
Enzymatic Construction of DARPin-Based Targeted Delivery Systems Using Protein Farnesyltransferase and a Capture and Release Strategy
 , and
, and
Abstract
:
1. Introduction
2. Results and Discussion
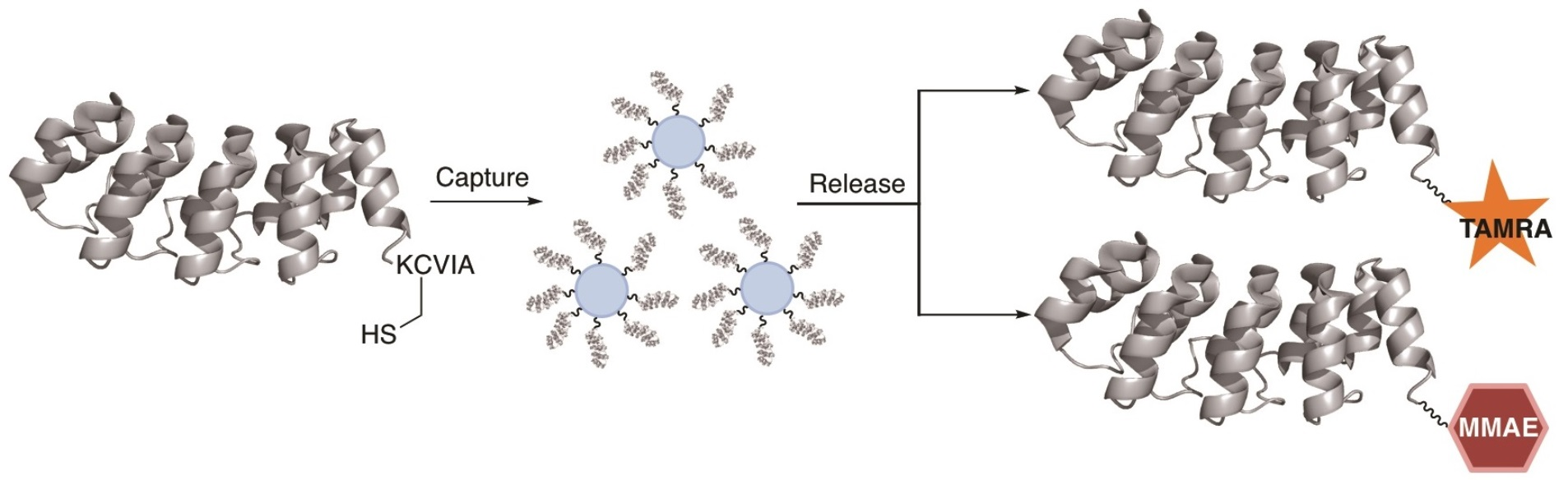
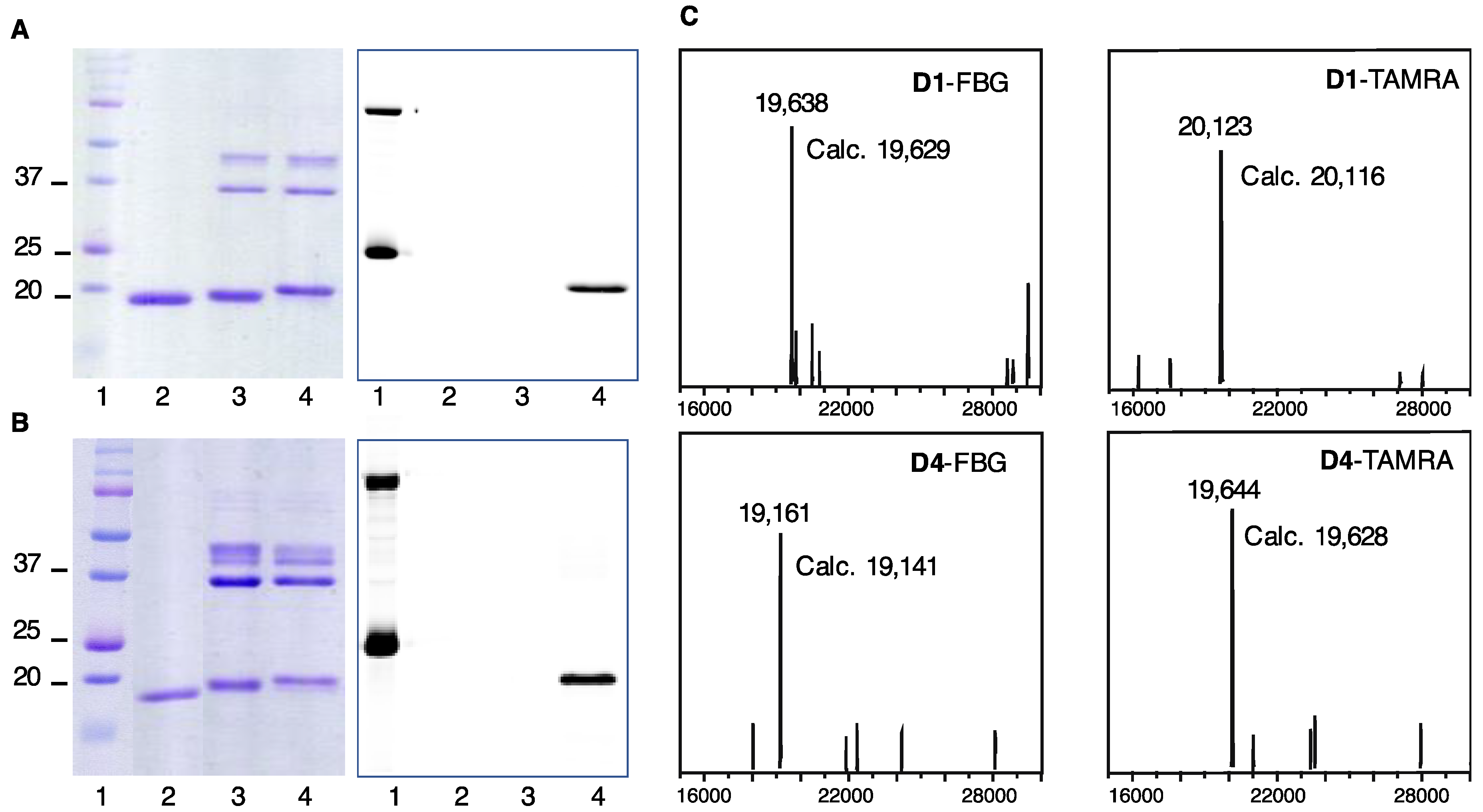
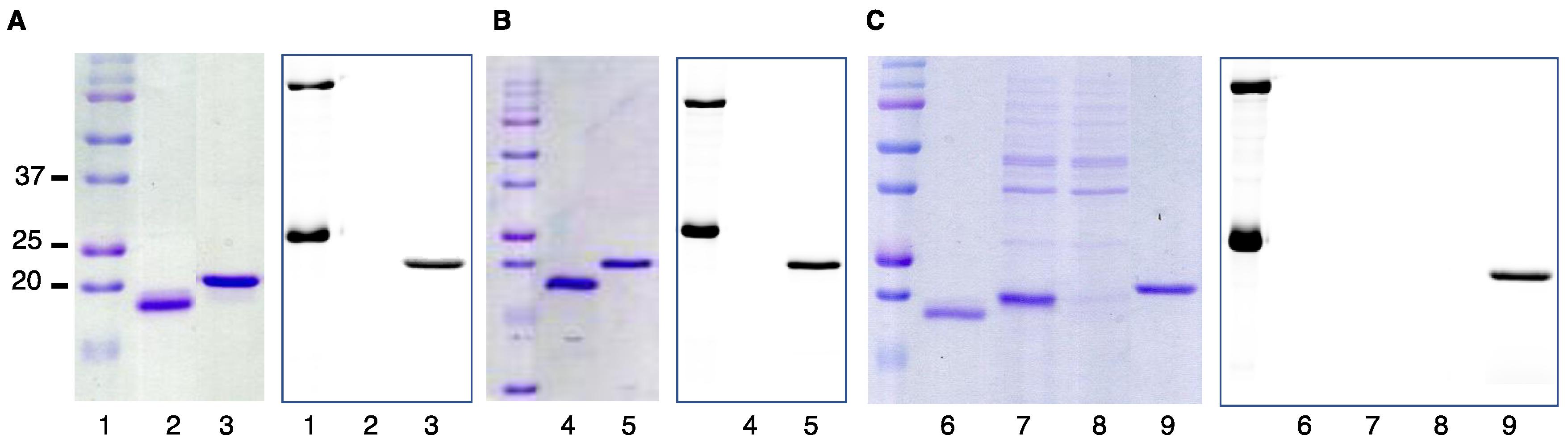
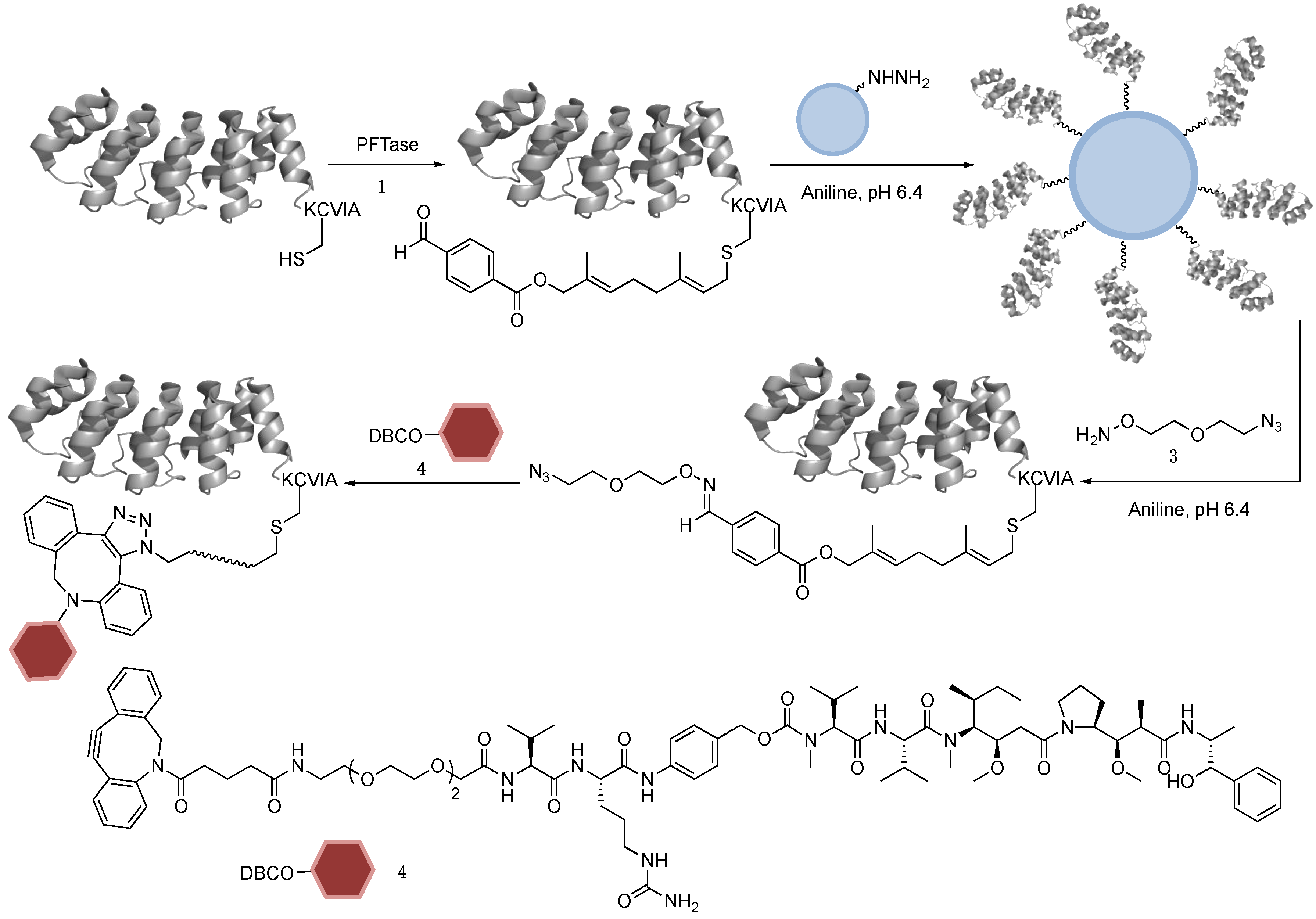
2.1. DARPin Modification by PFTase
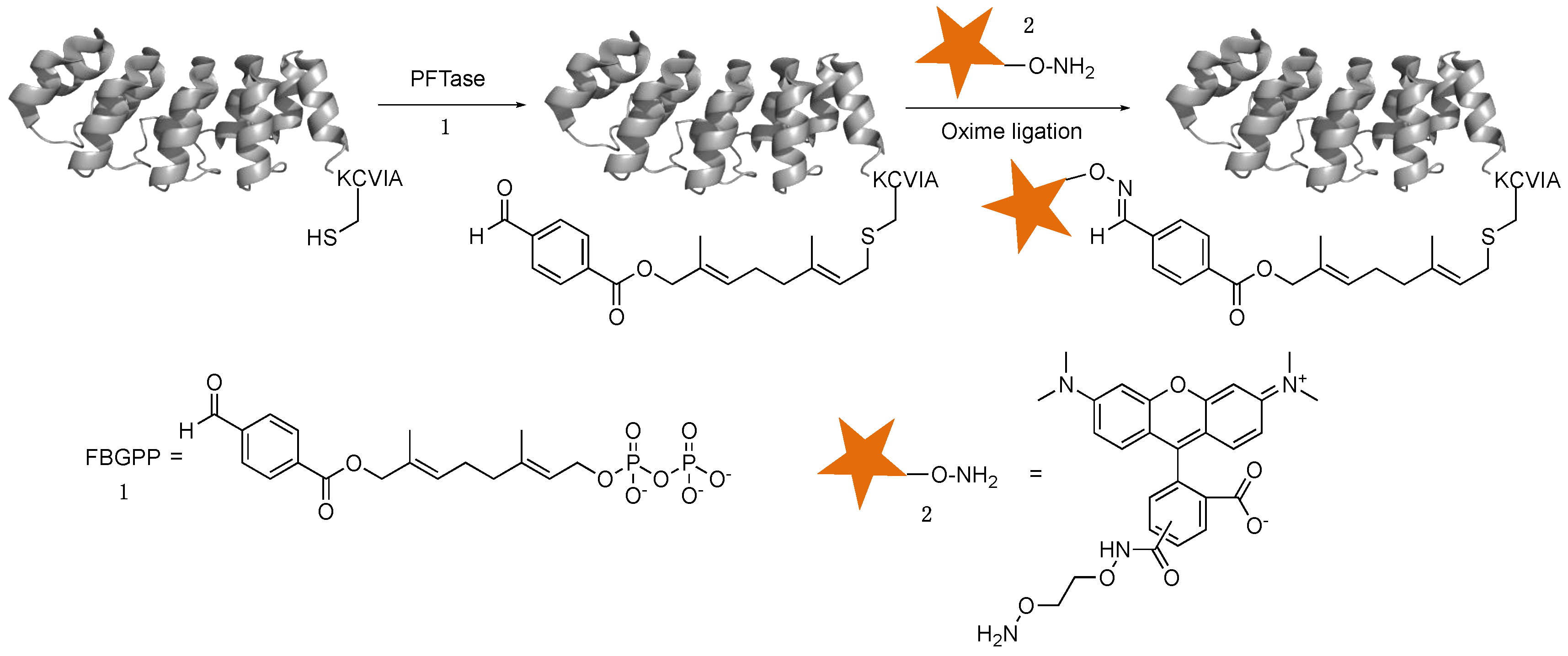
2.2. Enzymatic Incorporation of Aldehyde Functionality and Fluorophore Conjugation
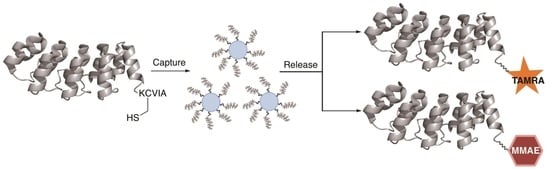
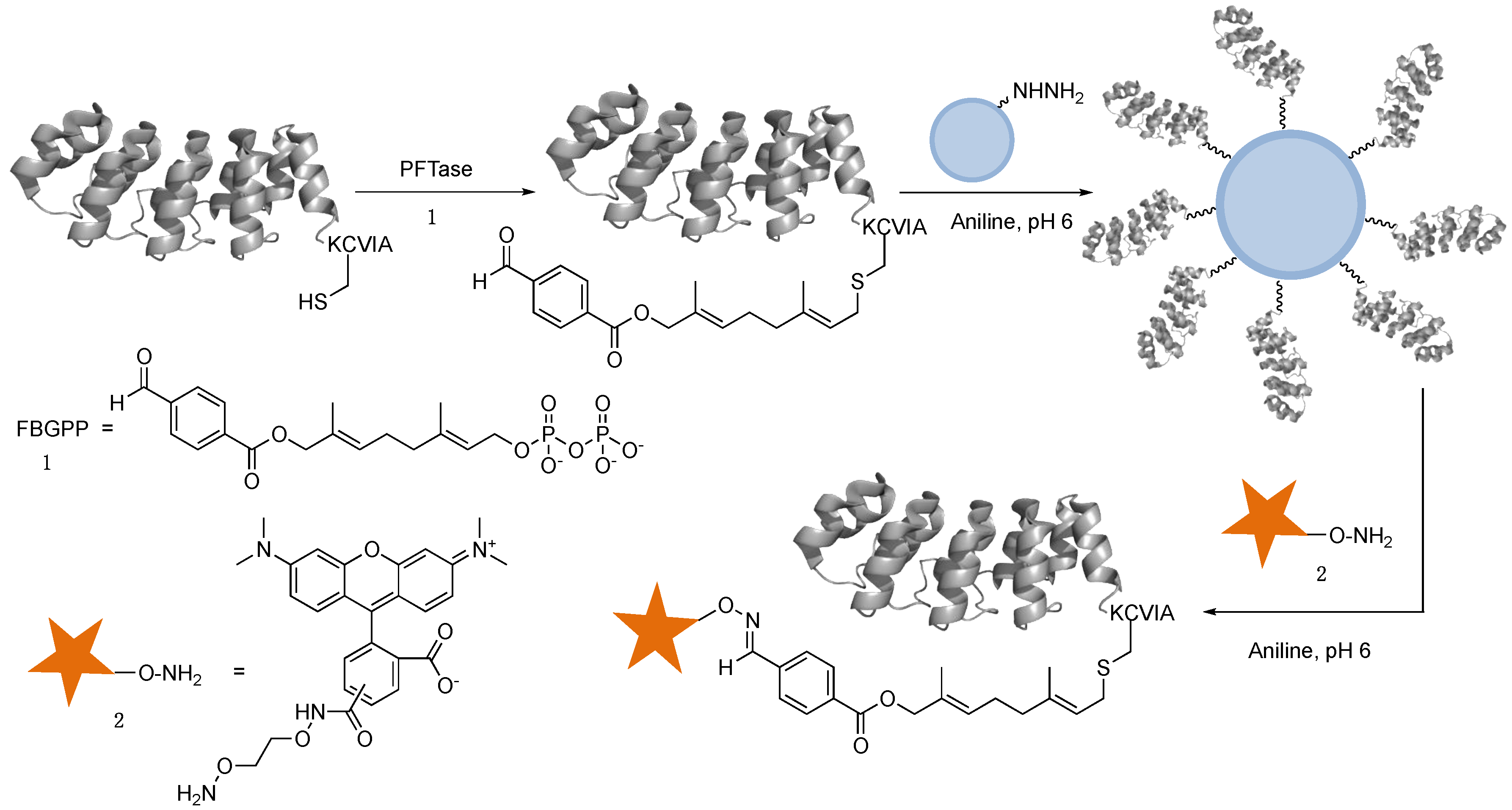
2.3. Capture and Release Strategy Allows Facile Construction of DARPin-TAMRA Conjugates
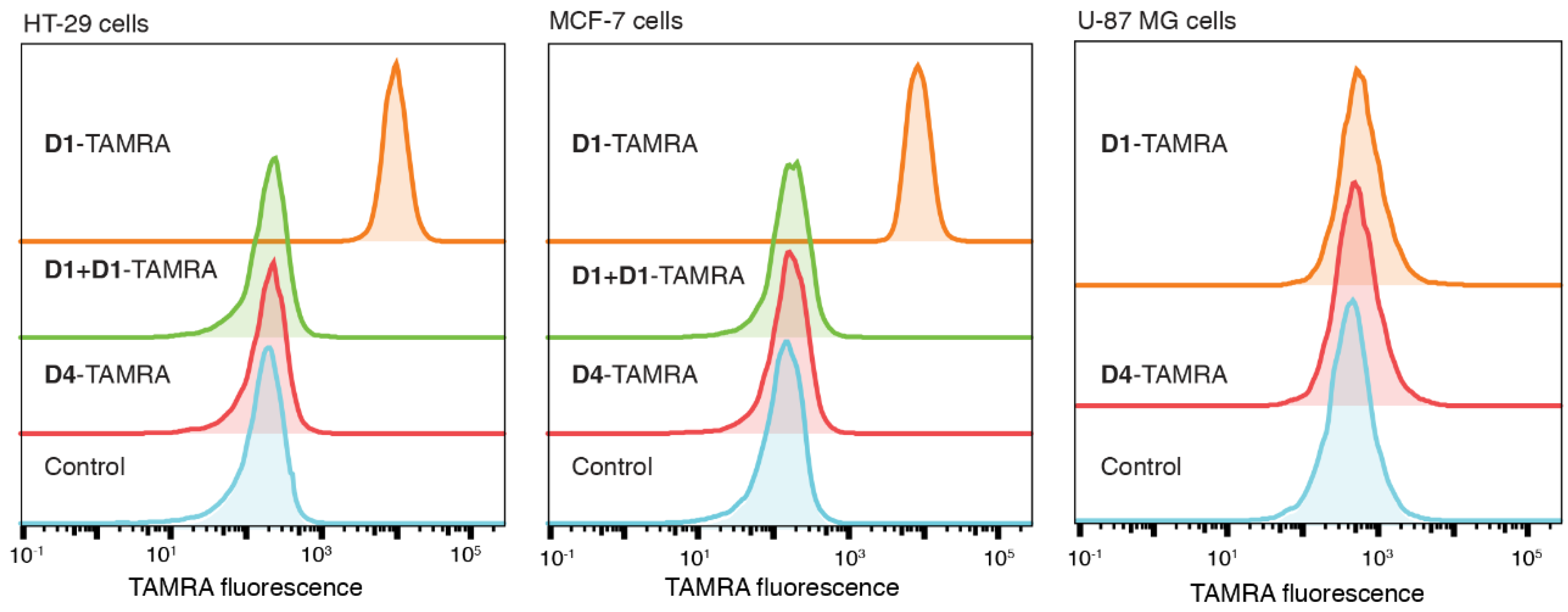
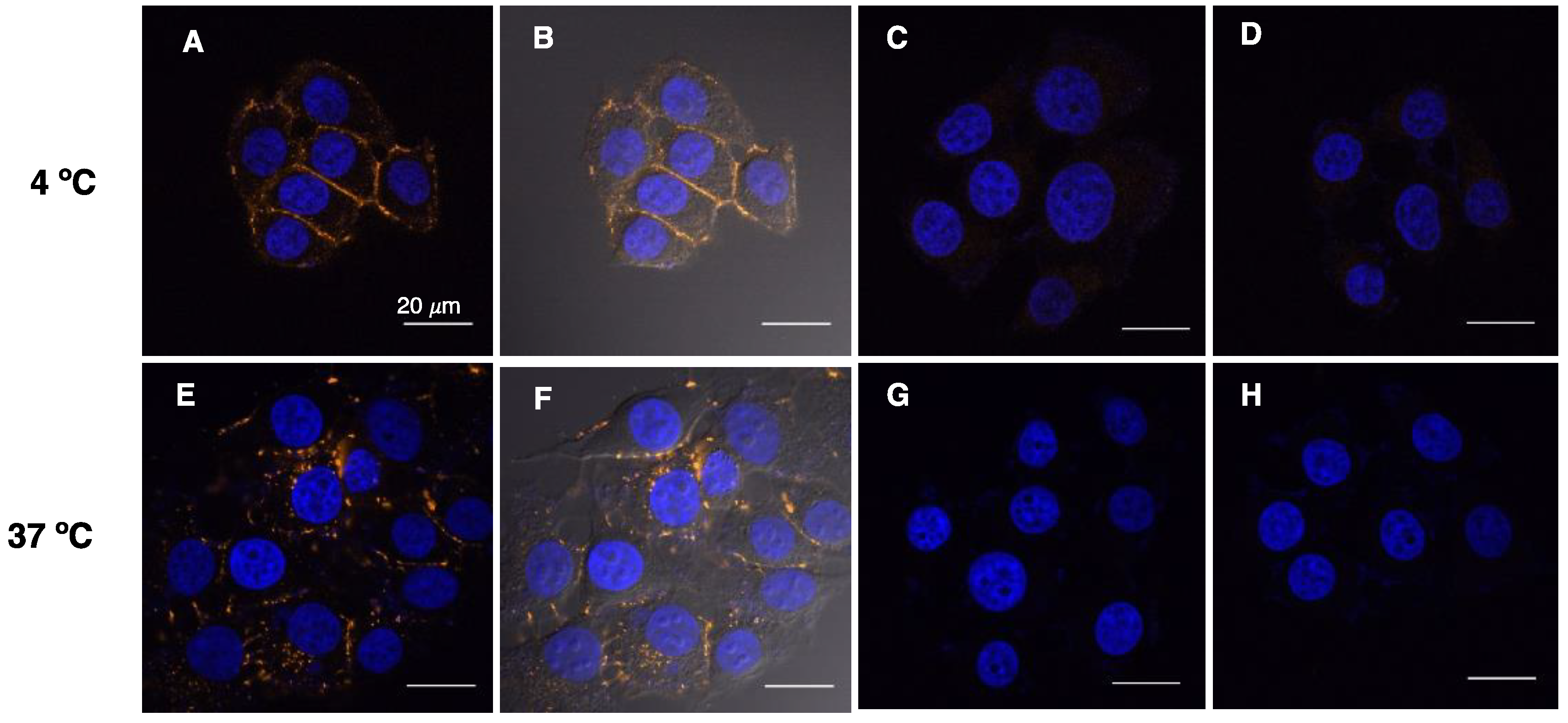
2.4. D1-TAMRA Retains Selective Binding to Cell-Surface EpCAM
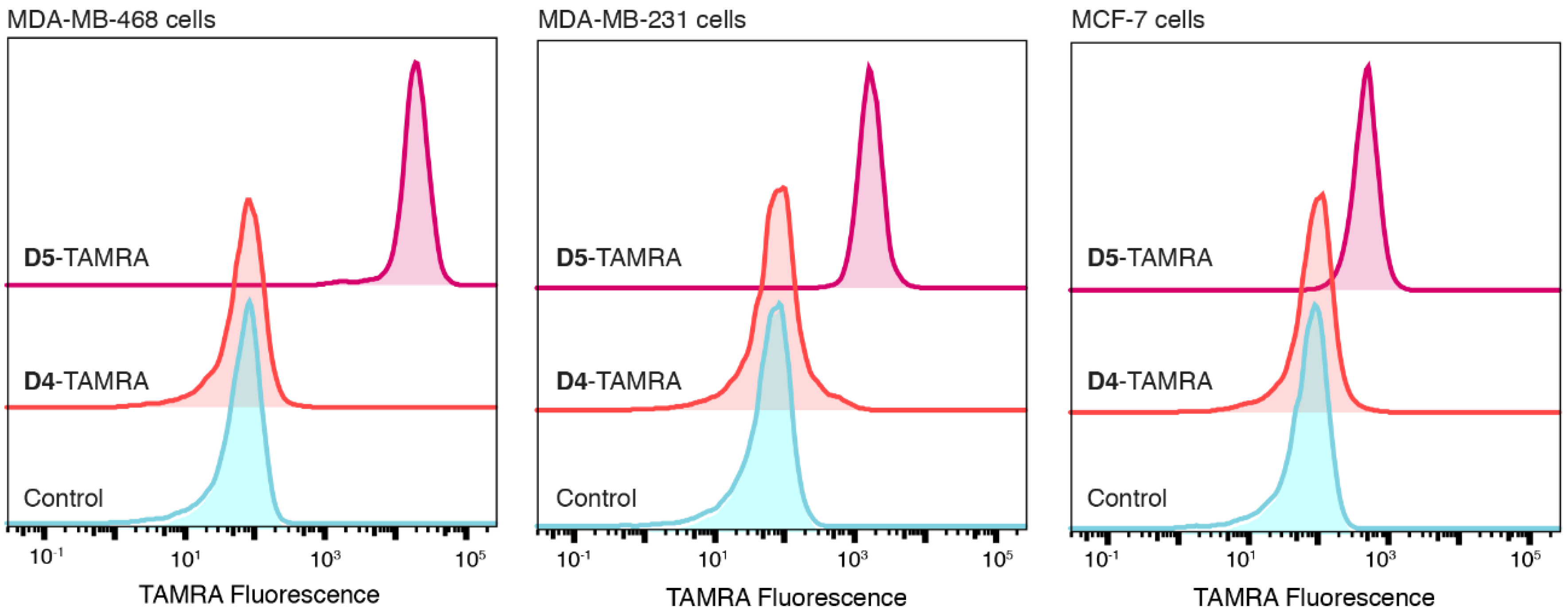
2.5. Application of PFTase Labeling to a DARPin Binding another Target
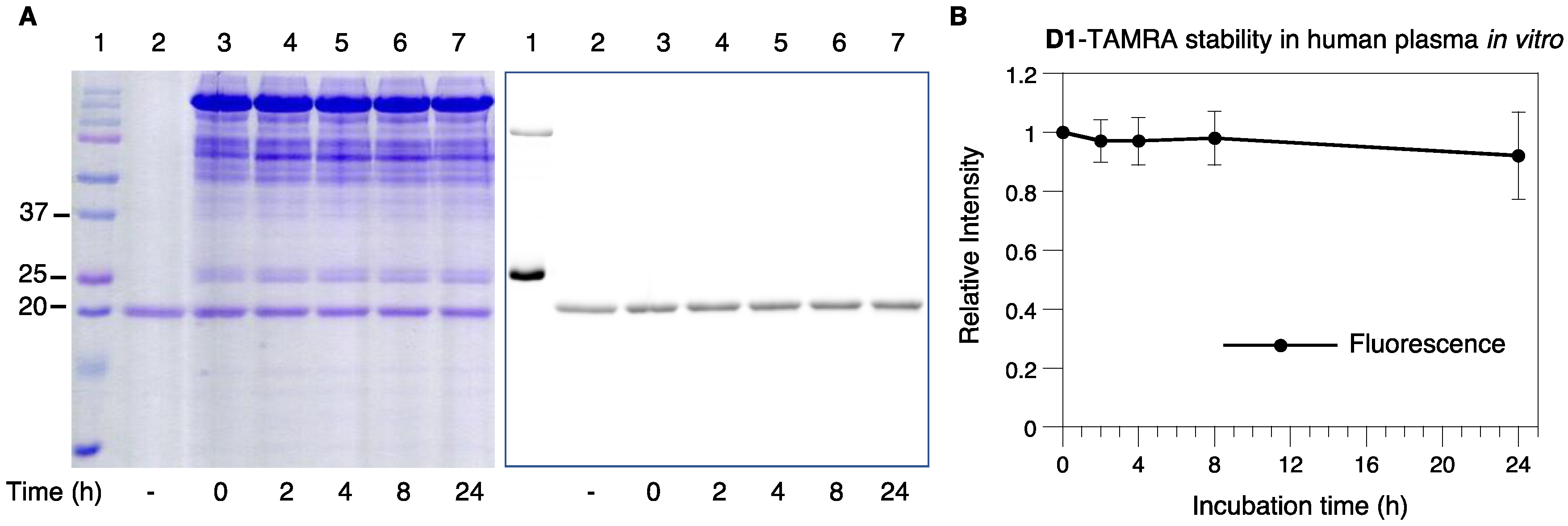
2.6. Serum Stability of D1-TAMRA
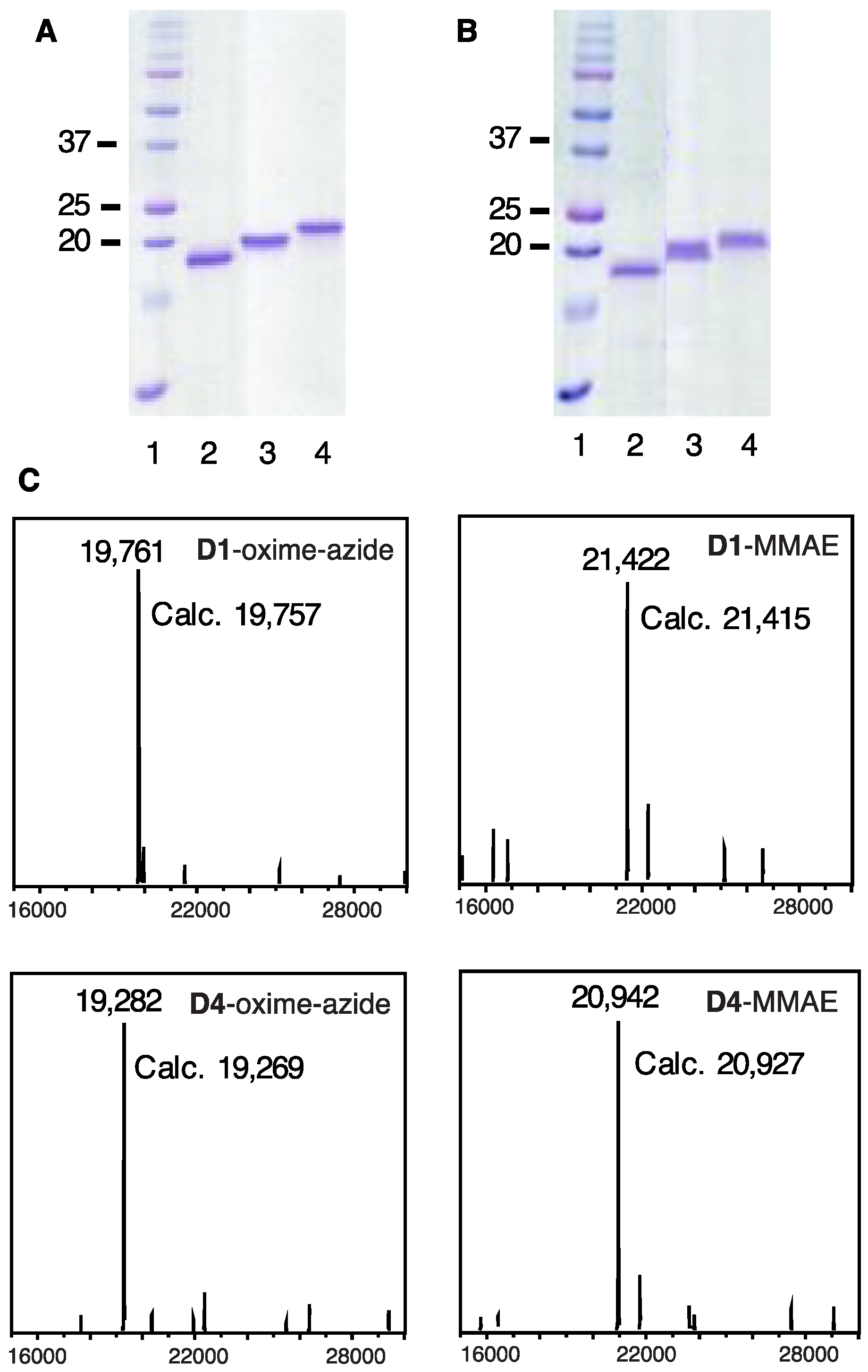
2.7. Construction of DARPin-MMAE Conjugates
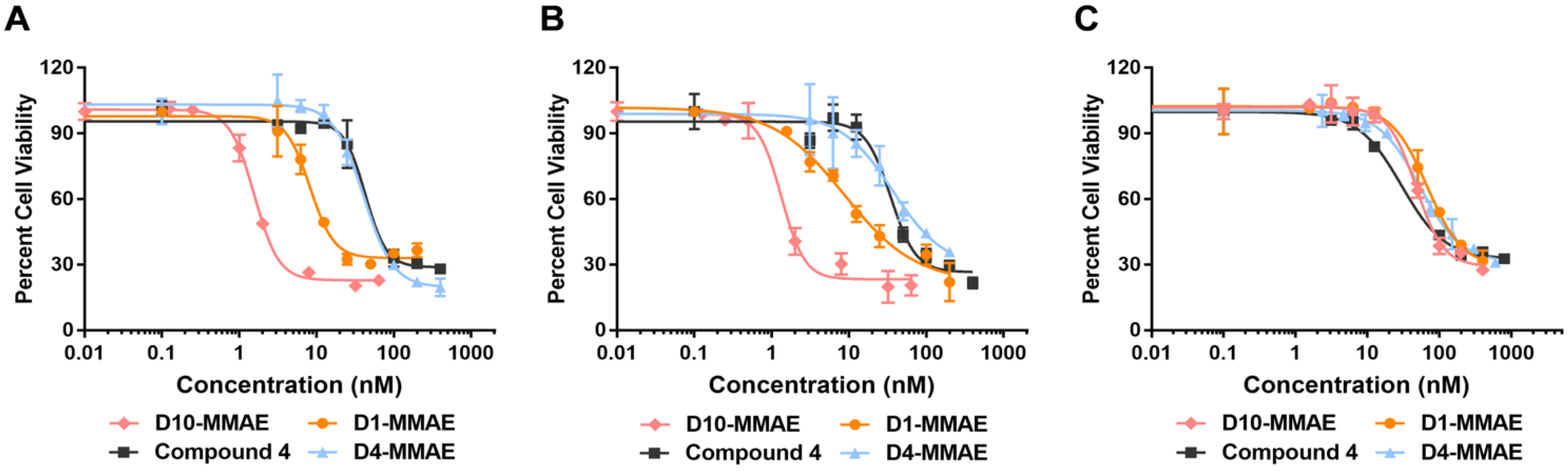
2.8. Cytotoxicity of DARPin-MMAE Conjugates In Vitro
3. Materials and Methods
3.1. Materials
3.2. Enzymatic Modification of DARPins
3.3. DARPin-FBG Conjugation to TAMRA-Aminooxy (2)
3.4. Capture and Release Strategy to Construct DARPin-TAMRA Conjugates
3.5. Flow Cytometry Analysis of D1-TAMRA Binding to Cell Surface EpCAM
3.6. Visualization of D1-TAMRA Binding and Internalization to MCF-7 Cells
3.7. Serum Stability of D1-TAMRA In Vitro
3.8. Construction of DARPin-MMAE Conjugates
3.9. DARPin-MMAE Cytotoxicity Assay in Cell Cultures
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dozier, J.K.; Distefano, M.D. Site-Specific Pegylation of Therapeutic Proteins. Int. J. Mol. Sci. 2015, 16, 25831–25864. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.; Bhavanasi, S.; Quadir, M.; Singh, K.; Ghosh, G.; Vasamreddy, K.; Ghosh, A.; Siahaan, T.J.; Banerjee, S.; Banerjee, S.K. Protein PEGylation for Cancer Therapy: Bench to Bedside. J. Cell Commun. Signal. 2019, 13, 319–330. [Google Scholar] [CrossRef] [PubMed]
- Stennicke, H.R.; Kjalke, M.; Karpf, D.M.; Balling, K.W.; Johansen, P.B.; Elm, T.; Øvlisen, K.; Möller, F.; Holmberg, H.L.; Gudme, C.N.; et al. A Novel B-Domain O-GlycoPEGylated FVIII (N8-GP) Demonstrates Full Efficacy and Prolonged Effect in Hemophilic Mice Models. Blood 2013, 121, 2108–2116. [Google Scholar] [CrossRef] [PubMed]
- Beck, A.; Goetsch, L.; Dumontet, C.; Corvaïa, N. Strategies and Challenges for the next Generation of Antibody-Drug Conjugates. Nat. Rev. Drug Discov. 2017, 16, 315–337. [Google Scholar] [CrossRef] [PubMed]
- Tai, Y.T.; Mayes, P.A.; Acharya, C.; Zhong, M.Y.; Cea, M.; Cagnetta, A.; Craigen, J.; Yates, J.; Gliddon, L.; Fieles, W.; et al. Novel Anti-B-Cell Maturation Antigen Antibody-Drug Conjugate (GSK2857916) Selectively Induces Killing of Multiple Myeloma. Blood 2014, 123, 3128–3138. [Google Scholar] [CrossRef]
- Zammarchi, F.; Corbett, S.; Adams, L.; Tyrer, P.C.; Kiakos, K.; Janghra, N.; Marafioti, T.; Britten, C.E.; Havenith, C.E.G.; Chivers, S.; et al. ADCT-402, a PBD Dimer–Containing Antibody Drug Conjugate Targeting CD19-Expressing Malignancies. Blood 2018, 131, 1094–1105. [Google Scholar] [CrossRef]
- Khongorzul, P.; Ling, C.J.; Khan, F.U.; Ihsan, A.U.; Zhang, J. Antibody-Drug Conjugates: A Comprehensive Review. Mol. Cancer Res. 2020, 18, 3–19. [Google Scholar] [CrossRef]
- Ogitani, Y.; Aida, T.; Hagihara, K.; Yamaguchi, J.; Ishii, C.; Harada, N.; Soma, M.; Okamoto, H.; Oitate, M.; Arakawa, S.; et al. DS-8201a, a Novel HER2-Targeting ADC with a Novel DNA Topoisomerase I Inhibitor, Demonstrates a Promising Antitumor Efficacy with Differentiation from T-DM1. Clin. Cancer Res. 2016, 22, 5097–5108. [Google Scholar] [CrossRef]
- Tolmachev, V.; Orlova, A. Affibody Molecules as Targeting Vectors for PET Imaging. Cancers 2020, 12, 651. [Google Scholar] [CrossRef]
- Rashidian, M.; Keliher, E.J.; Bilate, A.M.; Duarte, J.N.; Wojtkiewicz, G.R.; Jacobsen, J.T.; Cragnolini, J.; Swee, L.K.; Victora, G.D.; Weissleder, R.; et al. Noninvasive Imaging of Immune Responses. Proc. Natl. Acad. Sci. USA 2015, 112, 6146–6151. [Google Scholar] [CrossRef] [Green Version]
- Dammes, N.; Peer, D. Monoclonal Antibody-Based Molecular Imaging Strategies and Theranostic Opportunities. Theranostics 2020, 10, 938–955. [Google Scholar] [CrossRef]
- Wang, Y.; Rozumalski, L.; Lichtenfels, C.; Petersberg, J.R.; Kilic, O.; Distefano, M.D.; Wagner, C.R. Engineering Biomimetic Trogocytosis with Farnesylated Chemically Self-Assembled Nanorings. BioRxiv 2022. [Google Scholar] [CrossRef]
- Zhang, Y.; Park, K.Y.; Suazo, K.F.; Distefano, M.D. Recent Progress in Enzymatic Protein Labelling Techniques and Their Applications. Chem. Soc. Rev. 2018, 47, 9106–9136. [Google Scholar] [CrossRef]
- Walsh, S.J.; Bargh, J.D.; Dannheim, F.M.; Hanby, A.R.; Seki, H.; Counsell, A.J.; Ou, X.; Fowler, E.; Ashman, N.; Takada, Y.; et al. Site-Selective Modification Strategies in Antibody-Drug Conjugates. Chem. Soc. Rev. 2021, 50, 1305–1353. [Google Scholar] [CrossRef] [PubMed]
- Adusumalli, S.R.; Rawale, D.G.; Singh, U.; Tripathi, P.; Paul, R.; Kalra, N.; Mishra, R.K.; Shukla, S.; Rai, V. Single-Site Labeling of Native Proteins Enabled by a Chemoselective and Site-Selective Chemical Technology. J. Am. Chem. Soc. 2018, 140, 15114–15123. [Google Scholar] [CrossRef] [PubMed]
- Lobba, M.J.; Fellmann, C.; Marmelstein, A.M.; Maza, J.C.; Kissman, E.N.; Robinson, S.A.; Staahl, B.T.; Urnes, C.; Lew, R.J.; Mogilevsky, C.S.; et al. Site-Specific Bioconjugation through Enzyme-Catalyzed Tyrosine-Cysteine Bond Formation. ACS Cent. Sci. 2020, 6, 1564–1571. [Google Scholar] [CrossRef]
- Zhang, L.; Kang, J.; Liu, S.; Zhang, X.; Sun, J.; Hu, Y.; Yang, Y.; Chen, L. A Chemical Covalent Tactic for Bio-Thiol Sensing and Protein Labeling Agent Design. Chem. Commun. 2020, 56, 11485–11488. [Google Scholar] [CrossRef]
- Rashidian, M.; Dozier, J.K.; Distefano, M.D. Enzymatic Labeling of Proteins: Techniques and Approaches. Bioconjug. Chem. 2013, 24, 1277–1294. [Google Scholar] [CrossRef]
- Spycher, P.R.; Amann, C.A.; Wehrmüller, J.E.; Hurwitz, D.R.; Kreis, O.; Messmer, D.; Ritler, A.; Küchler, A.; Blanc, A.; Béhé, M.; et al. Dual, Site-Specific Modification of Antibodies by Using Solid-Phase Immobilized Microbial Transglutaminase. ChemBioChem 2017, 18, 1923–1927. [Google Scholar] [CrossRef]
- Wang, H.H.; Altun, B.; Nwe, K.; Tsourkas, A. Proximity-Based Sortase-Mediated Ligation. Angew. Chem. Int. Ed. 2017, 56, 5349–5352. [Google Scholar] [CrossRef] [Green Version]
- Bellucci, J.J.; Amiram, M.; Bhattacharyya, J.; McCafferty, D.; Chilkoti, A. Three-in-One Chromatography-Free Purification, Tag Removal, and Site-Specific Modification of Recombinant Fusion Proteins Using Sortase A and Elastin-like Polypeptides. Angew. Chem. Int. Ed 2013, 125, 3791–3796. [Google Scholar] [CrossRef]
- Policarpo, R.L.; Kang, H.; Liao, X.; Rabideau, A.E.; Simon, M.D.; Pentelute, B.L. Flow-Based Enzymatic Ligation by Sortase A. Angew. Chem. Int. Ed. 2014, 126, 9357–9362. [Google Scholar] [CrossRef]
- Chen, I.; Dorr, B.M.; Liu, D.R. A General Strategy for the Evolution of Bond-Forming Enzymes Using Yeast Display. Proc. Natl. Acad. Sci. USA 2011, 108, 11399–11404. [Google Scholar] [CrossRef] [PubMed]
- Heck, T.; Pham, P.H.; Yerlikaya, A.; Thöny-Meyer, L.; Richter, M. Sortase A Catalyzed Reaction Pathways: A Comparative Study with Six SrtA Variants. Catal. Sci. Technol. 2014, 4, 2946–2956. [Google Scholar] [CrossRef]
- Warden-Rothman, R.; Caturegli, I.; Popik, V.; Tsourkas, A. Sortase-Tag Expressed Protein Ligation: Combining Protein Purification and Site-Specific Bioconjugation into a Single Step. Anal. Chem. 2013, 85, 11090–11097. [Google Scholar] [CrossRef]
- Zhang, Y.; Auger, S.; Schaefer, J.V.; Plückthun, A.; Distefano, M.D. Site-Selective Enzymatic Labeling of Designed Ankyrin Repeat Proteins Using Protein Farnesyltransferase. In Methods in Molecular Biology: Bioconjugation; Massa, S., Ed.; Humana: New York, NY, USA, 2019; pp. 207–219. ISBN 9781452289830. [Google Scholar]
- Palsuledesai, C.C.; Distefano, M.D. Protein Prenylation: Enzymes, Therapeutics, and Biotechnology Applications. ACS Chem. Biol. 2015, 10, 51–62. [Google Scholar] [CrossRef]
- Kim, Y.; Park, T.; Woo, S.; Lee, H.; Kim, S.; Cho, J.; Jung, D.; Kim, Y.; Kwon, H.; Oh, K.; et al. Antibody-Active Agent Conjugates and Methods of Use. U.S. Patent No. 9669107 B2, 6 June 2017. [Google Scholar]
- Wang, Y.; Kilic, O.; Csizmar, C.M.; Ashok, S.; Hougland, J.L.; Distefano, M.D.; Wagner, C.R. Engineering Reversible Cell-Cell Interactions Using Enzymatically Lipidated Chemically Self-Assembled Nanorings. Chem. Sci. 2021, 12, 331–340. [Google Scholar] [CrossRef]
- Lee, J.J.; Choi, H.J.; Yun, M.; Kang, Y.; Jung, J.E.; Ryu, Y.; Kim, T.Y.; Cha, Y.J.; Cho, H.S.; Min, J.J.; et al. Enzymatic Prenylation and Oxime Ligation for the Synthesis of Stable and Homogeneous Protein-Drug Conjugates for Targeted Therapy. Angew. Chem. Int. Ed. 2015, 54, 12020–12024. [Google Scholar] [CrossRef]
- Rose, M.W.; Rose, N.D.; Boggs, J.; Lenevich, S.; Xu, J.; Barany, G.; Distefano, M.D. Evaluation of Geranylazide and Farnesylazide Diphosphate for Incorporation of Prenylazides into a CAAX Box-Containing Peptide Using Protein Farnesyltransferase. J. Pept. Res. 2005, 65, 529–537. [Google Scholar] [CrossRef]
- Hosokawa, A.; Wollack, J.W.; Zhang, Z.; Chen, L.; Barany, G.; Distefano, M.D. Evaluation of an Alkyne-Containing Analogue of Farnesyl Diphosphate as a Dual Substrate for Protein-Prenyltransferases. Int. J. Pept. Res. Ther. 2007, 13, 345–354. [Google Scholar] [CrossRef]
- Rashidian, M.; Song, J.M.; Pricer, R.E.; Distefano, M.D. Chemoenzymatic Reversible Immobilization and Labeling of Proteins without Prior Purification. J. Am. Chem. Soc. 2012, 134, 8455–8467. [Google Scholar] [CrossRef] [PubMed]
- Wollack, J.W.; Monson, B.J.; Dozier, J.K.; Dalluge, J.J.; Poss, K.; Hilderbrand, S.A.; Distefano, M.D. Site-Specific Labeling of Proteins and Peptides with Trans-Cyclooctene Containing Handles Capable of Tetrazine Ligation. Chem. Biol. Drug Des. 2014, 84, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Suazo, K.F.; Park, K.; Distefano, M.D. A Not-So-Ancient Grease History: Click Chemistry and Protein Lipid Modifications. Chem. Rev. 2021, 121, 7178–7248. [Google Scholar] [CrossRef] [PubMed]
- Plückthun, A. Designed Ankyrin Repeat Proteins (DARPins): Binding Proteins for Research, Diagnostics, and Therapy. Annu. Rev. Pharmacol. Toxicol. 2015, 55, 489–511. [Google Scholar] [CrossRef] [PubMed]
- Stumpp, M.T.; Dawson, K.M.; Binz, H.K. Beyond Antibodies: The DARPin® Drug Platform. BioDrugs 2020, 34, 423–433. [Google Scholar] [CrossRef]
- Bery, N.; Miller, A.; Rabbitts, T. A Potent KRAS Macromolecule Degrader Specifically Targeting Tumours with Mutant KRAS. Nat. Commun. 2020, 11, 1–14. [Google Scholar] [CrossRef]
- Tamaskovic, R.; Simon, M.; Stefan, N.; Schwill, M.; Plückthun, A. Designed Ankyrin Repeat Proteins (DARPins) from Research to Therapy. Methods Enzym. 2012, 503, 101–134. [Google Scholar]
- van den Brand, D.; van Lith, S.A.M.; de Jong, J.M.; Gorris, M.A.J.; Palacio-Castañeda, V.; Couwenbergh, S.T.; Goldman, M.R.G.; Ebisch, I.; Massuger, L.F.; Leenders, W.P.J.; et al. EpCAM-Binding Darpins for Targeted Photodynamic Therapy of Ovarian Cancer. Cancers 2020, 12, 1762. [Google Scholar] [CrossRef]
- Brandl, F.; Busslinger, S.; Zangemeister-Wittke, U.; Plückthun, A. Optimizing the Anti-Tumor Efficacy of Protein-Drug Conjugates by Engineering the Molecular Size and Half-Life. J. Control. Release 2020, 327, 186–197. [Google Scholar] [CrossRef]
- Deyev, S.; Vorobyeva, A.; Schulga, A.; Proshkina, G.; Güler, R.; Löfblom, J.; Mitran, B.; Garousi, J.; Altai, M.; Buijs, J.; et al. Comparative Evaluation of Two DARPin Variants: Effect of Affinity, Size, and Label on Tumor Targeting Properties. Mol. Pharm. 2019, 16, 995–1008. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, R.; Sosabowski, J.; Livanos, M.; Leyton, J.; Vigor, K.; Bhavsar, G.; Nagy-Davidescu, G.; Rashid, M.; Miranda, E.; Yeung, J.; et al. Development of the Designed Ankyrin Repeat Protein (DARPin) G3 for HER2 Molecular Imaging. Eur. J. Nucl. Med. Mol. Imaging 2015, 42, 288–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorenzer, C.; Streußnig, S.; Tot, E.; Winkler, A.M.; Merten, H.; Brandl, F.; Sayers, E.J.; Watson, P.; Jones, A.T.; Zangemeister-Wittke, U.; et al. Targeted Delivery and Endosomal Cellular Uptake of DARPin-SiRNA Bioconjugates: Influence of Linker Stability on Gene Silencing. Eur. J. Pharm. Biopharm. 2019, 141, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, G.A.; Mason, M.; Christie, L.A.; Hansen, C.; Hernandez, L.M.; Burke, J.; Luhrs, K.A.; Hohman, T.C. Functional Characterization of Abicipar-Pegol, an Anti-VEGF DARPin Therapeutic That Potently Inhibits Angiogenesis and Vascular Permeability. Investig. Ophthalmol. Vis. Sci. 2018, 59, 5836–5846. [Google Scholar] [CrossRef]
- Sokolova, E.; Proshkina, G.; Kutova, O.; Shilova, O.; Ryabova, A.; Schulga, A.; Stremovskiy, O.; Zdobnova, T.; Balalaeva, I.; Deyev, S. Recombinant Targeted Toxin Based on HER2-Specific DARPin Possesses a Strong Selective Cytotoxic Effect in Vitro and a Potent Antitumor Activity in Vivo. J. Control. Release 2016, 233, 48–56. [Google Scholar] [CrossRef]
- Vorobyeva, A.; Konovalova, E.; Xu, T.; Schulga, A.; Altai, M.; Garousi, J.; Rinne, S.S.; Orlova, A.; Tolmachev, V.; Deyev, S. Feasibility of Imaging Epcam Expression in Ovarian Cancer Using Radiolabeled Darpin Ec1. Int. J. Mol. Sci. 2020, 21, 3310. [Google Scholar] [CrossRef]
- Stefan, N.; Martin-Killias, P.; Wyss-Stoeckle, S.; Honegger, A.; Zangemeister-Wittke, U.; Plückthun, A. DARPins Recognizing the Tumor-Associated Antigen EpCAM Selected by Phage and Ribosome Display and Engineered for Multivalency. J. Mol. Biol. 2011, 413, 826–843. [Google Scholar] [CrossRef]
- Simon, M.; Stefan, N.; Plückthun, A.; Zangemeister-Wittke, U. Epithelial Cell Adhesion Molecule-Targeted Drug Delivery for Cancer Therapy. Expert Opin Drug Deliv. 2013, 10, 451–468. [Google Scholar] [CrossRef]
- Patriarca, C.; Macchi, R.M.; Marschner, A.K.; Mellstedt, H. Epithelial Cell Adhesion Molecule Expression (CD326) in Cancer: A Short Review. Cancer Treat. Rev. 2012, 38, 68–75. [Google Scholar] [CrossRef]
- Bryant, K.L.; Mancias, J.D.; Kimmelman, A.C.; Der, C.J. KRAS: Feeding Pancreatic Cancer Proliferation. Trends Biochem. Sci. 2014, 39, 91–100. [Google Scholar] [CrossRef]
- Binz, H.K.; Stumpp, M.T.; Forrer, P.; Amstutz, P.; Plückthun, A. Designing Repeat Proteins: Well-Expressed, Soluble and Stable Proteins from Combinatorial Libraries of Consensus Ankyrin Repeat Proteins. J. Mol. Biol. 2003, 332, 489–503. [Google Scholar] [CrossRef]
- Boersma, Y.L.; Chao, G.; Steiner, D.; Wittrup, K.D.; Plückthun, A. Bispecific Designed Ankyrin Repeat Proteins (DARPins) Targeting Epidermal Growth Factor Receptor Inhibit A431 Cell Proliferation and Receptor Recycling. J. Biol. Chem. 2011, 286, 41273–41285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rae, J.M.; Scheys, J.O.; Clark, K.M.; Chadwick, R.B.; Kiefer, M.C.; Lippman, M.E. EGFR and EGFRvIII Expression in Primary Breast Cancer and Cell Lines. Breast Cancer Res. Treat. 2004, 87, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Maderna, A.; Leverett, C.A. Recent Advances in the Development of New Auristatins: Structural Modifications and Application in Antibody Drug Conjugates. Mol. Pharm. 2015, 12, 1798–1812. [Google Scholar] [CrossRef]
- Doronina, S.O.; Toki, B.E.; Torgov, M.Y.; Mendelsohn, B.A.; Cerveny, C.G.; Chace, D.F.; Deblanc, R.L.; Gearing, R.P.; Bovee, T.D.; Siegall, C.B.; et al. Development of Potent Monoclonal Antibody Auristatin Conjugates for Cancer Therapy. Nat. Biotech. 2003, 21, 778–784. [Google Scholar] [CrossRef]
- Miller, J.T.; Vitro, C.N.; Fang, S.; Benjamin, S.R.; Tumey, L.N. Enzyme-Agnostic Lysosomal Screen Identifies New Legumain-Cleavable ADC Linkers. Bioconjug. Chem. 2021, 32, 842–858. [Google Scholar] [CrossRef]
- Rashidian, M.; Mahmoodi, M.M.; Shah, R.; Dozier, J.K.; Wagner, C.R.; Distefano, M.D. A Highly Efficient Catalyst for Oxime Ligation and Hydrazone-Oxime Exchange Suitable for Bioconjugation. Bioconjug. Chem. 2013, 24, 333–342. [Google Scholar] [CrossRef]
- Dozier, J.K.; Distefano, M.D. An Enzyme-Coupled Continuous Fluorescence Assay for Farnesyl Diphosphate Synthases. Anal. Biochem. 2012, 421, 158–163. [Google Scholar] [CrossRef] [Green Version]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule | IC50 (nM) | Selectivity Ratio * | |||
|---|---|---|---|---|---|
| HT29 | MCF-7 | U87-MG | HT-29 | MCF-7 | |
| 4 | 42.8 ± 6.2 | 34.8 ± 8.1 | 29.7 ± 3.9 | n.a. | n.a. |
| D4-MMAE | 40.6 ± 3.0 | 36.9 ± 5.7 | 53.6 ± 3.9 | n.a. | n.a. |
| D1-MMAE | 8.3 ± 0.8 | 8.8 ± 2.2 | 67.8 ± 4.1 | 8 | 8 |
| D10-MMAE | 1.6 ± 0.1 | 1.3 ± 0.2 | 47.4 ± 3.4 | 30 | 36 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Wang, Y.; Uslu, S.; Venkatachalapathy, S.; Rashidian, M.; Schaefer, J.V.; Plückthun, A.; Distefano, M.D. Enzymatic Construction of DARPin-Based Targeted Delivery Systems Using Protein Farnesyltransferase and a Capture and Release Strategy. Int. J. Mol. Sci. 2022, 23, 11537. https://doi.org/10.3390/ijms231911537
Zhang Y, Wang Y, Uslu S, Venkatachalapathy S, Rashidian M, Schaefer JV, Plückthun A, Distefano MD. Enzymatic Construction of DARPin-Based Targeted Delivery Systems Using Protein Farnesyltransferase and a Capture and Release Strategy. International Journal of Molecular Sciences. 2022; 23(19):11537. https://doi.org/10.3390/ijms231911537
Chicago/Turabian StyleZhang, Yi, Yiao Wang, Safak Uslu, Sneha Venkatachalapathy, Mohammad Rashidian, Jonas V. Schaefer, Andreas Plückthun, and Mark D. Distefano. 2022. "Enzymatic Construction of DARPin-Based Targeted Delivery Systems Using Protein Farnesyltransferase and a Capture and Release Strategy" International Journal of Molecular Sciences 23, no. 19: 11537. https://doi.org/10.3390/ijms231911537