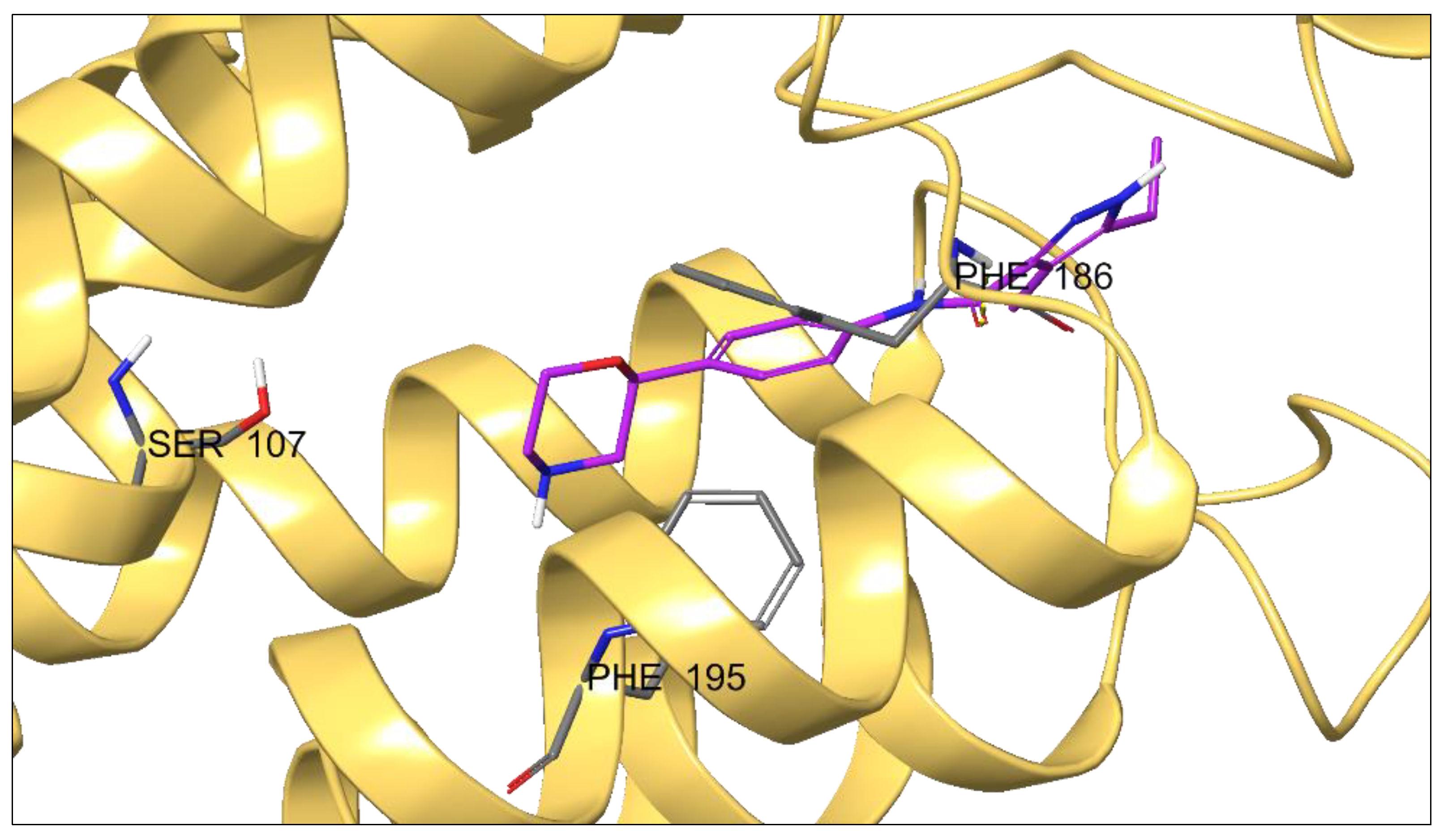
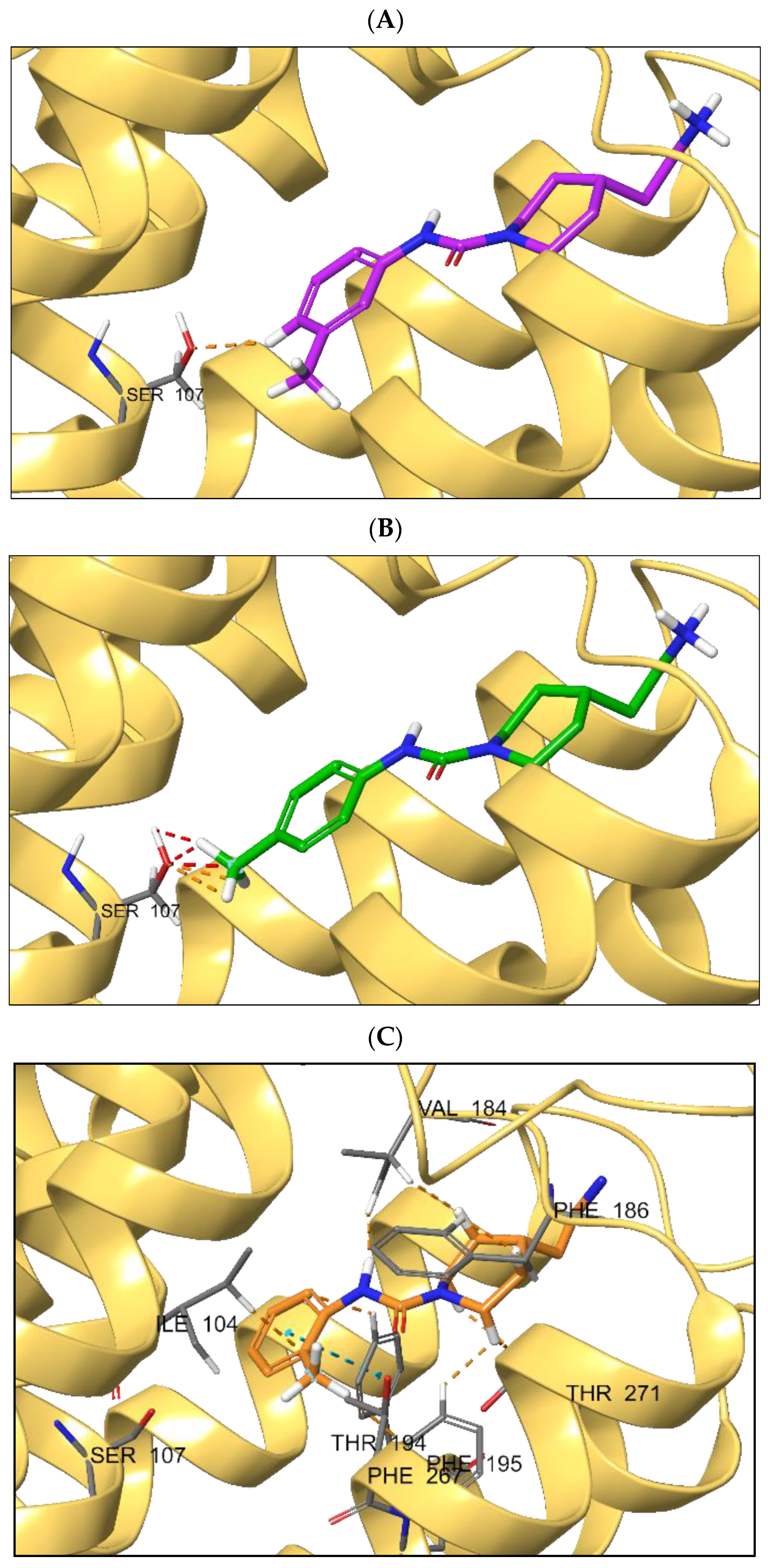
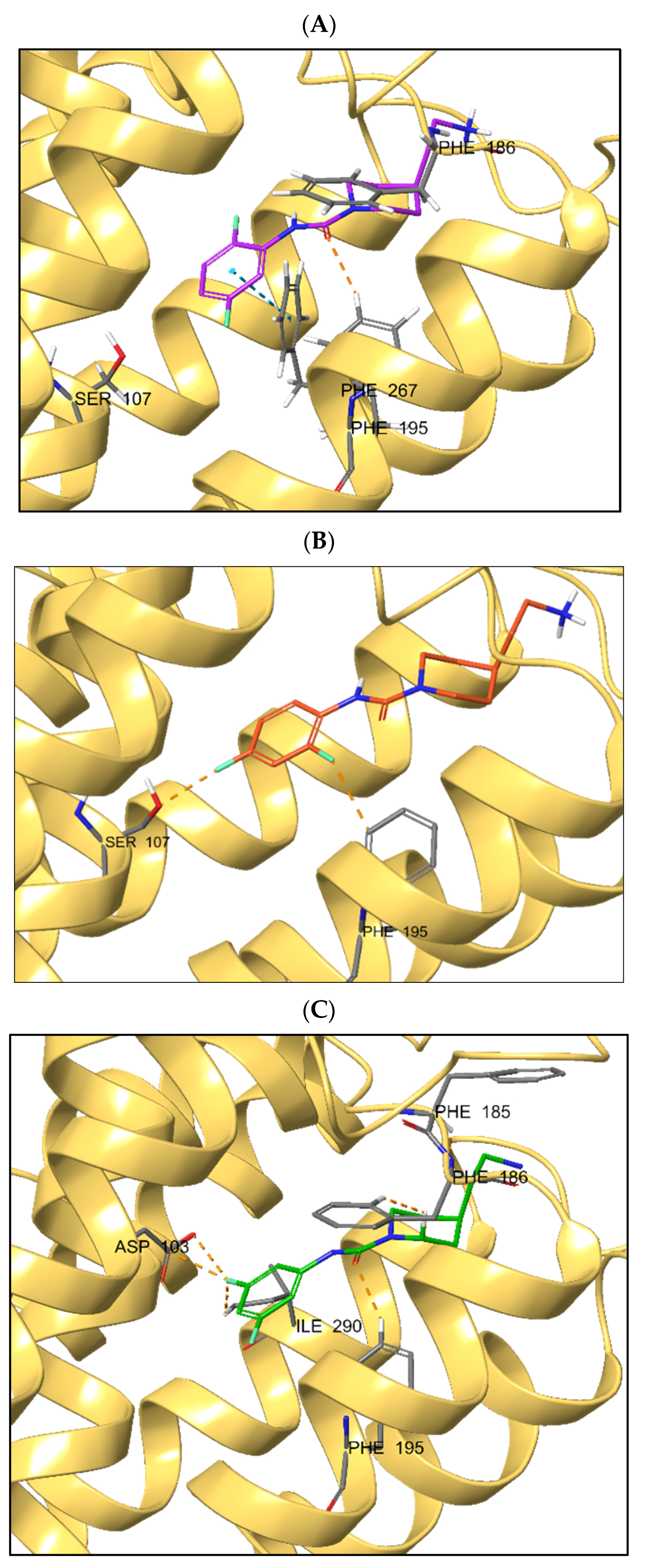
2.3. Molecular Modeling
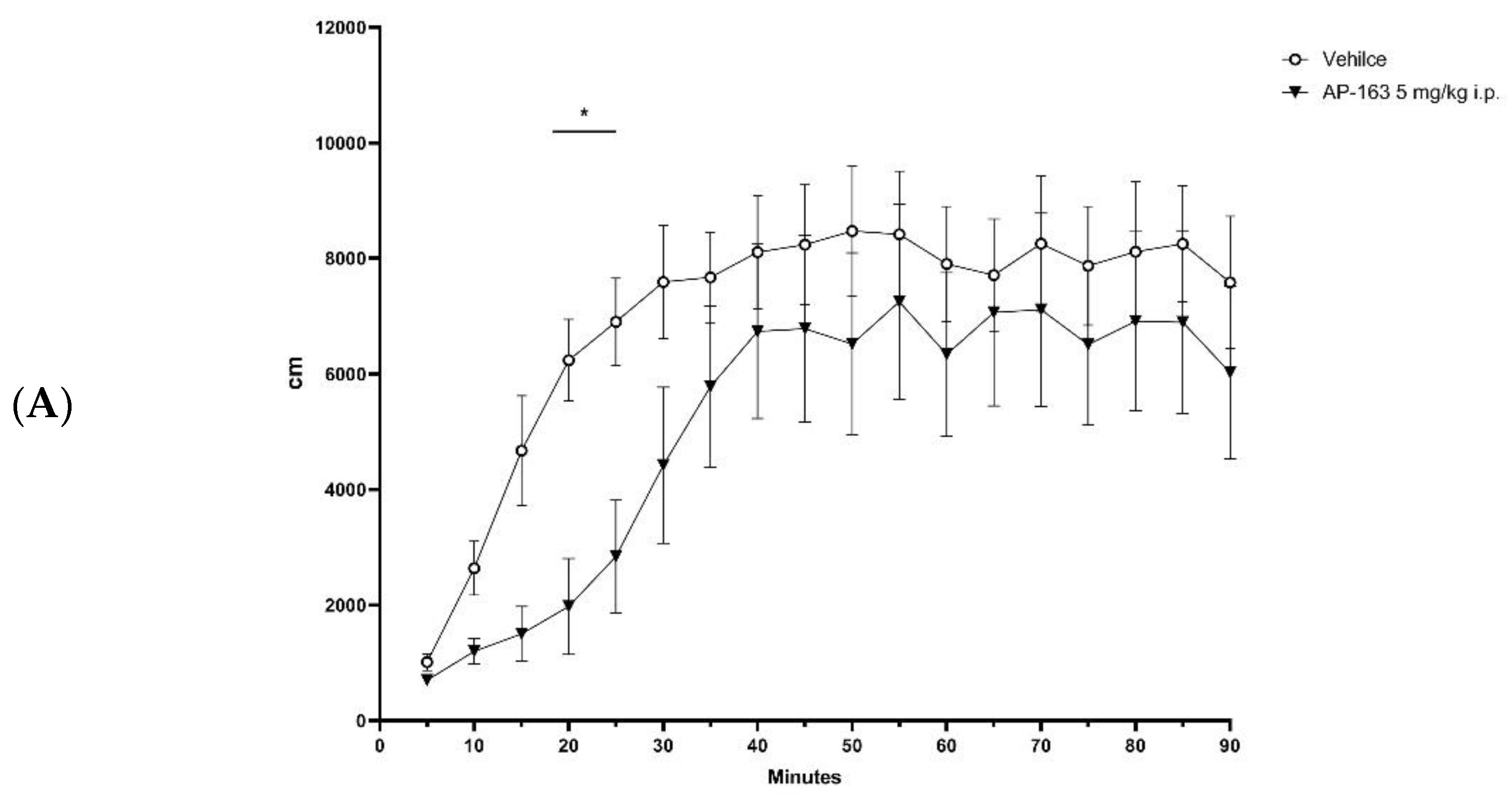
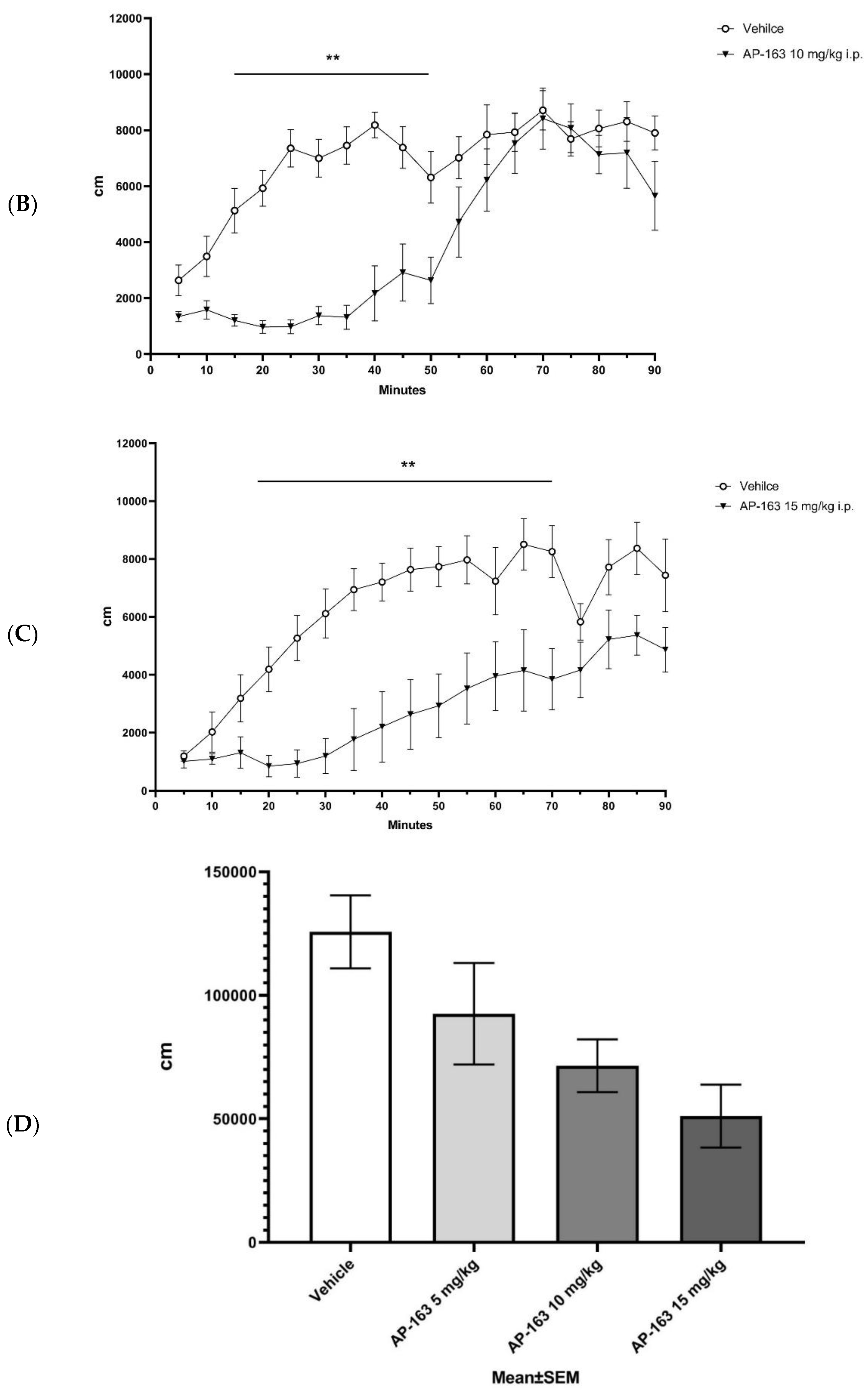
2.4. In Vivo Efficacy
3. Experimental Procedures
3.1. Compound Synthesis-General
3.1.1. Synthesis of Diethyl (Cyanomethyl)phosphonate (3)
3.1.2. Synthesis of tert-Butyl 4-(Cyanomethylene)piperidine-1-carboxylate (4)
3.1.3. Synthesis of tert-Butyl 4-(2-Aminoethyl)piperidine-1-carboxylate (5)
3.1.4. Synthesis of tert-Butyl 4-(2-(((Benzyloxy)carbonyl)amino)ethyl)piperidine-1-carboxylate (6)
3.1.5. Synthesis of Benzyl (2-(Piperidin-4-yl)ethyl)carbamate (2)
3.1.6. Synthesis of Benzyl (2-(1-(Arylcarbamoyl)piperidin-4-yl)ethyl)carbamates 1, 7–23
- (1) 4-(2-Aminoethyl)-N-phenylpiperidine-1-carboxamide hydrochloride (1). Yield 96 mg (68%). 1H NMR (300 MHz, DMSO-d6) δ 8.49 (s, 1H), 7.96 (br.s, 3H), 7.50–7.41 (m, 2H), 7.25–7.15 (m, 2H), 6.96–6.85 (m, 1H), 4.18–4.06 (m, 2H), 2.87–2.66 (m, 4H), 1.73–1.61 (m, 2H), 1.61–1.44 (m, 3H), 1.14–0.96 (m, 2H). 13C NMR (75 MHz, DMSO-d6) δ 154.9, 140.8, 128.2, 121.5, 119.6, 43.9, 36.4, 33.5, 32.6, 31.5. HRMS (ESI) calcd for C14H22N3O+ [M+H+] 248.1757; found 248.1757.
- (2) 4-(2-Aminoethyl)-N-(4-methylphenyl)piperidine-1-carboxamide hydrochloride (7). Yield 87 mg (59%). 1H NMR (300 MHz, DMSO-d6) δ 8.39 (s, 1H), 7.98 (br.s, 3H), 7.33 (d, J = 8.4 Hz, 2H), 7.01 (d, J = 8.3 Hz, 2H), 4.18–4.03 (m, 2H), 2.87–2.65 (m, 4H), 2.21 (s, 3H), 1.72–1.60 (m, 2H), 1.60–1.44 (m, 3H), 1.13–0.94 (m, 2H). 13C NMR (75 MHz, DMSO-d6) δ 155.0, 138.1, 130.3, 128.6, 119.8, 43.8, 36.4, 33.5, 32.6, 31.5, 20.3. HRMS (ESI) calcd for C15H24N3O+ [M+H+] 262.1914; found 262.1912.
- (3) 4-(2-Aminoethyl)-N-(3-fluorophenyl)piperidine-1-carboxamide hydrochloride (8). Yield 123 g (82%). 1H NMR (300 MHz, DMSO-d6) δ 8.73 (s, 1H), 7.91 (br.s, 3H), 7.50–7.39 (m, 1H), 7.30–7.16 (m, 2H), 6.77–6.65 (m, 1H), 4.19–4.05 (m, 2H), 2.88–2.68 (m, 4H), 1.74–1.62 (m, 2H), 1.61–1.45 (m, 3H), 1.14–0.97 (m, 2H). 13C NMR (75 MHz, DMSO-d6) δ 162.1 (d, J = 239.6 Hz), 154.5, 142.8 (d, J = 11.4 Hz), 129.6 (d, J = 9.7 Hz), 115.0 (d, J = 2.4 Hz), 107.7 (d, J = 21.2 Hz), 105.9 (d, J = 26.3 Hz), 43.9, 36.4, 33.5, 32.6, 31.5. HRMS (ESI) calcd for C14H21FN3O+ [M+H+] 266.1663; found 266.1662.
- (4) 4-(2-Aminoethyl)-N-(3-methylphenyl)piperidine-1-carboxamide hydrochloride (9). Yield 107 mg (72%). 1H NMR (300 MHz, DMSO-d6) δ 8.41 (s, 1H), 7.96 (br.s, 3H), 7.32–7.20 (m, 2H), 7.13–7.03 (m, 1H), 6.77–6.68 (m, 1H), 4.18–4.04 (m, 2H), 2.87–2.65 (m, 4H), 2.23 (s, 3H), 1.72–1.61 (m, 2H), 1.59–1.44 (m, 3H), 1.12–0.95 (m, 2H). 13C NMR (75 MHz, DMSO-d6) δ 154.9, 140.7, 137.2, 128.0, 122.2, 120.2, 116.8, 43.9, 36.4, 33.5, 32.6, 31.5, 21.2. HRMS (ESI) calcd for C15H24N3O+ [M+H+] 262.1914; found 262.1912.
- (5) 4-(2-Aminoethyl)-N-(2-methylphenyl)piperidine-1-carboxamide hydrochloride (10). Yield 98 mg (66%). 1H NMR (300 MHz, DMSO-d6) δ 8.15–7.82 (m, 4H), 7.20–6.98 (m, 4H), 4.15–4.01 (m, 2H), 2.87–2.68 (m, 4H), 2.14 (s, 3H), 1.72–1.61 (m, 2H), 1.61–1.46 (m, 3H), 1.15–0.97 (m, 2H). 13C NMR (75 MHz, DMSO-d6) δ 155.5, 138.2, 133.1, 130.0, 126.0, 125.7, 124.4, 44.1, 36.4, 33.6, 32.7, 31.5, 18.0. HRMS (ESI) calcd for C15H24N3O+ [M+H+] 262.1914; found 262.1913.
- (6) 4-(2-Aminoethyl)-N-(4-fluorophenyl)piperidine-1-carboxamide hydrochloride (11). Yield 120 mg (79%). 1H NMR (300 MHz, DMSO-d6) δ 8.58 (s, 1H), 8.03 (br.s, 3H), 7.53–7.41 (m, 2H), 7.10–6.98 (m, 2H), 4.19–4.04 (m, 2H), 2.87–2.66 (m, 4H), 1.72–1.61 (m, 2H), 1.60–1.45 (m, 3H), 1.13–0.94 (m, 2H). 13C NMR (75 MHz, DMSO-d6) δ 157.3 (d, J = 237.7 Hz), 155.0, 137.1 (d, J = 2.4 Hz), 121.4 (d, J = 7.6 Hz), 114.6 (d, J = 21.9 Hz), 43.9, 36.4, 33.5, 32.6, 31.5. HRMS (ESI) calcd for C14H21FN3O+ [M+H+] 266.1663; found 266.1663.
- (7) 4-(2-Aminoethyl)-N-(2,5-difluorophenyl)piperidine-1-carboxamide hydrochloride (12). Yield 110 mg (69%). 1H NMR (300 MHz, DMSO-d6) δ 8.42 (s, 1H), 8.02 (br.s, 3H), 7.44–7.34 (m, 1H), 7.27–7.15 (m, 1H), 6.94–6.83 (m, 1H), 4.14–4.01 (m, 2H), 2.87–2.70 (m, 4H), 1.73–1.62 (m, 2H), 1.60–1.45 (m, 3H), 1.14–0.97 (m, 2H). 13C NMR (75 MHz, DMSO-d6) δ 157.6 (dd, J = 238.0, 1.9 Hz), 154.5, 150.9 (dd, J = 240.4, 2.5 Hz), 129.4 (dd, J = 13.5, 11.8 Hz), 116.1 (dd, J = 22.8, 10.0 Hz), 111.4 (dd, J = 27.2, 2.2 Hz), 110.0 (dd, J = 24.1, 7.8 Hz), 44.1, 36.4, 33.4, 32.5, 31.4. HRMS (ESI) calcd for C14H20F2N3O+ [M+H+] 284.1569; found 284.1573.
- (8) 4-(2-Aminoethyl)-N-(2,4-difluorophenyl)piperidine-1-carboxamide hydrochloride (13). Yield 108 mg (68%). 1H NMR (300 MHz, DMSO-d6) δ 8.29 (s, 1H), 7.99 (br.s, 3H), 7.42–7.30 (m, 1H), 7.27–7.15 (m, 1H), 7.05–6.94 (m, 1H), 4.13–4.00 (m, 2H), 2.87–2.68 (m, 4H), 1.72–1.61 (m, 2H), 1.61–1.45 (m, 3H), 1.14–0.96 (m, 2H). 13C NMR (75 MHz, DMSO-d6) δ 158.6 (dd, J = 242.6, 11.5 Hz), 155.8 (dd, J = 248.2, 12.6 Hz), 155.1, 127.8 (dd, J = 9.5, 3.1 Hz), 124.4 (dd, J = 12.0, 3.6 Hz), 110.6 (dd, J = 21.8, 3.6 Hz), 103.9 (dd, J = 26.5, 24.6 Hz), 44.0, 36.4, 33.5, 32.6, 31.4. HRMS (ESI) calcd for C14H20F2N3O+ [M+H+] 284.1569; found 284.1530.
- (9) 4-(2-Aminoethyl)-N-(3,5-difluorophenyl)piperidine-1-carboxamide hydrochloride (14). Yield 114 mg (71%). 1H NMR (300 MHz, DMSO-d6) δ 9.07–8.94 (m, 1H), 7.99 (br.s, 3H), 7.38–7.21 (m, 2H), 6.77–6.61 (m, 1H), 4.20–4.07 (m, 2H), 2.89–2.68 (m, 4H), 1.74–1.62 (m, 2H), 1.62–1.43 (m, 3H), 1.13–0.95 (m, 2H). 13C NMR (75 MHz, DMSO-d6) δ 162.3 (dd, J = 241.1, 15.7 Hz), 154.3, 143.9 (t, J = 14.2 Hz), 101.9–101.6 (m), 96.1 (t, J = 26.2 Hz), 44.0, 36.5, 33.5, 32.6, 31.5. HRMS (ESI) calcd for C14H20F2N3O+ [M+H+] 284.1569; found 284.1568.
- (10) 4-(2-Aminoethyl)-N-(3-fluoro-4-methylphenyl)piperidine-1-carboxamide hydrochloride (15). Yield 107 mg (68%). 1H NMR (300 MHz, DMSO-d6) δ 8.63 (s, 1H), 8.00 (br.s, 3H), 7.44–7.34 (m, 1H), 7.20–7.03 (m, 2H), 4.18–4.05 (m, 2H), 2.87–2.66 (m, 4H), 2.13 (s, 3H), 1.73–1.61 (m, 2H), 1.61–1.45 (m, 3H), 1.13–0.95 (m, 2H). 13C NMR (75 MHz, DMSO-d6) δ 160.2 (d, J = 239.2 Hz), 154.7, 140.4 (d, J = 11.2 Hz), 130.7 (d, J = 6.5 Hz), 116.2 (d, J = 17.4 Hz), 115.0 (d, J = 2.8 Hz), 106.0 (d, J = 27.0 Hz), 43.9, 36.4, 33.5, 32.6, 31.5, 13.5 (d, J = 2.9 Hz). HRMS (ESI) calcd for C15H23FN3O+ [M+H+] 280.1820; found 280.1817.
- (11) 4-(2-Aminoethyl)-N-(4-fluoro-3-methylphenyl)piperidine-1-carboxamide hydrochloride (16). Yield 90 mg (57%). 1H NMR (400 MHz, DMSO-d6) δ 8.43 (s, 1H), 7.89 (br.s, 3H), 7.40–7.31 (m, 1H), 7.31–7.20 (m, 1H), 7.03–6.91 (m, 1H), 4.18–4.03 (m, 2H), 2.90–2.63 (m, 4H), 2.22–2.11 (m, 3H), 1.73–1.62 (m, 2H), 1.60–1.44 (m, 3H), 1.12–0.98 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 155.8 (d, J = 237.0 Hz), 154.9, 136.7 (d, J = 2.5 Hz).123.3 (d, J = 17.8 Hz), 122.7 (d, J = 4.3 Hz), 118.7 (d, J = 7.6 Hz), 114.2 (d, J = 22.8 Hz), 43.9, 36.8, 34.6, 32.7, 31.5, 14.3 (d, J = 3.0 Hz). HRMS (ESI) calcd for C15H23FN3O+ [M+H+] 280.1820; found 280.1820.
- (12) 4-(2-Aminoethyl)-N-(3,4-dimethylphenyl)piperidine-1-carboxamide hydrochloride (17). Yield 92 mg (59%). 1H NMR (400 MHz, DMSO-d6) δ 8.29 (s, 1H), 7.96 (br.s, 3H), 7.28–7.19 (m, 1H), 7.19–7.12 (m, 1H), 6.98–6.92 (m, 1H), 4.15–4.05 (m, 2H), 2.87–2.76 (m, 2H), 2.76–2.66 (m, 2H), 2.18–2.09 (m, 6H), 1.72–1.60 (m, 2H), 1.60–1.45 (m, 3H), 1.12–0.96 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 155.0, 138.3, 135.5, 129.1, 129.0, 121.1, 117.3, 43.8, 36.4, 33.5, 32.6, 31.5, 19.6, 18.6. HRMS (ESI) calcd for C16H26N3O+ [M+H+] 276.2070; found 276.2072.
- (13) 4-(2-Aminoethyl)-N-(3,5-dimethylphenyl)piperidine-1-carboxamide hydrochloride (18). Yield 95 mg (61%). 1H NMR (400 MHz, DMSO-d6) δ 8.30 (s, 1H), 7.92 (br.s, 3H), 7.08 (s, 2H), 6.56 (s, 1H), 4.15–4.04 (m, 2H), 2.87–2.76 (m, 2H), 2.76–2.66 (m, 2H), 2.19 (s, 6H), 1.73–1.60 (m, 2H), 1.60–1.45 (m, 3H), 1.12–0.96 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 154.9, 140.5, 137.0, 123.1, 117.4, 43.9, 36.4, 33.5, 32.6, 31.5, 21.1. HRMS (ESI) calcd for C16H26N3O+ [M+H+] 276.2070; found 276.2071.
- (14) 4-(2-Aminoethyl)-N-(2,5-dimethylphenyl)piperidine-1-carboxamide hydrochloride (19). Yield 80 mg (51%). 1H NMR (400 MHz, DMSO-d6) δ 8.00–7.78 (m, 4H), 7.06–7.00 (m, 1H), 6.98 (s, 1H), 6.88–6.79 (m, 1H), 4.14–3.99 (m, 2H), 2.89–2.68 (m, 4H), 2.23 (s, 3H), 2.09 (s, 3H), 1.72–1.62 (m, 2H), 1.61–1.46 (m, 3H), 1.14–0.98 (m, 2H). 13C NMR (126 MHz, DMSO-d6) δ 155.5, 137.9, 134.6, 129.9, 129.8, 126.6, 125.1, 44.0, 36.4, 33.6, 32.7, 31.5, 20.5, 17.5. HRMS (ESI) calcd for C16H26N3O+ [M+H+] 276.2070; found 276.2071.
- (15) 4-(2-Aminoethyl)-N-(2,3-dimethylphenyl)piperidine-1-carboxamide hydrochloride (20). Yield 75 mg (48%). 1H NMR (400 MHz, DMSO-d6) δ 8.70 (s, 1H), 8.11–7.76 (m, 3H), 7.07–6.84 (m, 3H), 4.15–4.00 (m, 2H), 2.96–2.65 (m, 4H), 2.23 (s, 3H), 2.02 (s, 3H), 1.74–1.47 (m, 5H), 1.16–0.99 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 155.8, 138.0, 136.4, 132.3, 126.2, 124.8, 124.3, 44.0, 36.4, 33.6, 32.7, 31.5, 20.2, 18.5. HRMS (ESI) calcd for C16H26N3O+ [M+H+] 276.2070; found 276.2075.
- (16) 4-(2-Aminoethyl)-N-(3-(trifluoromethyl)phenyl)piperidine-1-carboxamide hydrochloride (21). Yield 81 mg (46%). 1H NMR (400 MHz, DMSO-d6) δ 8.88 (s, 1H), 8.11–7.85 (m, 4H), 7.80–7.71 (m, 1H), 7.49–7.38 (m, 1H), 7.28–7.18 (m, 1H), 4.22–4.06 (m, 2H), 2.94–2.69 (m, 4H), 1.76–1.63 (m, 2H), 1.63–1.46 (m, 3H), 1.15–0.98 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 154.5, 141.7, 129.4, 129.1 (q, J = 31.2 Hz), 124.3 (q, J = 272.0 Hz), 122.8 (d, J = 1.1 Hz), 117.6 (q, J = 3.9 Hz), 115.4 (q, J = 4.0 Hz), 43.9, 36.5, 33.5, 32.6, 31.4. HRMS (ESI) calcd for C15H21F3N3O+ [M+H+] 316.1631; found 316.1629.
- (17) 4-(2-Aminoethyl)-N-(2-methoxy-5-methylphenyl)piperidine-1-carboxamide hydrochloride (22). Yield 95 mg (58%). 1H NMR (400 MHz, DMSO-d6) δ 7.93 (br.s, 3H), 7.60–7.40 (m, 2H), 6.89–6.83 (m, 1H), 6.82–6.75 (m, 1H), 4.06–3.96 (m, 2H), 3.76 (s, 3H), 2.87–2.71 (m, 4H), 2.20 (s, 3H), 1.74–1.61 (m, 2H), 1.61–1.45 (m, 3H), 1.14–0.99 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 154.7, 147.8, 128.8, 128.4, 123.2, 122.7, 110.7, 55.8, 43.9, 36.4, 33.5, 32.5, 31.3, 20.5. HRMS (ESI) calcd for C16H26N3O2+ [M+H+] 292.2020; found 292.2021.
- (18) 4-(2-Aminoethyl)-N-(3-chlorophenyl)piperidine-1-carboxamide hydrochloride (23). Yield 71 mg (45%). 1H NMR (300 MHz, DMSO-d6) δ 8.79 (s, 1H), 8.08 (br.s, 3H), 7.68 (s, 1H), 7.48–7.38 (m, 1H), 7.27–7.16 (m, 1H), 6.99–6.88 (m, 1H), 4.22–4.04 (m, 2H), 2.88–2.66 (m, 4H), 1.74–1.47 (m, 5H), 1.14–0.95 (m, 2H). 13C NMR (75 MHz, DMSO-d6) δ 154.5, 142.5, 132.6, 129.8, 121.0, 118.8, 117.7, 43.9, 36.4, 33.4, 32.6, 31.5. HRMS (ESI) calcd for C14H21ClN3O+ [M+H+] 282.1368; found 282.1366.
3.1.7. Synthesis of Benzyl (2-(1-Benzoylpiperidin-4-yl)ethyl)carbamates 24–25
- (1) (4-(2-Aminoethyl)piperidin-1-yl)(phenyl)methanone hydrochloride (24). Yield 79 mg (59%). 1H NMR (400 MHz, DMSO-d6) δ 7.96 (br.s, 3H), 7.47–7.40 (m, 3H), 7.40–7.32 (m, 2H), 4.46 (br.s, 1H), 3.55 (br.s, 1H), 3.09–2.65 (m, 4H), 1.84–1.46 (m, 5H), 1.19–0.99 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 168.9, 136.4, 129.3, 128.4, 126.6, 47.1 (br.s), 41.5 (br.s), 36.3, 33.3, 32.6, 31.8, 31.2. HRMS (ESI) calcd for C14H21N2O+ [M+H+] 233.1648; found 233.1644.
- (2) (4-(2-Aminoethyl)piperidin-1-yl)(m-tolyl)methanone (25). Yield 82 mg (58%). 1H NMR (400 MHz, DMSO-d6) δ 7.91 (br.s, 3H), 7.35–7.28 (m, 1H), 7.28–7.22 (m, 1H), 7.19–7.10 (m, 2H), 4.45 (br.s, 1H), 3.56 (br.s, 1H), 3.08–2.64 (m, 4H), 2.33 (s, 3H), 1.83–1.45 (m, 5H), 1.16–0.99 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 169.0, 137.7, 136.4, 129.8, 128.2, 127.1, 123.6, 47.0 (br.s), 41.4 (br.s), 36.3, 33.3, 32.6, 31.8, 31.1, 20.9. HRMS (ESI) calcd for C15H23N2O+ [M+H+] 247.1805; found 247.1805.
3.1.8. Synthesis of Benzyl (2-(1-(Arylsulfonyl)piperidin-4-yl)ethyl)carbamates 26–28
- (1) 2-[1-(Phenylsulfonyl)piperidin-4-yl]ethanamine hydrochloride (26). Yield 85 mg (56%). 1H NMR (300 MHz, DMSO-d6) δ 8.08 (br.s, 3H), 7.77–7.60 (m, 5H), 3.66–3.56 (m, 2H), 2.79–2.64 (m, 2H), 2.23–2.07 (m, 2H), 1.75–1.63 (m, 2H), 1.52–1.38 (m, 2H), 1.38–1.21 (m, 1H), 1.21–1.05 (m, 2H). 13C NMR (75 MHz, DMSO-d6) δ 135.4, 133.1, 129.4, 127.4, 46.0, 36.2, 32.9, 31.4, 30.5. HRMS (ESI) calcd for C13H21N2O2S+ [M+H+] 269.1318; found 269.1317.
- (2) 2-{1-[(4-Fluorophenyl)sulfonyl]piperidin-4-yl}ethanamine hydrochloride (27). Yield 125 mg (78%). 1H NMR (300 MHz, DMSO-d6) δ 8.05 (br.s, 3H), 7.85–7.76 (m, 2H), 7.54–7.44 (m, 2H), 3.67–3.53 (m, 2H), 2.81–2.64 (m, 2H), 2.24–2.09 (m, 2H), 1.76–1.63 (m, 2H), 1.53–1.39 (m, 2H), 1.39–1.22 (m, 1H), 1.22–1.05 (m, 2H). 13C NMR (75 MHz, DMSO-d6) δ 164.6 (d, J = 251.7 Hz), 131.9 (d, J = 2.9 Hz), 130.5 (d, J = 9.6 Hz), 116.6 (d, J = 22.6 Hz), δ 45.9, 36.2, 32.9, 31.4, 30.5. HRMS (ESI) calcd for C13H20FN2O2S+ [M+H+] 287.1224; found 287.1224.
- (3) 2-{1-[(4-Methylphenyl)sulfonyl]piperidin-4-yl}ethanamine hydrochloride (28). Yield 115 mg (72%). 1H NMR (300 MHz, DMSO-d6) δ 8.05 (br.s, 3H), 7.61 (d, J = 8.2 Hz, 2H), 7.44 (d, J = 8.1 Hz, 2H), 3.64–3.52 (m, 2H), 2.80–2.64 (m, 2H), 2.40 (s, 3H), 2.19–2.04 (m, 2H), 1.74–1.62 (m, 2H), 1.51–1.38 (m, 2H), 1.36–1.21 (m, 1H), 1.21–1.04 (m, 2H). 13C NMR (75 MHz, DMSO-d6) δ 143.5, 132.4, 129.8, 127.5, 45.9, 36.2, 32.9, 31.4, 30.5, 21.0. HRMS (ESI) calcd for C14H23N2O2S+ [M+H+] 283.1475; found 283.1474.
3.1.9. Synthesis of 2-(1-Methylpiperidin-4-yl)ethan-1-amine dihydrochloride (29)
3.2. BRET Analysis
3.3. Molecular Modeling
3.3.1. Protein Structure Preparation
3.3.2. Ligand Structure Preparation
3.3.3. Molecular Docking Procedure
3.3.4. Molecular Docking: Best-Fitting Structure Selection
3.3.5. Molecular Mechanics with Generalized Born and Surface Area Solvation (MM-GBSA)
3.4. In Vivo Evaluation of Compounds Activity on DAT-KO Rat Hyperlocomotion
3.4.1. Animals
3.4.2. Compound Administration
3.4.3. Locomotion Tests
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kahn, R.S.; Sommer, I.E.; Murray, R.M.; Meyer-Lindenberg, A.; Weinberger, D.R.; Cannon, T.D.; O’Donovan, M.; Correll, C.U.; Kane, J.M.; van Os, J.; et al. Schizophrenia. Nat. Rev. Dis. Prim. 2015, 1, 15067. [Google Scholar] [CrossRef] [PubMed]
- Huhn, M.; Nikolakopoulou, A.; Schneider-Thoma, J.; Krause, M.; Samara, M.; Peter, N.; Arndt, T.; Bäckers, L.; Rothe, P.; Cipriani, A.; et al. Comparative efficacy and tolerability of 32 oral antipsychotics for the acute treatment of adults with multi-episode schizophrenia: A systematic review and network meta-analysis. Lancet 2019, 394, 939–951. [Google Scholar] [CrossRef]
- Lieberman, J.A.; Stroup, T.S.; McEvoy, J.P.; Swartz, M.S.; Rosenheck, R.A.; Perkins, D.O.; Keefe, R.S.; Davis, S.M.; Davis, C.E.; Lebowitz, B.D.; et al. Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) Investigators. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N. Engl. J. Med. 2005, 353, 1209–1223. [Google Scholar] [CrossRef] [PubMed]
- Gainetdinov, R.R.; Hoener, M.C.; Berry, M.D. Trace Amines and Their Receptors. Pharmacol. Rev. 2018, 70, 549–620. [Google Scholar] [CrossRef] [PubMed]
- Borowsky, B.; Adham, N.; Jones, K.A.; Raddatz, R.; Artymyshyn, R.; Ogozalek, K.L.; Durkin, M.M.; Lakhlani, P.P.; Bonini, J.A.; Pathirana, S.; et al. Trace amines: Identification of a family of mammalian G protein-coupled receptors. Proc. Natl. Acad. Sci. USA 2001, 98, 8966–8971. [Google Scholar] [CrossRef]
- Bunzow, J.R.; Sonders, M.S.; Arttamangkul, S.; Harrison, L.M.; Zhang, G.; Quigley, D.I.; Darland, T.; Suchland, K.L.; Pasumamula, S.; Kennedy, J.L.; et al. Amphetamine, 3,4-methylenedioxymethamphetamine, lysergic acid diethylamide, and metabolites of the catecholamine neurotransmitters are agonists of a rat trace amine receptor. Mol. Pharmacol. 2001, 60, 1181–1188. [Google Scholar] [CrossRef]
- Berry, M.D.; Gainetdinov, R.R.; Hoener, M.C.; Shahid, M. Pharmacology of human trace amine-associated receptors: Therapeutic opportunities and challenges. Pharmacol. Ther. 2017, 180, 161–180. [Google Scholar] [CrossRef]
- Dedic, N.; Jones, P.G.; Hopkins, S.C.; Lew, R.; Shao, L.; Campbell, J.E.; Spear, K.L.; Large, T.H.; Campbell, U.C.; Hanania, T.; et al. SEP-363856, a Novel Psychotropic Agent with a Unique, Non-D2 Receptor Mechanism of Action. J. Pharmacol. Exp. Ther. 2019, 371, 1–14. [Google Scholar] [CrossRef]
- Heffernan, M.L.R.; Herman, L.W.; Brown, S.; Jones, P.G.; Shao, L.; Hewitt, M.C.; Campbell, J.E.; Dedic, N.; Hopkins, S.C.; Koblan, K.S.; et al. Ulotaront: A TAAR1 Agonist for the Treatment of Schizophrenia. ACS Med. Chem. Lett. 2021, 13, 92–98. [Google Scholar] [CrossRef]
- Revel, F.G.; Moreau, J.L.; Gainetdinov, R.R.; Ferragud, A.; Velázquez-Sánchez, C.; Sotnikova, T.D.; Morairty, S.R.; Harmeier, A.; Groebke Zbinden, K.; Norcross, R.D.; et al. Trace amine-associated receptor 1 partial agonism reveals novel paradigm for neuropsychiatric therapeutics. Biol. Psychiatry 2012, 72, 934–942. [Google Scholar] [CrossRef]
- Revel, F.G.; Moreau, J.-L.; Pouzet, B.; Mory, R.; Bradaia, A.; Buchy, D.; Metzler, V.; Chaboz, S.; Groebke Zbinden, K.; Galley, G.; et al. A new perspective for schizophrenia: TAAR1 agonists reveal antipsychotic- and antidepressant-like activity, improve cognition and control body weight. Mol. Psychiatry 2013, 18, 543–556. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wu, R.; Li, J.X. TAAR1 and Psychostimulant Addiction. Cell. Mol. Neurobiol. 2020, 40, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Sukhanov, I.; Dorotenko, A.; Dolgorukova, A.; Hoener, M.C.; Gainetdinov, R.R.; Bespalov, A.Y. Activation of trace amine-associated receptor 1 attenuates schedule-induced polydipsia in rats. Neuropharmacology 2019, 144, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Galley, G.; Stalder, H.; Goergler, A.; Hoener, M.C.; Norcross, R.D. Optimisation of imidazole compounds as selective TAAR1 agonists: Discovery of RO5073012. Bioorg. Med. Chem. Lett. 2012, 22, 5244–5248. [Google Scholar] [CrossRef]
- Galley, G.; Beurier, A.; Décoret, G.; Goergler, A.; Hutter, R.; Mohr, S.; Pähler, A.; Schmid, P.; Türck, D.; Unger, R.; et al. Discovery and Characterization of 2-Aminooxazolines as Highly Potent, Selective, and Orally Active TAAR1 Agonists. ACS Med. Chem. Lett. 2015, 7, 192–197. [Google Scholar] [CrossRef]
- Guariento, S.; Tonelli, M.; Espinoza, S.; Gerasimov, A.S.; Gainetdinov, R.R.; Cichero, E. Rational design, chemical synthesis and biological evaluation of novel biguanides exploring species-specificity responsiveness of TAAR1 agonists. Eur. J. Med. Chem. 2018, 146, 171–184. [Google Scholar] [CrossRef]
- Krasavin, M.; Lukin, A.; Sukhanov, I.; Gerasimov, A.; Kuvarzin, S.; Efimova, E.V.; Dorofeikova, M.; Nichugovskaya, A.; Matveev, A.; Onokhin, K.; et al. Department of Medicinal Chemistry, Institute of Chemistry, Saint Petersburg State University, Saint Petersburg 199034, Russia. Eur. J. Med. Chem. 2022; submitting. [Google Scholar]
- Espinoza, S.; Masri, B.; Salahpour, A.; Gainetdinov, R.R. BRET approaches to characterize dopamine and TAAR1 receptor phar-macology and signaling. Methods Mol. Biol. 2013, 964, 107–122. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Leo, D.; Sukhanov, I.; Zoratto, F.; Illiano, P.; Caffino, L.; Sanna, F.; Messa, G.; Emanuele, M.; Esposito, A.; Dorofeikova, M.; et al. Pronounced Hyperactivity, Cognitive Dysfunctions, and BDNF Dysregulation in Dopamine Transporter Knock-out Rats. J. Neurosci. 2018, 38, 1959–1972. [Google Scholar] [CrossRef]
- Efimova, E.V.; Gainetdinov, R.R.; Budygin, E.A.; Sotnikova, T.D. Dopamine transporter mutant animals: A translational perspective. J. Neurogenet. 2016, 30, 5–15. [Google Scholar] [CrossRef] [Green Version]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Bateman, A.; Martin, M.J.; O’Donovan, C.; Magrane, M.; Alpi, E.; Antunes, R.; Bely, B.; Bingley, M.; Bonilla, C.; Britto, R.; et al. The UniProt Consortium, UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [Google Scholar] [CrossRef]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Lu, C.; Wu, C.; Ghoreishi, D.; Chen, W.; Wang, L.; Damm, W.; Ross, G.A.; Dahlgren, M.K.; Russell, E.; Von Bargen, C.D.; et al. OPLS4: Improving Force Field Accuracy on Challenging Regimes of Chemical Space. J. Chem. Theory Comput. 2021, 17, 4291–4300. [Google Scholar] [CrossRef] [PubMed]
- Greenwood, J.R.; Calkins, D.; Sullivan, A.P.; Shelley, J.C. Towards the comprehensive, rapid, and accurate prediction of the favorable tautomeric states of drug-like molecules in aqueous solution. J. Comput. Aided Mol. Des. 2010, 24, 591–604. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Tonelli, M.; Espinoza, S.; Gainetdinov, R.R.; Cichero, E. Novel biguanide-based derivatives scouted as TAAR1 agonists: Synthesis, biological evaluation, ADME prediction and molecular docking studies. Eur. J. Med. Chem. 2017, 127, 781–792. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Ar | Agonistic Activity a at 1 μM, % | EC50, μM |
|---|---|---|---|
| 1 |  | 65 | 0.507 |
| 7 |  | Inactive b | - |
| 8 |  | Inactive | - |
| 9 |  | 89 | 0.052 |
| 10 |  | Inactive | - |
| 11 |  | 84 | 0.206 |
| 12 |  | 75 | 0.273 |
| 13 |  | Inactive | - |
| 14 |  | Inactive | - |
| 15 |  | Inactive | inactive |
| 16 |  | 107 c | 0.143 |
| 17 |  | 84 c | 0.033 |
| 18 (AP163) |  | 85 c | 0.112 |
| 19 |  | 80 c | 0.042 |
| 20 |  | 82 c | 0.914 |
| 21 |  | 96 c | 0.035 |
| 22 |  | 84c | 1.90 |
| 23 |  | Inactive | - |
| 24 |  | 31 c | 0.635 |
| 25 |  | 41 c | 0.634 |
| 26 |  | Inactive | - |
| 27 |  | Inactive | - |
| 28 |  | Inactive | - |
| 29 | - | Inactive | - |
| Compound | GScore | ΔG | ‘Correctness’ of Ligand Binding | TAAR1 Activity |
|---|---|---|---|---|
| Ralmitaront | −8.91 | −36.85 | Reference ligand | Active |
| 7 | −8.26 | −30.93 | Incorrect | Inactive |
| 9 | −8.26 | −33.41 | Fully correct | Active |
| 10 | −7.87 | −29.50 | Incorrect | Inactive |
| 12 | −8.26 | −42.94 | Fully correct | Active |
| 13 | −8.45 | −27.27 | Partially correct | Inactive |
| 14 | −5.92 | −33.08 | Incorrect | Inactive |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krasavin, M.; Peshkov, A.A.; Lukin, A.; Komarova, K.; Vinogradova, L.; Smirnova, D.; Kanov, E.V.; Kuvarzin, S.R.; Murtazina, R.Z.; Efimova, E.V.; et al. Discovery and In Vivo Efficacy of Trace Amine-Associated Receptor 1 (TAAR1) Agonist 4-(2-Aminoethyl)-N-(3,5-dimethylphenyl)piperidine-1-carboxamide Hydrochloride (AP163) for the Treatment of Psychotic Disorders. Int. J. Mol. Sci. 2022, 23, 11579. https://doi.org/10.3390/ijms231911579
Krasavin M, Peshkov AA, Lukin A, Komarova K, Vinogradova L, Smirnova D, Kanov EV, Kuvarzin SR, Murtazina RZ, Efimova EV, et al. Discovery and In Vivo Efficacy of Trace Amine-Associated Receptor 1 (TAAR1) Agonist 4-(2-Aminoethyl)-N-(3,5-dimethylphenyl)piperidine-1-carboxamide Hydrochloride (AP163) for the Treatment of Psychotic Disorders. International Journal of Molecular Sciences. 2022; 23(19):11579. https://doi.org/10.3390/ijms231911579
Chicago/Turabian StyleKrasavin, Mikhail, Anatoly A. Peshkov, Alexey Lukin, Kristina Komarova, Lyubov Vinogradova, Daria Smirnova, Evgeny V. Kanov, Savelii R. Kuvarzin, Ramilya Z. Murtazina, Evgeniya V. Efimova, and et al. 2022. "Discovery and In Vivo Efficacy of Trace Amine-Associated Receptor 1 (TAAR1) Agonist 4-(2-Aminoethyl)-N-(3,5-dimethylphenyl)piperidine-1-carboxamide Hydrochloride (AP163) for the Treatment of Psychotic Disorders" International Journal of Molecular Sciences 23, no. 19: 11579. https://doi.org/10.3390/ijms231911579