
Transgenerationally Transmitted DNA Demethylation of a Spontaneous Epialleles Using CRISPR/dCas9-TET1cd Targeted Epigenetic Editing in Arabidopsis
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
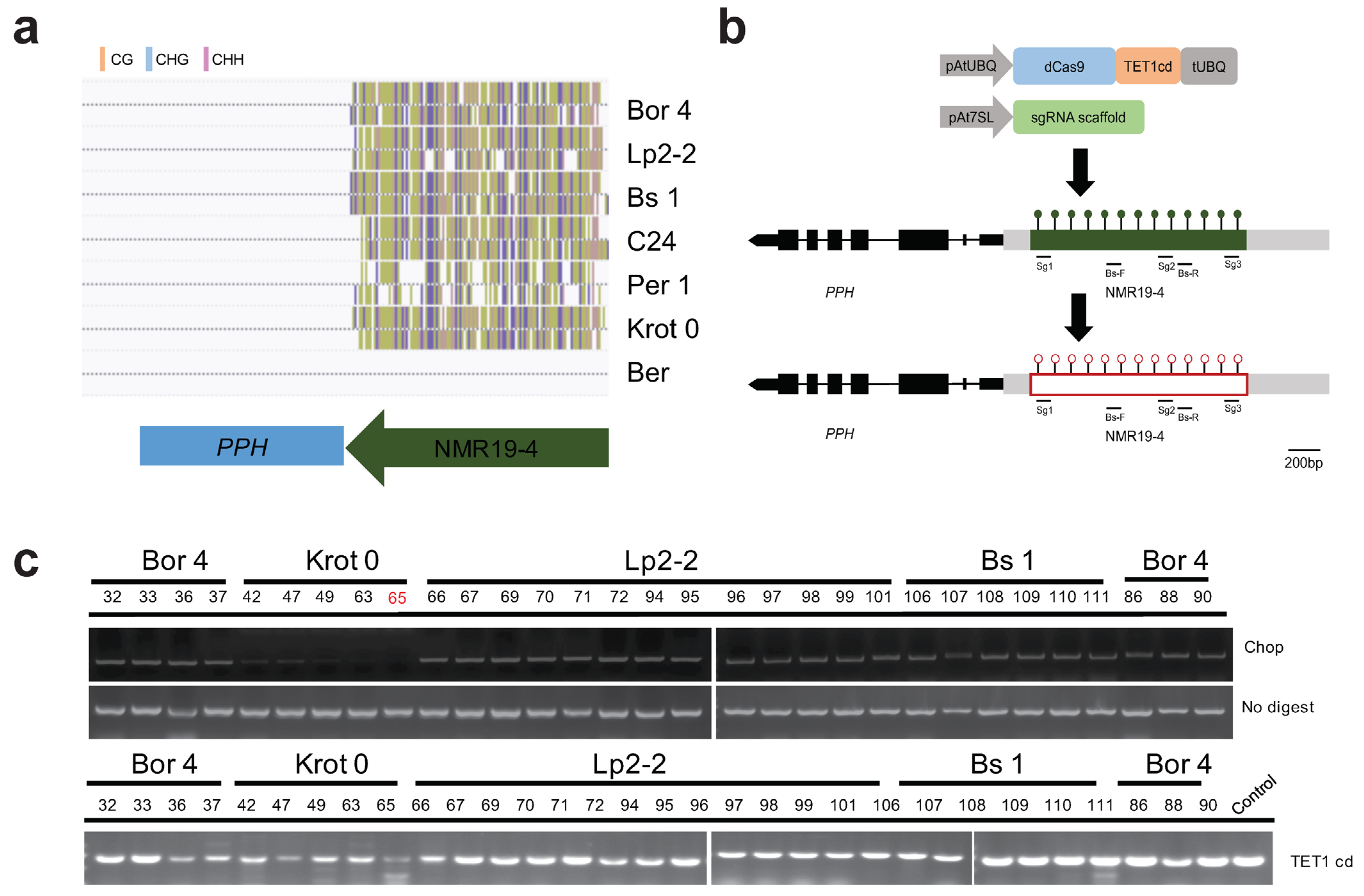
2.1. Identification of NMR19-4-Related CRISPR/dCas9-TET1cd Epimutation
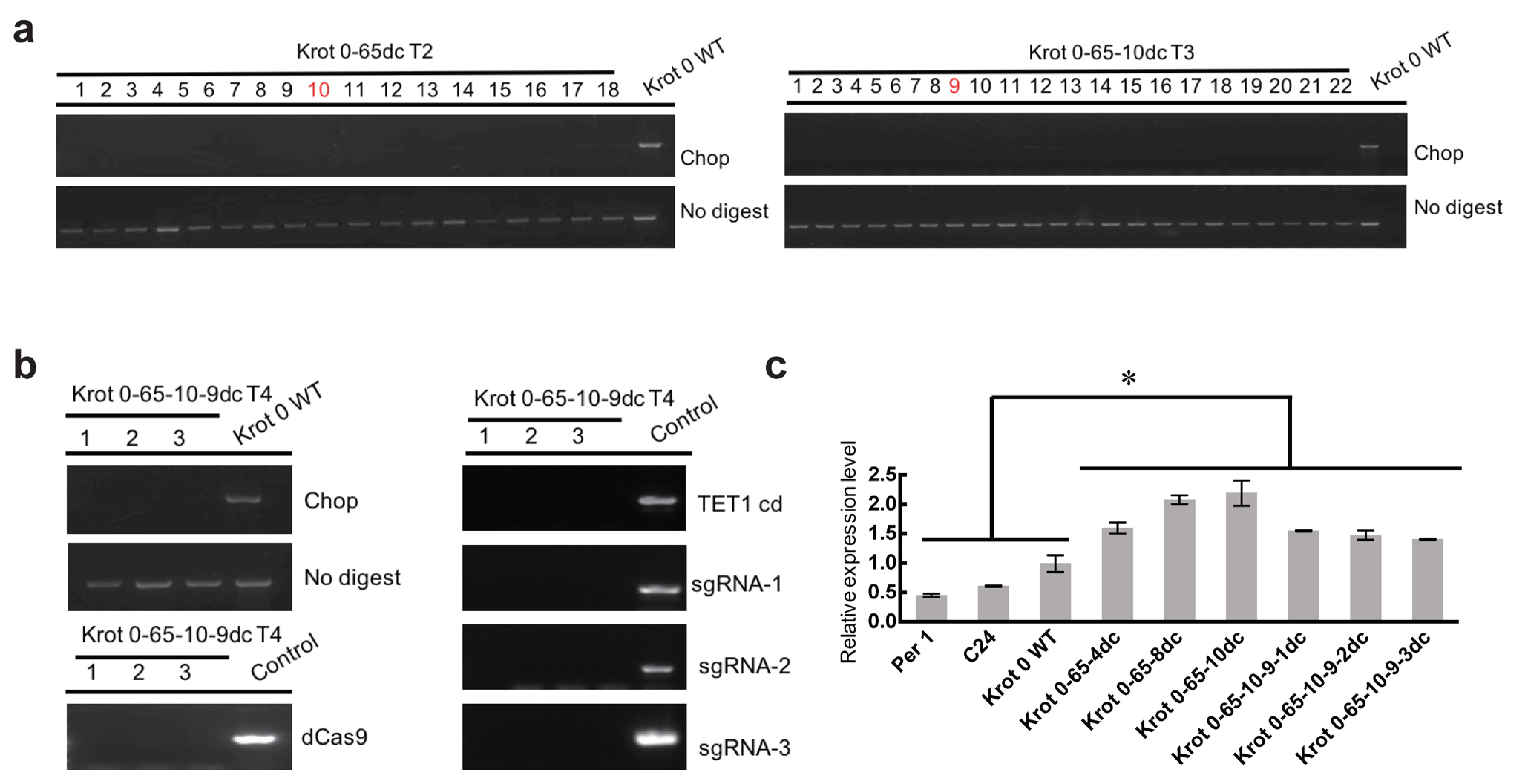
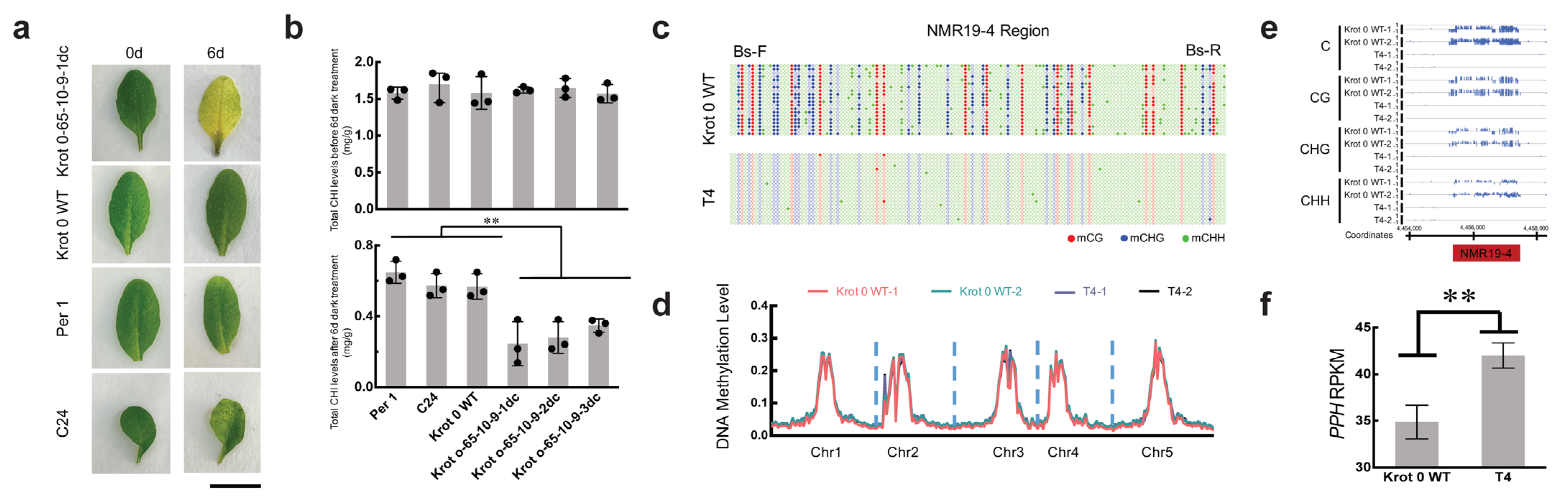
2.2. Inheritance of the Epimutation in Krot 0-65dc
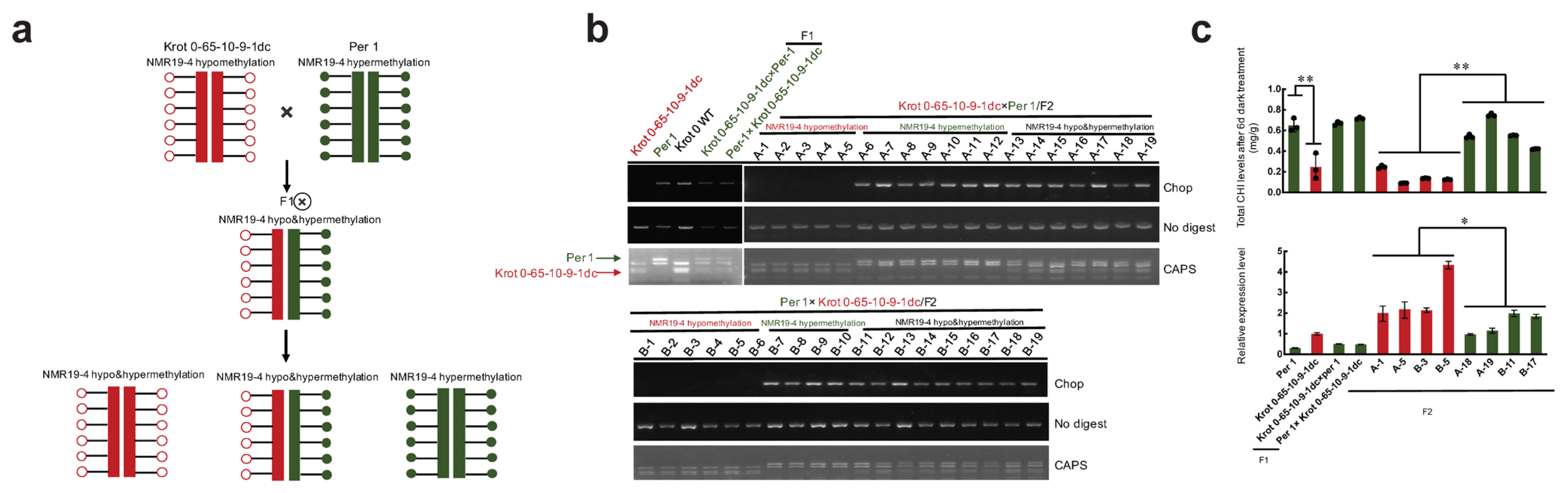
2.3. NMR19-4 Methylation Status after CRISPR/dCas9 Editing Is Mendelian Inheritance
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Growth Conditions
4.2. Vector Construction and PCR Assay
4.3. Whole-Genome Bisulfite Sequencing
4.4. Bisulfite Sequencing
4.5. qRT-PCR Analysis
4.6. RNA-Seq Analysis
4.7. ATAC-Seq Analysis
4.8. Darkness-Induced Leaf Senescence Assay and Quantification of Chlorophyll Content
4.9. Published Data
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Reinberg, D.; Allis, D.; Jenuwein, T. Epigenetics; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2007. [Google Scholar]
- Holliday, R.; Pugh, J.E. DNA Modification Mechanisms and Gene Activity During Development: Developmental clocks may depend on the enzymic modification of specific bases in repeated DNA sequences. Science 1975, 187, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Riggs, A.D. X inactivation, differentiation, and DNA methylation. Cytogenet. Genome Res. 1975, 14, 9–25. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Johnson, L.M.; Jacobsen, S.E.; Patel, D.J. DNA methylation pathways and their crosstalk with histone methylation. Nat. Rev. Mol. Cell Biol. 2015, 16, 519–532. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Cokus, S.J.; Zhang, X.; Chen, P.-Y.; Bostick, M.; Goll, M.G.; Hetzel, J.; Jain, J.; Strauss, S.H.; Halpern, M.E.; et al. Conservation and divergence of methylation patterning in plants and animals. Proc. Natl. Acad. Sci. USA 2010, 107, 8689–8694. [Google Scholar] [CrossRef] [PubMed]
- Zilberman, D.; Gehring, M.; Tran, R.K.; Ballinger, T.; Henikoff, S. Genome-wide analysis of Arabidopsis thaliana DNA methylation uncovers an interdependence between methylation and transcription. Nat. Genet. 2007, 39, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Bartels, A.; Han, Q.; Nair, P.; Stacey, L.; Gaynier, H.; Mosley, M.; Huang, Q.Q.; Pearson, J.K.; Hsieh, T.-F.; An, Y.-Q.C.; et al. Dynamic DNA methylation in plant growth and development. Int. J. Mol. Sci. 2018, 19, 2144. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Lang, Z.; Zhu, J.-K. Dynamics and function of DNA methylation in plants. Nat. Rev. Mol. Cell Biol. 2018, 19, 489–506. [Google Scholar] [CrossRef]
- Law, J.A.; Jacobsen, S.E. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 2010, 11, 204–220. [Google Scholar] [CrossRef]
- Blevins, T.; Podicheti, R.; Mishra, V.; Marasco, M.; Wang, J.; Rusch, D.; Tang, H.; Pikaard, C.S. Identification of Pol IV and RDR2-dependent precursors of 24 nt siRNAs guiding de novo DNA methylation in Arabidopsis. eLife 2015, 4, e09591. [Google Scholar] [CrossRef]
- Zhai, J.; Bischof, S.; Wang, H.; Feng, S.; Lee, T.-F.; Teng, C.; Chen, X.; Park, S.Y.; Liu, L.; Gallego-Bartolome, J.; et al. A one precursor one siRNA model for Pol IV-dependent siRNA biogenesis. Cell 2015, 163, 445–455. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Vandivier, L.E.; Tu, B.; Gao, L.; Won, S.Y.; Li, S.; Zheng, B.; Gregory, B.D.; Chen, X. Detection of Pol IV/RDR2-dependent transcripts at the genomic scale in Arabidopsis reveals features and regulation of siRNA biogenesis. Genome Res. 2015, 25, 235–245. [Google Scholar] [CrossRef]
- Xie, Z.; Johansen, L.K.; Gustafson, A.M.; Kasschau, K.D.; Lellis, A.D.; Zilberman, D.; Jacobsen, S.E.; Carrington, J.C.; Weigel, D. Genetic and functional diversification of small RNA pathways in plants. PLoS Biol. 2004, 2, e104. [Google Scholar] [CrossRef]
- Haag, J.R.; Ream, T.S.; Marasco, M.; Nicora, C.D.; Norbeck, A.D.; Pasa-Tolic, L.; Pikaard, C.S. In vitro transcription activities of Pol IV, Pol V, and RDR2 reveal coupling of Pol IV and RDR2 for dsRNA synthesis in plant RNA silencing. Mol. Cell 2012, 48, 811–818. [Google Scholar] [CrossRef]
- Qi, Y.; Denli, A.M.; Hannon, G.J. Biochemical specialization within Arabidopsis RNA silencing pathways. Mol. Cell 2005, 19, 421–428. [Google Scholar] [CrossRef]
- Zilberman, D.; Cao, X.; Jacobsen, S.E. ARGONAUTE4 control of locus-specific siRNA accumulation and DNA and histone methylation. Science 2003, 299, 716–719. [Google Scholar] [CrossRef]
- Li, C.F.; Pontes, O.; El-Shami, M.; Henderson, I.R.; Bernatavichute, Y.V.; Chan, S.W.-L.; Lagrange, T.; Pikaard, C.S.; Jacobsen, S.E. An ARGONAUTE4-containing nuclear processing center colocalized with Cajal bodies in Arabidopsis thaliana. Cell 2006, 126, 93–106. [Google Scholar] [CrossRef]
- Qi, Y.; He, X.; Wang, X.-J.; Kohany, O.; Jurka, J.; Hannon, G.J. Distinct catalytic and non-catalytic roles of ARGONAUTE4 in RNA-directed DNA methylation. Nature 2006, 443, 1008–1012. [Google Scholar] [CrossRef]
- Wierzbicki, A.T.; Haag, J.R.; Pikaard, C.S. Noncoding transcription by RNA polymerase Pol IVb/Pol V mediates transcriptional silencing of overlapping and adjacent genes. Cell 2008, 135, 635–648. [Google Scholar] [CrossRef]
- Zhong, X.; Du, J.; Hale, C.J.; Gallego-Bartolome, J.; Feng, S.; Vashisht, A.A.; Chory, J.; Wohlschlegel, J.A.; Patel, D.J.; Jacobsen, S.E. Molecular mechanism of action of plant DRM de novo DNA methyltransferases. Cell 2014, 157, 1050–1060. [Google Scholar] [CrossRef]
- Liu, W.; Duttke, S.H.; Hetzel, J.; Groth, M.; Feng, S.; Gallego-Bartolome, J.; Zhong, Z.; Kuo, H.Y.; Wang, Z.; Zhai, J.; et al. RNA-directed DNA methylation involves co-transcriptional small-RNA-guided slicing of polymerase V transcripts in Arabidopsis. Nat. Plants 2018, 4, 181–188. [Google Scholar] [CrossRef]
- Stroud, H.; Do, T.; Du, J.; Zhong, X.; Feng, S.; Johnson, L.; Patel, D.J.; Jacobsen, S.E. Non-CG methylation patterns shape the epigenetic landscape in Arabidopsis. Nat. Struct. Mol. Biol. 2014, 21, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Zemach, A.; Kim, M.Y.; Hsieh, P.H.; Coleman-Derr, D.; Eshed-Williams, L.; Thao, K.; Harmer, S.L.; Zilberman, D. The Arabidopsis nucleosome remodeler DDM1 allows DNA methyltransferases to access H1-containing heterochromatin. Cell 2013, 153, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Stroud, H.; Greenberg, M.V.; Feng, S.; Bernatavichute, Y.V.; Jacobsen, S.E. Comprehensive analysis of silencing mutants reveals complex regulation of the Arabidopsis methylome. Cell 2013, 152, 352–364. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Kapoor, A.; Sridhar, V.V.; Agius, F.; Zhu, J.K. The DNA glycosylase/lyase ROS1 functions in pruning DNA methylation patterns in Arabidopsis. Curr. Biol. 2007, 17, 54–59. [Google Scholar] [CrossRef]
- Zhang, H.; Zhu, J.K. Cold Spring Harbor symposia on quantitative biology. In Active DNA Demethylation in Plants and Animals; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2012; pp. 161–173. [Google Scholar]
- Kankel, M.W.; Ramsey, D.E.; Stokes, T.L.; Flowers, S.K.; Haag, J.R.; Jeddeloh, J.A.; Riddle, N.C.; Verbsky, M.L.; Richards, E.J. Arabidopsis MET1 cytosine methyltransferase mutants. Genetics 2003, 163, 1109–1122. [Google Scholar] [CrossRef]
- Baubec, T.; Pecinka, A.; Rozhon, W.; Scheid, O.M. Effective, homogeneous and transient interference with cytosine methylation in plant genomic DNA by zebularine. Plant J. 2009, 57, 542–554. [Google Scholar] [CrossRef]
- Griffin, P.T.; Niederhuth, C.E.; Schmitz, R.J. A comparative analysis of 5-azacytidine-and zebularine-induced DNA demethylation. G3 Genes Genomes Genet. 2016, 6, 2773–2780. [Google Scholar] [CrossRef]
- Gallego-Bartolomé, J.; Liu, W.; Kuo, P.H.; Feng, S.; Ghoshal, B.; Gardiner, J.; Zhao, J.M.C.; Park, S.Y.; Chory, J.; Jacobsen, S.E. Co-targeting RNA polymerases IV and V promotes efficient de novo DNA methylation in Arabidopsis. Cell 2019, 176, 1068–1082.e19. [Google Scholar] [CrossRef]
- Pickar-Oliver, A.; Gersbach, C.A. The next generation of CRISPR–Cas technologies and applications. Nat. Rev. Mol. Cell Biol. 2019, 20, 490–507. [Google Scholar] [CrossRef]
- Qi, L.S.; Larson, M.H.; Gilbert, L.A.; Doudna, J.A.; Weissman, J.S.; Arkin, A.P.; Lim, W.A. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 2013, 152, 1173–1183. [Google Scholar] [CrossRef] [Green Version]
- Makarova, K.S.; Haft, D.H.; Barrangou, R.; Brouns, S.J.J.; Charpentier, E.; Horvath, P.; Moineau, S.; Mojica, F.J.M.; Wolf, Y.I.; Yakunin, A.F.; et al. Evolution and classification of the CRISPR–Cas systems. Nat. Rev. Microbiol. 2011, 9, 467–477. [Google Scholar] [CrossRef]
- McCarty, N.S.; Graham, A.E.; Studená, L.; Ledesma-Amaro, R. Multiplexed CRISPR technologies for gene editing and transcriptional regulation. Nat. Commun. 2020, 11, 1–13. [Google Scholar] [CrossRef]
- Dominguez, A.A.; Lim, W.A.; Qi, L.S. Beyond editing: Repurposing CRISPR–Cas9 for precision genome regulation and interrogation. Nat. Rev. Mol. Cell Biol. 2016, 17, 5–15. [Google Scholar] [CrossRef]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef]
- Ito, S.; D’Alessio, A.C.; Taranova, O.V.; Hong, K.; Sowers, L.C.; Zhang, Y. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature 2010, 466, 1129–1133. [Google Scholar] [CrossRef]
- Wu, X.; Zhang, Y. TET-mediated active DNA demethylation: Mechanism, function and beyond. Nat. Rev. Genet. 2017, 18, 517–534. [Google Scholar] [CrossRef]
- Kohli, R.M.; Zhang, Y. TET enzymes, TDG and the dynamics of DNA demethylation. Nature 2013, 502, 472–479. [Google Scholar] [CrossRef]
- Gallego-Bartolomé, J.; Gardiner, J.; Liu, W.; Papikian, A.; Ghoshal, B.; Kuo, H.Y.; Zhao, J.M.C.; Segal, D.J.; Jacobsen, S.E. Targeted DNA demethylation of the Arabidopsis genome using the human TET1 catalytic domain. Proc. Natl. Acad. Sci. USA 2018, 115, E2125–E2134. [Google Scholar] [CrossRef]
- Li, J.; Yang, D.L.; Huang, H.; Zhang, G.; He, L.; Pang, J.; Lozano-Durán, R.; Lang, Z.; Zhu, J.K. Epigenetic memory marks determine epiallele stability at loci targeted by de novo DNA methylation. Nat. Plants 2020, 6, 661–674. [Google Scholar] [CrossRef]
- Ji, L.; Jordan, W.T.; Shi, X.; Hu, L.; He, C.; Schmitz, R.J. TET-mediated epimutagenesis of the Arabidopsis thaliana methylome. Nat. Commun. 2018, 9, 895. [Google Scholar] [CrossRef] [Green Version]
- Tang, S.; Yang, C.; Wang, D.; Deng, X.; Cao, X.; Song, X. Targeted DNA demethylation produces heritable epialleles in rice. Sci. China Life Sci. 2021, 65, 753–756. [Google Scholar] [CrossRef]
- Johannes, F.; Schmitz, R.J. Spontaneous epimutations in plants. New Phytol. 2018, 221, 1253–1259. [Google Scholar] [CrossRef]
- Richards, E.J. Inherited epigenetic variation—revisiting soft inheritance. Nat. Rev. Genet. 2006, 7, 395–401. [Google Scholar] [CrossRef]
- Jacobsen, S.E.; Meyerowitz, E.M. Hypermethylated SUPERMAN epigenetic alleles in Arabidopsis. Science 1997, 277, 1100–1103. [Google Scholar] [CrossRef]
- Song, Q.; Zhang, T.; Stelly, D.M.; Chen, Z.J. Epigenomic and functional analyses reveal roles of epialleles in the loss of photoperiod sensitivity during domestication of allotetraploid cottons. Genome Biol. 2017, 18, 1–14. [Google Scholar] [CrossRef]
- Wei, X.; Song, X.; Wei, L.; Tang, S.; Sun, J.; Hu, P.; Cao, X.J. An epiallele of rice AK1 affects photosynthetic capacity. J. Integr. Plant Biol. 2017, 59, 158–163. [Google Scholar] [CrossRef]
- Quadrana, L.; Almeida, J.; Asís, R.; Duffy, T.; Dominguez, P.G.; Bermúdez, L.; Conti, G.; da Silva, J.V.C.; Peralta, I.E.; Colot, V. Natural occurring epialleles determine vitamin E accumulation in tomato fruits. Nat. Commun. 2014, 5, 4027. [Google Scholar] [CrossRef]
- Manning, K.; Tör, M.; Poole, M.; Hong, Y.; Thompson, A.J.; King, G.J.; Giovannoni, J.J.; Seymour, G.B. A naturally occurring epigenetic mutation in a gene encoding an SBP-box transcription factor inhibits tomato fruit ripening. Nat. Genet. 2006, 38, 948–952. [Google Scholar] [CrossRef]
- Martin, A.; Troadec, C.; Boualem, A.; Rajab, M.; Fernandez, R.; Morin, H.; Pitrat, M.; Dogimont, C.; Bendahmane, A.J.N. A transposon-induced epigenetic change leads to sex determination in melon. Nature 2009, 461, 1135–1138. [Google Scholar] [CrossRef]
- He, L.; Wu, W.; Zinta, G.; Yang, L.; Wang, D.; Liu, R.; Zhang, H.; Zheng, Z.; Huang, H.; Zhang, Q.; et al. A naturally occurring epiallele associates with leaf senescence and local climate adaptation in Arabidopsis accessions. Nat. Commun. 2018, 9, 460. [Google Scholar] [CrossRef] [Green Version]
- Weigel, D.; Colot, V. Epialleles in plant evolution. Genome Biol. 2012, 13, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Kawakatsu, T.; Huang, S.S.C.; Jupe, F.; Sasaki, E.; Schmitz, R.J.; Urich, M.A.; Castanon, R.; Nery, J.R.; Barragan, C.; He, Y.; et al. Epigenomic diversity in a global collection of Arabidopsis thaliana accessions. Cell 2016, 166, 492–505. [Google Scholar] [CrossRef] [PubMed]
- Vidalis, A.; Živković, D.; Wardenaar, R.; Roquis, D.; Tellier, A.; Johannes, F. Methylome evolution in plants. Genome Biol. 2016, 17, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Springer, N.M.; Schmitz, R.J. Exploiting induced and natural epigenetic variation for crop improvement. Nat. Rev. Genet. 2017, 18, 563–575. [Google Scholar] [CrossRef]
- Liu, W.; Gallego-Bartolomé, J.; Zhou, Y.; Zhong, Z.; Wang, M.; Wongpalee, S.P.; Gardiner, J.; Feng, S.; Kuo, P.H.; Jacobsen, S.E. Ectopic targeting of CG DNA methylation in Arabidopsis with the bacterial SssI methyltransferase. Nat. Commun. 2021, 12, 1–14. [Google Scholar] [CrossRef]
- Zhang, Z.; Mao, Y.; Ha, S.; Liu, W.; Botella, J.R.; Zhu, J.-K. A multiplex CRISPR/Cas9 platform for fast and efficient editing of multiple genes in Arabidopsis. Plant Cell Rep. 2016, 35, 1519–1533. [Google Scholar] [CrossRef]
- Greaves, I.K.; Groszmann, M.; Ying, H.; Taylor, J.M.; Peacock, W.J.; Dennis, E.S. Trans chromosomal methylation in Arabidopsis hybrids. Proc. Natl. Acad. Sci. USA 2012, 109, 3570–3575. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, D.; Lang, Z.; He, L.; Yang, L.; Zeng, L.; Li, Y.; Zhao, C.; Huang, H.; Zhang, H. Methylation interactions in Arabidopsis hybrids require RNA-directed DNA methylation and are influenced by genetic variation. Proc. Natl. Acad. Sci. USA 2016, 113, E4248–E4256. [Google Scholar] [CrossRef]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA–guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Jinek, M.; Jiang, F.; Taylor, D.W.; Sternberg, S.H.; Kaya, E.; Ma, E.; Anders, C.; Hauer, M.; Zhou, K.; Lin, S.; et al. Structures of Cas9 endonucleases reveal RNA-mediated conformational activation. Science 2014, 343, 1247997. [Google Scholar] [CrossRef] [Green Version]
- Nishimasu, H.; Cong, L.; Yan, W.X.; Ran, F.A.; Zetsche, B.; Li, Y.; Kurabayashi, A.; Ishitani, R.; Zhang, F.; Nureki, O. Crystal structure of Staphylococcus aureus Cas9. Cell 2015, 162, 1113–1126. [Google Scholar] [CrossRef]
- Barman, A.; Deb, B.; Chakraborty, S. A glance at genome editing with CRISPR–Cas9 technology. Curr. Genet. 2020, 66, 447–462. [Google Scholar] [CrossRef]
- Papikian, A.; Liu, W.; Gallego-Bartolomé, J.; Jacobsen, S.E. Site-specific manipulation of Arabidopsis loci using CRISPR-Cas9 SunTag systems. Nat. Commun. 2019, 10, 729. [Google Scholar] [CrossRef]
- Gilbert, L.A.; Larson, M.H.; Morsut, L.; Liu, Z.; Brar, G.A.; Torres, S.E.; Stern-Ginossar, N.; Brandman, O.; Whitehead, E.H.; Doudna, J.A.; et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 2013, 154, 442–451. [Google Scholar] [CrossRef]
- Hilton, I.B.; D’ippolito, A.M.; Vockley, C.M.; Thakore, P.I.; Crawford, G.E.; Reddy, T.E.; Gersbach, C.A. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat. Biotechnol. 2015, 33, 510–517. [Google Scholar] [CrossRef]
- Liu, J.; Sun, M.; Cho, K.B.; Gao, X.; Guo, B. A CRISPR-Cas9 repressor for epigenetic silencing of KRAS. Pharmacol. Res. 2010, 164, 105304. [Google Scholar] [CrossRef]
- Chen, X.; Wei, M.; Liu, X.; Song, S.; Wang, L.; Yang, X.; Song, Y. Construction and validation of the CRISPR/dCas9-EZH2 system for targeted H3K27Me3 modification. Biochem. Biophys. Res. Commun. 2019, 511, 246–252. [Google Scholar] [CrossRef]
- Guhathakurta, S.; Kim, J.; Adams, L.; Basu, S.; Song, M.K.; Adler, E.; Je, G.; Fiadeiro, M.B.; Kim, Y.S. Targeted attenuation of elevated histone marks at SNCA alleviates α-synuclein in Parkinson’s disease. EMBO Mol. Med. 2021, 13, e12188. [Google Scholar] [CrossRef]
- Vojta, A.; Dobrinić, P.; Tadić, V.; Bočkor, L.; Korać, P.; Julg, B.; Klasić, M.; Zoldoš, V. Repurposing the CRISPR-Cas9 system for targeted DNA methylation. Nucleic Acids Res. 2016, 44, 5615–5628. [Google Scholar] [CrossRef]
- Liu, R.; Lang, Z.J. The mechanism and function of active DNA demethylation in plants. J. Integr. Plant Biol. 2019, 62, 148–159. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Tao, Y.; Gao, X.; Zhang, L.; Li, X.; Zou, W.; Ruan, K.; Wang, F.; Xu, G.l.; Hu, R. A CRISPR-based approach for targeted DNA demethylation. Cell Discov. 2016, 2, 16009. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.S.; Pan, Y.J.; Zhao, X.Q.; Dwivedi, D.; Zhu, L.H.; Ali, J.; Fu, B.Y.; Li, Z.K. Drought-induced site-specific DNA methylation and its association with drought tolerance in rice (Oryza sativa L.). J. Exp. Bot. 2011, 62, 1951–1960. [Google Scholar] [CrossRef]
- Labra, M.; Grassi, F.; Imazio, S.; di Fabio, T.; Citterio, S.; Sgorbati, S.; Agradi, E. Genetic and DNA-methylation changes induced by potassium dichromate in Brassica napus L. Chemosphere 2004, 54, 1049–1058. [Google Scholar] [CrossRef] [PubMed]
- Aina, R.; Sgorbati, S.; Santagostino, A.; Labra, M.; Ghiani, A.; Citterio, S. Specific hypomethylation of DNA is induced by heavy metals in white clover and industrial hemp. Physiol. Plant. 2004, 121, 472–480. [Google Scholar] [CrossRef]
- Sabbah, S.; Raise, M.; Tal, M. Methylation of DNA in NaCl-adapted cells of potato. Plant Cell Rep. 1995, 14, 467–470. [Google Scholar] [CrossRef] [PubMed]
- Al-Lawati, A.; Al-Bahry, S.; Victor, R.; Al-Lawati, A.; Yaish, M. Salt stress alters DNA methylation levels in alfalfa (Medicago spp). Genet. Mol. Res. 2016, 15, 15018299. [Google Scholar] [CrossRef] [PubMed]
- Naydenov, M.; Baev, V.; Apostolova, E.; Gospodinova, N.; Sablok, G.; Gozmanova, M.; Yahubyan, G. High-temperature effect on genes engaged in DNA methylation and affected by DNA methylation in Arabidopsis. Plant Physiol. Biochem. 2015, 87, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Steward, N.; Kusano, T.; Sano, H. Expression of ZmMET1, a gene encoding a DNA methyltransferase from maize, is associated not only with DNA replication in actively proliferating cells, but also with altered DNA methylation status in cold-stressed quiescent cells. Nucleic Acids Res. 2000, 28, 3250–3259. [Google Scholar] [CrossRef]
- Ay, N.; Irmler, K.; Fischer, A.; Uhlemann, R.; Reuter, G.; Humbeck, K. Epigenetic programming via histone methylation at WRKY53 controls leaf senescence in Arabidopsis thaliana. Plant J. 2009, 58, 333–346. [Google Scholar] [CrossRef]
- Ay, N.; Raum, U.; Balazadeh, S.; Seidensticker, T.; Fischer, A.; Reuter, G.; Humbeck, K. Regulatory factors of leaf senescence are affected in Arabidopsis plants overexpressing the histone methyltransferase SUVH2. J. Plant Growth Regul. 2014, 33, 119–136. [Google Scholar] [CrossRef]
- Chinnusamy, V.; Zhu, J.-K. Epigenetic regulation of stress responses in plants. Curr. Opin. Plant Biol. 2009, 12, 133–139. [Google Scholar] [CrossRef]
- Gruntman, E.; Qi, Y.; Slotkin, R.K.; Roeder, T.; Martienssen, R.A.; Sachidanandam, R. Kismeth: Analyzer of plant methylation states through bisulfite sequencing. BMC Bioinform. 2008, 9, 371. [Google Scholar] [CrossRef]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Tarasov, A.; Vilella, A.J.; Cuppen, E.; Nijman, I.J.; Prins, P. Sambamba: Fast processing of NGS alignment formats. Bioinformatics 2015, 31, 2032–2034. [Google Scholar] [CrossRef]
- Ni, Z.; Kim, E.-D.; Chen, Z.J. Chlorophyll and starch assays. Protoc. Exch. 2009. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, M.; He, L.; Chen, B.; Wang, Y.; Wang, L.; Zhou, W.; Zhang, T.; Cao, L.; Zhang, P.; Xie, L.; et al. Transgenerationally Transmitted DNA Demethylation of a Spontaneous Epialleles Using CRISPR/dCas9-TET1cd Targeted Epigenetic Editing in Arabidopsis. Int. J. Mol. Sci. 2022, 23, 10492. https://doi.org/10.3390/ijms231810492
Wang M, He L, Chen B, Wang Y, Wang L, Zhou W, Zhang T, Cao L, Zhang P, Xie L, et al. Transgenerationally Transmitted DNA Demethylation of a Spontaneous Epialleles Using CRISPR/dCas9-TET1cd Targeted Epigenetic Editing in Arabidopsis. International Journal of Molecular Sciences. 2022; 23(18):10492. https://doi.org/10.3390/ijms231810492
Chicago/Turabian StyleWang, Min, Li He, Bowei Chen, Yanwei Wang, Lishan Wang, Wei Zhou, Tianxu Zhang, Lesheng Cao, Peng Zhang, Linan Xie, and et al. 2022. "Transgenerationally Transmitted DNA Demethylation of a Spontaneous Epialleles Using CRISPR/dCas9-TET1cd Targeted Epigenetic Editing in Arabidopsis" International Journal of Molecular Sciences 23, no. 18: 10492. https://doi.org/10.3390/ijms231810492