

The Genetic Architecture of the Etiology of Lower Extremity Peripheral Artery Disease: Current Knowledge and Future Challenges in the Era of Genomic Medicine
 ,
,  , , , ,
, , , ,
Abstract
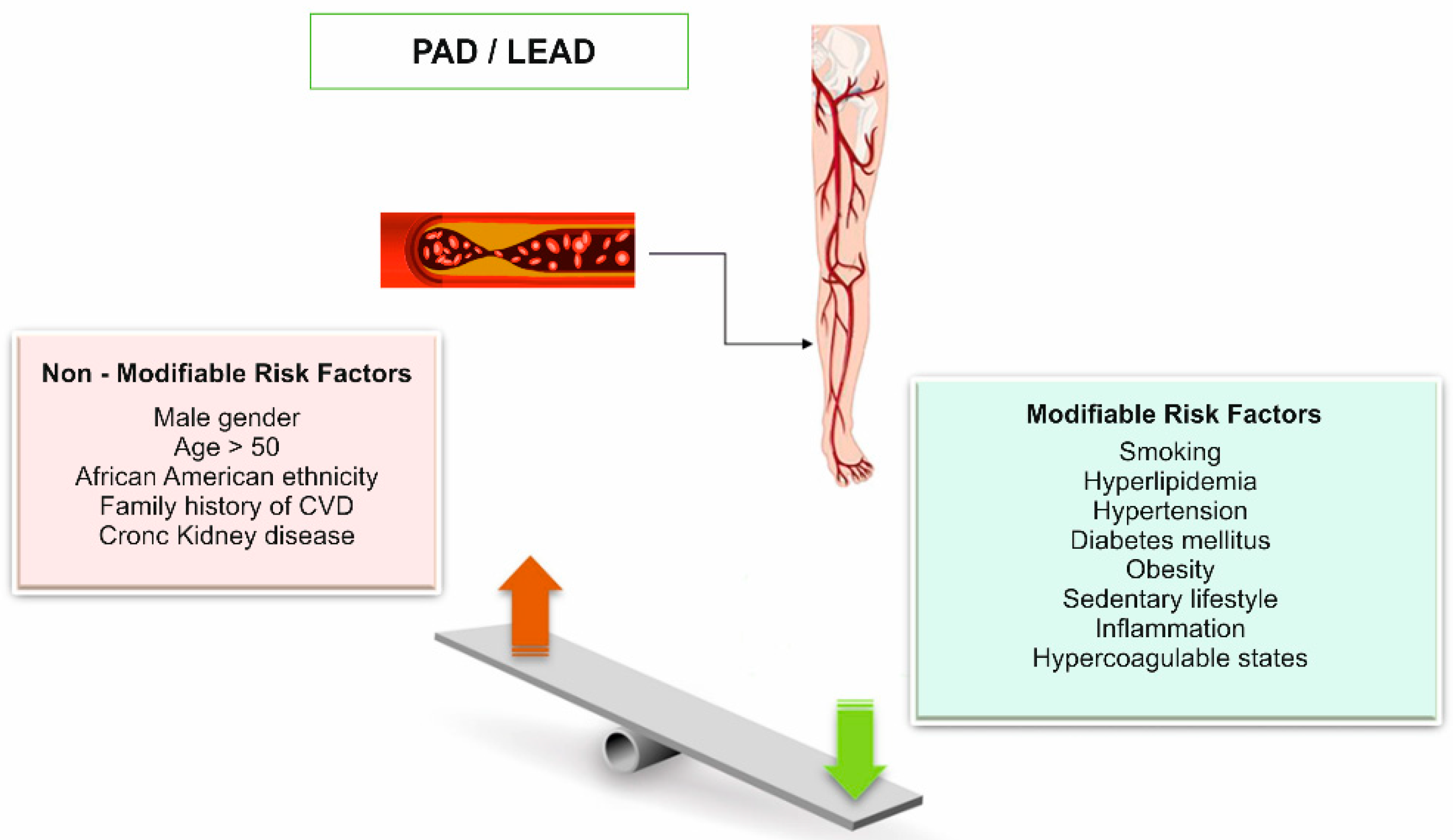
:1. Introduction
2. Literature Search Strategies and Data Collection
3. Phenotypic Variability of Lower Extremity Peripheral Artery Disease
4. Heritability of Atherosclerotic Lower Extremity Peripheral Artery Disease: Current Knowledge
4.1. Linkage Analysis Studies
4.2. Association Studies
4.2.1. Candidate Gene-Based Studies (CGS)
4.2.2. Genome-Wide Association Studies (GWASs)
4.2.3. Sequencing-Based Association Analysis for LEAD
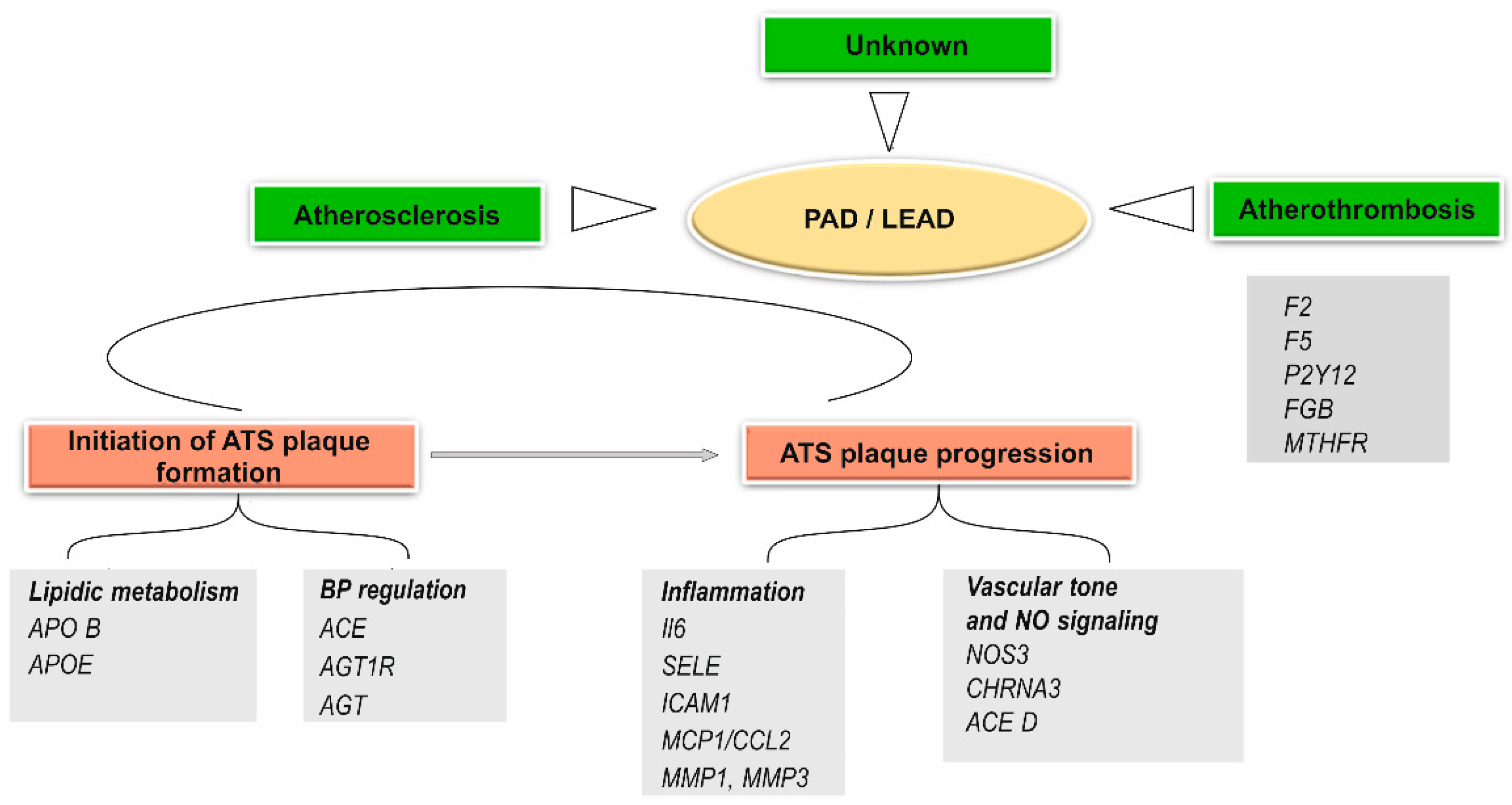
4.2.4. Other Genetic Polymorphisms Associated with the Pathophysiological Mechanisms of Peripheral Arterial Disease (LEAD)
- a.
- Polymorphism of Genes Involved in Lipid Metabolism
- b.
- Polymorphism of Genes Involved in the Mechanism of Inflammation in the Vascular Wall
- c.
- Genes Involved in Blood Pressure Regulation
Angiotensin-Converting Enzyme (ACE), Angiotensin II Type I Receptor (AGTR1), and Angiotensinogen (AGT) Genes Polymorphism
- d.
- Genes Involved in the Function of Vascular Smooth Muscle Cells (VSMc)
Endothelial Cell Nitric Oxide Synthase 3 (NOS3) Gene Polymorphism
- e.
- Genes Involved in Vascular Homeostasis
- f.
- Gene Polymorphisms Contributing to Atherothrombosis in LEAD
- g.
- Hyperhomocysteinemia
5. Discussions and Future Challenges
5.1. Challenges for the Future in the Post-GWAS Era
5.1.1. Interactions between Genes (Epistasis) and between Genetic and Environmental Factors
5.1.2. Rare Allelic Variant Association Studies
5.1.3. Epigenetics, Differential Gene Expression, Modifier Genes, Pleiotropy
5.1.4. Translating the Results of GWASs into Clinical Practice and The Importance of Polygenic Risk Scores (PRS) for Prevention of LEAD
5.1.5. Mendelian Randomization
5.1.6. Microbiome
5.1.7. Prophylactic Measures for Patients and Families at High Risk for LEAD
5.1.8. Genetic Counseling in Peripheral Artery Disease Patients
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| LEAD | Lower extremities artery disease |
| CAD | Coronary artery disease |
| DM | Diabetes mellitus |
| LEA | Lower extremity amputation |
| MZ | Monozygotic twins |
| DZ | Dizygotic twins |
| T2DM | Type 2 diabetes mellitus |
| AAA | Abdominal aortic aneurysm |
| LAS | Large cerebral artery stroke |
| VTE | Venous thromboembolism |
| VSMc | Vascular smooth muscle cell |
| PAOD | Peripheral artery occlusive disease |
| RAS | Renin−angiotensin system |
| ESRD | End-stage renal disease |
| CNV | Copy number variation |
| LD | Linkage disequilibrium |
References
- Aboyans, V.; Ricco, J.-B.; Bartelink, M.E.L.; Björck, M.; Brodmann, M.; Cohnert, T.; Collet, J.P.; Czerny, M.; De Carlo, M.; Debus, S.; et al. 2017 ESC Guidelines on the Diagnosis and Treatment of Peripheral Arterial Diseases, in collaboration with the European Society for Vascular Surgery (ESVS): Document covering atherosclerotic disease of extracranial carotid and vertebral, mesenteric, renal, upper and lower extremity arteries. Endorsed by: The European Stroke Organization (ESO)The Task Force for the Diagnosis and Treatment of Peripheral Arterial Diseases of the European Society of Cardiology (ESC) and of the European Society for Vascular Surgery (ESVS). Eur. Heart J. 2018, 39, 763–816. [Google Scholar] [CrossRef] [PubMed]
- Gerhard-Herman, M.D.; Gornik, H.L.; Barrett, C.; Barshes, N.R.; Corriere, M.A.; Drachman, D.E.; Fleisher, L.A.; Fowkes, F.G.; Hamburg, N.M.; Kinlay, S.; et al. 2016 AHA/ACC Guideline on the Management of Patients with Lower Extremity Peripheral Artery Disease: Executive Summary: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation 2017, 135, e686–e725, Erratum in Circulation 2017, 135, e790. [Google Scholar] [CrossRef] [PubMed]
- Brunton, S.; Anderson, J.; Vacalis, S. Updates in the Management of Peripheral Arterial Disease: Focus on Reduction of Atherothrombotic Risk. J. Fam. Pract. 2021, 70, S1e–S8e. [Google Scholar] [CrossRef]
- Hazarika, S.; Annex, B.H. Biomarkers and Genetics in Peripheral Artery Disease. Clin Chem. 2017, 63, 236–244. [Google Scholar] [CrossRef]
- Kithcart, A.P.; Beckman, J.A. ACC/AHA Versus ESC Guidelines for Diagnosis and Management of Peripheral Artery Disease: JACC Guideline Comparison. J. Am. Coll. Cardiol. 2018, 72, 2789–2801. [Google Scholar] [CrossRef]
- Criqui, M.H.; Matsushita, K.; Aboyans, V.; Hess, C.N.; Hicks, C.W.; Kwan, T.W.; McDermott, M.M.; Misra, S.; Ujueta, F.; American Heart Association Council on Epidemiology and Prevention; et al. Lower Extremity Peripheral Artery Disease: Contemporary Epidemiology, Management Gaps, and Future Directions: A Scientific Statement from the American Heart Association. Circulation 2021, 144, e171–e191, Erratum in Circulation 2021, 144, e193. [Google Scholar] [CrossRef]
- Abramson, B.L.; Al-Omran, M.; Anand, S.S.; Albalawi, Z.; Coutinho, T.; de Mestral, C.; Dubois, L.; Gill, H.L.; Greco, E.; Guzman, R.; et al. Canadian Cardiovascular Society 2022 Guidelines for Peripheral Arterial Disease. Can. J. Cardiol. 2022, 38, 560–587. [Google Scholar] [CrossRef]
- Grøndal, N.; Søgaard, R.; Lindholt, J.S. Baseline prevalence of abdominal aortic aneurysm, peripheral arterial disease and hypertension in men aged 65–74 years from a population screening study (VIVA trial). Br. J. Surg. 2015, 102, 902–906. [Google Scholar] [CrossRef]
- Sigvant, B.; Wiberg-Hedman, K.; Bergqvist, D.; Rolandsson, O.; Andersson, B.; Persson, E.; Wahlberg, E. A population-based study of peripheral arterial disease prevalence with special focus on critical limb ischemia and sex differences. J. Vasc. Surg. 2007, 45, 1185–1191. [Google Scholar] [CrossRef] [PubMed]
- Diehm, C.; Schuster, A.; Allenberg, J.R.; Darius, H.; Haberl, R.; Lange, S.; Pittrow, D.; von Stritzky, B.; Tepohl, G.; Trampisch, H.J. High prevalence of peripheral arterial disease and co-morbidity in 6880 primary care patients: Cross-sectional study. Atherosclerosis 2004, 172, 95–105. [Google Scholar] [CrossRef]
- Kochar, A.; Mulder, H.; Rockhold, F.W.; Baumgartner, I.; Berger, J.S.; Blomster, J.I.; Fowkes, F.G.R.; Katona, B.G.; Lopes, R.D.; Al-Khalidi, H.R.; et al. Cause of Death Among Patients with Peripheral Artery Disease: Insights from the EUCLID Trial. Circ. Cardiovasc. Qual. Outcomes 2020, 13, e006550. [Google Scholar] [CrossRef] [PubMed]
- Franey, E.G.; Kritz-Silverstein, D.; Richard, E.L.; Alcaraz, J.E.; Nievergelt, C.M.; Shaffer, R.A.; Bhatnagar, V. Association of Race and Peripheral Artery Disease: The Atherosclerosis Risk in Communities (ARIC) Cohort. J. Hypertens. Manag. 2020, 6, 047. [Google Scholar] [CrossRef]
- Dokun, A.O.; Annex, B.H. Genetics and Genomics of Peripheral Arterial Disease. In Genomic and Precision Medicine: Cardiovascular Disease, 3rd ed.; Ginsburg, G.S., Willard, H.F., Eds.; Academic Press, Elsevier Inc.: Cambridge, MA, USA, 2017; pp. 197–219. [Google Scholar] [CrossRef]
- Hinchliffe, R.J.; Forsythe, R.O.; Apelqvist, J.; Boyko, E.J.; Fitridge, R.; Hong, J.P.; Katsanos, K.; Mills, J.L.; Nikol, S.; Reekers, J.; et al. Guidelines on diagnosis, prognosis, and management of peripheral artery disease in patients with foot ulcers and diabetes (IWGDF 2019 update). Diabetes Metab. Res. Rev. 2020, 36, e3276. [Google Scholar] [CrossRef] [PubMed]
- Ceasovschih, A.; Sorodoc, V.; Onofrei Aursulesei, V.; Tesloianu, D.; Tuchilus, C.; Anisie, E.; Petris, A.; Statescu, C.; Jaba, E.; Stoica, A.; et al. Biomarker Utility for Peripheral Artery Disease Diagnosis in Real Clinical Practice: A Prospective Study. Diagnostics 2020, 10, 723. [Google Scholar] [CrossRef]
- Ziegler, L.; Hedin, U.; Gottsäter, A. Circulating Biomarkers in Lower Extremity Artery Disease. Eur. Cardiol. 2022, 17, e09. [Google Scholar] [CrossRef]
- Belkin, N.; Damrauer, S.M. Peripheral Arterial Disease Genetics: Progress to Date and Challenges Ahead. Curr. Cardiol. Rep. 2017, 19, 131. [Google Scholar] [CrossRef]
- Kullo, I.J.; Leeper, N.J. The genetic basis of peripheral arterial disease: Current knowledge, challenges, and future directions. Circ. Res. 2015, 116, 1551–1560. [Google Scholar] [CrossRef]
- Shemirani, A.-H.; Zsóri, K.S.; Jávor, A.; Csiki, Z. Genetics in Peripheral Artery Disease. In Peripheral Arterial Disease; Nishtha Sareen, N., Ojha, A., Eds.; IntechOpen: London, UK, 2018; pp. 107–116. [Google Scholar] [CrossRef]
- Golledge, J.; Biros, E.; Bingley, J.; Iyer, V.; Krishna, S.M. Epigenetics and Peripheral Artery Disease. Curr. Atheroscler. Rep. 2016, 18, 15. [Google Scholar] [CrossRef]
- Soler-Botija, C.; Gálvez-Montón, C.; Bayés-Genís, A. Epigenetic Biomarkers in Cardiovascular Diseases. Front. Genet. 2019, 10, 950. [Google Scholar] [CrossRef]
- Klarin, D.; Tsao, P.S.; Damrauer, S.M. Genetic Determinants of Peripheral Artery Disease. Circ. Res. 2021, 128, 1805–1817. [Google Scholar] [CrossRef]
- Gudmundsson, G.; Matthiasson, S.E.; Arason, H.; Johannsson, H.; Runarsson, F.; Bjarnason, H.; Helgadottir, K.; Thorisdottir, S.; Ingadottir, G.; Lindpaintner, K.; et al. Localization of a gene for peripheral arterial occlusive disease to chromosome 1p31. Am. J. Hum. Genet. 2002, 70, 586–592. [Google Scholar] [CrossRef] [PubMed]
- Kullo, I.J.; Turner, S.T.; Kardia, S.L.; Mosley, T.H., Jr.; Boerwinkle, E.; de Andrade, M. A genome-wide linkage scan for ankle-brachial index in African American and non-Hispanic white subjects participating in the GENOA study. Atherosclerosis 2006, 187, 433–438. [Google Scholar] [CrossRef] [PubMed]
- OMIM—Online Mendelian Inheritance in Man. Available online: https://www.omim.org (accessed on 6 July 2022).
- Wassel, C.L.; Lamina, C.; Nambi, V.; Coassin, S.; Mukamal, K.J.; Ganesh, S.K.; Jacobs, D.R., Jr.; Franceschini, N.; Papanicolaou, G.J.; Gibson, Q.; et al. Genetic determinants of the ankle-brachial index: A meta-analysis of a cardiovascular candidate gene 50K SNP panel in the candidate gene association resource (CARe) consortium. Atherosclerosis 2012, 222, 138–147. [Google Scholar] [CrossRef] [PubMed]
- Kardia, S.L.; Greene, M.T.; Boerwinkle, E.; Turner, S.T.; Kullo, I.J. Investigating the complex genetic architecture of ankle-brachial index, a measure of peripheral arterial disease, in non-Hispanic whites. BMC Med. Genom. 2008, 1, 16. [Google Scholar] [CrossRef]
- Murabito, J.M.; White, C.C.; Kavousi, M.; Sun, Y.V.; Feitosa, M.F.; Nambi, V.; Lamina, C.; Schillert, A.; Coassin, S.; Bis, J.C.; et al. Association between chromosome 9p21 variants and the ankle-brachial index identified by a meta-analysis of 21 genome-wide association studies. Circ. Cardiovasc. Genet. 2012, 5, 100–112. [Google Scholar] [CrossRef]
- Helgadottir, A.; Thorleifsson, G.; Magnusson, K.P.; Grétarsdottir, S.; Steinthorsdottir, V.; Manolescu, A.; Jones, G.T.; Rinkel, G.J.; Blankensteijn, J.D.; Ronkainen, A.; et al. The same sequence variant on 9p21 associates with myocardial infarction, abdominal aortic aneurysm and intracranial aneurysm. Nat. Genet. 2008, 40, 217–224. [Google Scholar] [CrossRef]
- Cluett, C.; McDermott, M.M.; Guralnik, J.; Ferrucci, L.; Bandinelli, S.; Miljkovic, I.; Zmuda, J.M.; Li, R.; Tranah, G.; Harris, T.; et al. The 9p21 myocardial infarction risk allele in-creases risk of peripheral artery disease in older people: Cluett—9p21 alleles and peripheral artery disease. Circ. Cardiovasc. Genet. 2009, 2, 347–353. [Google Scholar] [CrossRef]
- Thorgeirsson, T.E.; Geller, F.; Sulem, P.; Rafnar, T.; Wiste, A.; Magnusson, K.P.; Manolescu, A.; Thorleifsson, G.; Stefansson, H.; Ingason, A.; et al. A variant associated with nicotine dependence, lung cancer and peripheral arterial disease. Nature 2008, 452, 638–642. [Google Scholar] [CrossRef] [Green Version]
- Gretarsdottir, S.; Baas, A.F.; Thorleifsson, G.; Holm, H.; den Heijer, M.; de Vries, J.P.; Kranendonk, S.E.; Zeebregts, C.J.; van Sterkenburg, S.M.; Geelkerken, R.H.; et al. Genome-wide association study identifies a sequence variant within the DAB2IP gene conferring susceptibility to abdominal aortic aneurysm. Nat. Genet. 2010, 42, 692–697. [Google Scholar] [CrossRef]
- Shi, Z.F.; Fang, Q.B.; Limu, S.; Jiareke, T.; Ge, X.H. Association Between Three SNPs and Thromboangiitis Obliterans in Xinjiang Uyghur Population. Genet. Test. Mol. Biomark. 2016, 20, 55–62. [Google Scholar] [CrossRef]
- Małecki, R.; Zdrojowy, K.; Adamiec, R. Thromboangiitis obliterans in the 21st century—A new face of disease. Atherosclerosis 2009, 206, 328–334. [Google Scholar] [CrossRef] [PubMed]
- Lett, R.L.; Wang, W.; O’Connor, T.P. Semaphorin 5B is a novel inhibitory cue for corticofugal axons. Cereb. Cortex 2009, 19, 1408–1421. [Google Scholar] [CrossRef] [PubMed]
- Koriyama, H.; Nakagami, H.; Katsuya, T.; Sugimoto, K.; Yamashita, H.; Takami, Y.; Maeda, S.; Kubo, M.; Takahashi, A.; Nakamura, Y.; et al. Identification of evidence suggestive of an association with peripheral arterial disease at the OSBPL10 locus by genome-wide investigation in the Japanese population. J. Atheroscler. Thromb. 2010, 17, 1054–1062. [Google Scholar] [CrossRef]
- Matsukura, M.; Ozaki, K.; Takahashi, A.; Onouchi, Y.; Morizono, T.; Komai, H.; Shigematsu, H.; Kudo, T.; Inoue, Y.; Kimura, H.; et al. Genome-Wide Association Study of Peripheral Arterial Disease in a Japanese Population. PLoS ONE 2015, 10, e0139262. [Google Scholar] [CrossRef] [PubMed]
- van Zuydam, N.R.; Stiby, A.; Abdalla, M.; Austin, E.; Dahlström, E.H.; McLachlan, S.; Vlachopoulou, E.; Ahlqvist, E.; Di Liao, C.; Sandholm, N.; et al. Genome-Wide Association Study of Peripheral Artery Disease. Circ. Genom. Precis. Med. 2021, 14, e002862. [Google Scholar] [CrossRef]
- Yaseen, N.R.; Blobel, G. Cloning and characterization of human karyopherin beta3. Proc. Natl. Acad. Sci. USA 1997, 94, 4451–4456. [Google Scholar] [CrossRef]
- Kullo, I.J.; Shameer, K.; Jouni, H.; Lesnick, T.G.; Pathak, J.; Chute, C.G.; de Andrade, M. The ATXN2-SH2B3 locus is associated with peripheral arterial disease: An electronic medical record-based genome-wide association study. Front. Genet. 2014, 5, 166. [Google Scholar] [CrossRef]
- Monsalve, M.V.; Young, R.; Jobsis, J.; Wiseman, S.A.; Dhamu, S.; Powell, J.T.; Greenhalgh, R.M.; Humphries, S.E. DNA polymorphisms of the gene for apolipoprotein B in patients with peripheral arterial disease. Atherosclerosis 1988, 70, 123–129. [Google Scholar] [CrossRef]
- Koopal, C.; Geerlings, M.I.; Muller, M.; de Borst, G.J.; Algra, A.; van der Graaf, Y.; Visseren, F.L.; SMART Study Group. The relation between apolipoprotein E (APOE) genotype and peripheral artery disease in patients at high risk for cardiovascular disease. Atherosclerosis 2016, 246, 187–192. [Google Scholar] [CrossRef]
- Yin, Y.W.; Li, J.C.; Zhang, M.; Wang, J.Z.; Li, B.H.; Liu, Y.; Liao, S.Q.; Zhang, M.J.; Gao, C.Y.; Zhang, L.L. Influence of interleukin-6 gene-174G>C polymorphism on development of atherosclerosis: A meta-analysis of 50 studies involving 33,514 subjects. Gene 2013, 529, 94–103, Erratum in Gene 2014, 534, 456. [Google Scholar] [CrossRef]
- Flex, A.; Gaetani, E.; Pola, R.; Santoliquido, A.; Aloi, F.; Papaleo, P.; Dal Lago, A.; Pola, E.; Serricchio, M.; Tondi, P.; et al. The-174 G/C polymorphism of the interleukin-6 gene promoter is associated with peripheral artery occlusive disease. Eur. J. Vasc. Endovasc. Surg. 2002, 24, 264–268. [Google Scholar] [CrossRef] [PubMed]
- Flex, A.; Gaetani, E.; Angelini, F.; Sabusco, A.; Chillà, C.; Straface, G.; Biscetti, F.; Pola, P.; Castellot, J.J., Jr.; Pola, R. Pro-inflammatory genetic profiles in subjects with peripheral arterial occlusive disease and critical limb ischemia. J. Intern. Med. 2007, 262, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Butnariu, L.I.; Florea, L.; Badescu, M.C.; Țarcă, E.; Costache, I.-I.; Gorduza, E.V. Etiologic Puzzle of Coronary Artery Disease: How Important Is Genetic Component? Life 2022, 12, 865. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, A.T.; Duprez, D. The potential role of angiotensin-converting enzyme inhibition in peripheral arterial disease. Vasc. Med. 2003, 8, 273–278. [Google Scholar] [CrossRef]
- Han, C.; Han, X.K.; Liu, F.C.; Huang, J.F. Ethnic differences in the association between angiotensin-converting enzyme gene insertion/deletion polymorphism and peripheral vascular disease: A meta-analysis. Chronic. Dis. Transl. Med. 2017, 3, 230–241, Erratum in Chronic. Dis. Transl. Med. 2018, 4, 67–68. [Google Scholar] [CrossRef]
- Başar, Y.; Salmayenli, N.; Aksoy, M.; Seçkin, S.; Aydin, M.; Ozkök, E. ACE gene polymorphism in peripheral vascular disease. Horm. Metab. Res. 2007, 39, 534–537. [Google Scholar] [CrossRef]
- Fatini, C.; Sticchi, E.; Sofi, F.; Said, A.A.; Pratesi, G.; Pulli, R.; Pratesi, C.; Abbate, R. Multilocus analysis in candidate genes ACE, AGT, and AGTR1 and predisposition to peripheral arterial disease: Role of ACE D/-240T haplotype. J. Vasc. Surg. 2009, 50, 1399–1404. [Google Scholar] [CrossRef]
- Ismaeel, A.; Papoutsi, E.; Miserlis, D.; Lavado, R.; Haynatzki, G.; Casale, G.P.; Bohannon, W.T.; Smith, R.S.; Eidson, J.L.; Brumberg, R.; et al. The Nitric Oxide System in Peripheral Artery Disease: Connection with Oxidative Stress and Biopterins. Antioxidants 2020, 9, 590. [Google Scholar] [CrossRef]
- Aimo, A.; Botto, N.; Vittorini, S.; Emdin, M. Polymorphisms in the eNOS gene and the risk of coronary artery disease: Making the case for genome-wide association studies. Eur. J. Prev. Cardiol. 2019, 26, 157–159. [Google Scholar] [CrossRef]
- Hingorani, A.D. Polymorphisms in endothelial nitric oxide synthase and atherogenesis: John French Lecture 2000. Atherosclerosis 2001, 154, 521–527. [Google Scholar] [CrossRef]
- Fowkes, F.G.; Lee, A.J.; Hau, C.M.; Cooke, A.; Connor, J.M.; Lowe, G.D. Methylene tetrahydrofolate reductase (MTHFR) and nitric oxide synthase (ecNOS) genes and risks of peripheral arterial disease and coronary heart disease: Edinburgh Artery Study. Atherosclerosis 2000, 150, 179–185. [Google Scholar] [CrossRef]
- Sticchi, E.; Sofi, F.; Romagnuolo, I.; Pratesi, G.; Pulli, R.; Pratesi, C.; Abbate, R.; Fatini, C. eNOS and ACE genes influence peripheral arterial disease predisposition in smokers. J. Vasc. Surg. 2010, 52, 97–102.e1. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.D.; Chang, Y.C.; Chiu, Y.F.; Chang, T.J.; Li, H.Y.; Lin, W.H.; Yuan, H.Y.; Chen, Y.T.; Chuang, L.M. SLC2A10 genetic polymorphism predicts development of peripheral arterial disease in patients with type 2 diabetes. SLC2A10 and PAD in type 2 diabetes. BMC Med. Genet. 2010, 11, 126. [Google Scholar] [CrossRef] [PubMed]
- Herm, J.; Hoppe, B.; Siegerink, B.; Nolte, C.H.; Koscielny, J.; Haeusler, K.G. A Prothrombotic Score Based on Genetic Polymorphisms of the Hemostatic System Differs in Patients with Ischemic Stroke, Myocardial Infarction, or Peripheral Arterial Occlusive Disease. Front. Cardiovasc. Med. 2017, 4, 39. [Google Scholar] [CrossRef] [PubMed]
- Reny, J.L.; Alhenc-Gelas, M.; Fontana, P.; Bissery, A.; Julia, P.L.; Fiessinger, J.N.; Aiach, M.; Emmerich, J. The factor II G20210A gene polymorphism, but not factor V Arg506Gln, is associated with peripheral arterial disease: Results of a case-control study. J. Thromb. Haemost. 2004, 2, 1334–1340. [Google Scholar] [CrossRef] [PubMed]
- Malik, R.; Chauhan, G.; Traylor, M.; Sargurupremraj, M.; Okada, Y.; Mishra, A.; Rutten-Jacobs, L.; Giese, A.K.; van der Laan, S.W.; Gretarsdottir, S.; et al. Multiancestry genome-wide association study of 520,000 subjects identifies 32 loci associated with stroke and stroke subtypes. Nat. Genet. 2018, 50, 524–537, Erratum in Nat. Genet. 2019, 51, 1192–1193. [Google Scholar] [CrossRef]
- Scavone, M.; Femia, E.A.; Cattaneo, M. P2Y₁₂ receptor gene mutations associated with bleeding. Platelets 2017, 28, 421–423. [Google Scholar] [CrossRef]
- Fontana, P.; Gaussem, P.; Aiach, M.; Fiessinger, J.N.; Emmerich, J.; Reny, J.L. P2Y12 H2 haplotype is associated with peripheral arterial disease: A case-control study. Circulation 2003, 108, 2971–2973. [Google Scholar] [CrossRef] [Green Version]
- Behague, I.; Poirier, O.; Nicaud, V.; Evans, A.; Arveiler, D.; Luc, G.; Cambou, J.P.; Scarabin, P.Y.; Bara, L.; Green, F.; et al. Beta fibrinogen gene polymorphisms are associated with plasma fibrinogen and coronary artery disease in patients with myocardial infarction. The ECTIM Study. Etude Cas-Temoins sur l’Infarctus du Myocarde. Circulation 1996, 93, 440–449. [Google Scholar] [CrossRef]
- Fowkes, F.G.; Connor, J.M.; Smith, F.B.; Wood, J.; Donnan, P.T.; Lowe, G.D. Fibrinogen genotype and risk of peripheral atherosclerosis. Lancet 1992, 339, 693–696. [Google Scholar] [CrossRef]
- Archetti, S.; Martini, M.; Botteri, E.; Di Lorenzo, D.; Cervi, E.; Bonardelli, S. Influence of genetic and environmental factors in peripheral arterial disease natural history: Analysis from six years follow up. Int. J. Appl. Basic Med. Res. 2012, 2, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Khandanpour, N.; Willis, G.; Meyer, F.J.; Armon, M.P.; Loke, Y.K.; Wright, A.J.; Finglas, P.M.; Jennings, B.A. Peripheral arterial disease and methylenetetrahydrofolate reductase (MTHFR) C677T mutations: A case-control study and meta-analysis. J. Vasc. Surg. 2009, 49, 711–718. [Google Scholar] [CrossRef] [PubMed]
- Rutherford, R.B.; Baker, J.D.; Ernst, C.; Johnston, K.W.; Porter, J.M.; Ahn, S.; Jones, D.N. Recommended standards for reports dealing with lower extremity ischemia: Revised version. J. Vasc. Surg. 1997, 26, 517–538, Erratum in J. Vasc. Surg. 2001, 33, 805. [Google Scholar] [CrossRef]
- Hardman, R.L.; Jazaeri, O.; Yi, J.; Smith, M.; Gupta, R. Overview of classification systems in peripheral artery disease. Semin. Interv. Radiol. 2014, 31, 378–388. [Google Scholar] [CrossRef]
- Hughes, W.; Goodall, R.; Salciccioli, J.D.; Marshall, D.C.; Davies, A.H.; Shalhoub, J. Editor’s Choice—Trends in Lower Extremity Amputation Incidence in European Union 15+ Countries 1990–2017. Eur. J. Vasc. Endovasc. Surg. 2020, 60, 602–612. [Google Scholar] [CrossRef]
- Jensen, P.S.; Petersen, J.; Kirketerp-Møller, K.; Poulsen, I.; Andersen, O. Progression of disease preceding lower extremity amputation in Denmark: A longitudinal registry study of diagnoses, use of medication and healthcare services 14 years prior to amputation. BMJ Open 2017, 7, e016030. [Google Scholar] [CrossRef]
- Criqui, M.H.; Vargas, V.; Denenberg, J.O.; Ho, E.; Allison, M.; Langer, R.D.; Gamst, A.; Bundens, W.P.; Fronek, A. Ethnicity and peripheral arterial disease: The San Diego Population Study. Circulation 2005, 112, 2703–2707. [Google Scholar] [CrossRef]
- Vitalis, A.; Lip, G.Y.; Kay, M.; Vohra, R.K.; Shantsila, A. Ethnic differences in the prevalence of peripheral arterial disease: A systematic review and meta-analysis. Expert Rev. Cardiovasc. Ther. 2017, 15, 327–338. [Google Scholar] [CrossRef]
- Hicks, C.W.; Ding, N.; Kwak, L.; Ballew, S.H.; Kalbaugh, C.A.; Folsom, A.R.; Heiss, G.; Coresh, J.; Black, J.H., 3rd; Selvin, E.; et al. Risk of peripheral artery disease according to race and sex: The Atherosclerosis Risk in Communities (ARIC) study. Atherosclerosis 2021, 324, 52–57. [Google Scholar] [CrossRef]
- Leeper, N.J.; Kullo, I.J.; Cooke, J.P. Genetics of peripheral artery disease. Circulation 2012, 125, 3220–3228. [Google Scholar] [CrossRef]
- Wahlgren, C.M.; Magnusson, P.K. Genetic influences on peripheral arterial disease in a twin population. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 678–682. [Google Scholar] [CrossRef] [PubMed]
- Meaney, F.J.; Taylor, C. Heritability; Encyclopedia Britannica: Chicago, IL, USA, 2018; Available online: https://www.britannica.com/science/heritability (accessed on 20 August 2022).
- Knowles, J.W.; Assimes, T.L.; Li, J.; Quertermous, T.; Cooke, J.P. Genetic susceptibility to peripheral arterial disease: A dark corner in vascular biology. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2068–2078. [Google Scholar] [CrossRef] [PubMed]
- Valentine, R.J.; Guerra, R.; Stephan, P.; Scoggins, E.; Clagett, G.P.; Cohen, J. Family history is a major determinant of subclinical peripheral arterial disease in young adults. J. Vasc. Surg. 2004, 39, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Carmelli, D.; Fabsitz, R.R.; Swan, G.E.; Reed, T.; Miller, B.; Wolf, P.A. Contribution of genetic and environmental influences to ankle-brachial blood pressure index in the NHLBI Twin Study. National Heart, Lung, and Blood Institute. Am. J. Epidemiol. 2000, 151, 452–458. [Google Scholar] [CrossRef]
- Murabito, J.M.; Guo, C.Y.; Fox, C.S.; D’Agostino, R.B. Heritability of the ankle-brachial index: The Framingham Offspring study. Am. J. Epidemiol. 2006, 164, 963–968. [Google Scholar] [CrossRef]
- Wassel, C.L.; Loomba, R.; Ix, J.H.; Allison, M.; Denenberg, J.O.; Criqui, M.H. Family history of peripheral artery disease is associated with prevalence and severity of peripheral artery disease: The San Diego population study. J. Am. Coll. Cardiol. 2011, 58, 1386–1392. [Google Scholar] [CrossRef]
- Khaleghi, M.; Isseh, I.N.; Bailey, K.R.; Kullo, I.J. Family history as a risk factor for peripheral arterial disease. Am. J. Cardiol. 2014, 114, 928–932. [Google Scholar] [CrossRef]
- Ford, E.; Carroll, J.A.; Smith, H.E.; Scott, D.; Cassell, J.A. Extracting information from the text of electronic medical records to improve case detection: A systematic review. J. Am. Med. Inform. Assoc. 2016, 23, 1007–1015. [Google Scholar] [CrossRef] [Green Version]
- McCarty, C.A.; Chisholm, R.L.; Chute, C.G.; Kullo, I.J.; Jarvik, G.P.; Larson, E.B.; Li, R.; Masys, D.R.; Ritchie, M.D.; Roden, D.M.; et al. The eMERGE Network: A consortium of biorepositories linked to electronic medical records data for conducting genomic studies. BMC Med. Genom. 2011, 4, 13. [Google Scholar] [CrossRef]
- Available online: https://www.genome.gov/Funded-Programs-Projects/eMERGE-Genomics-Risk-Assessment-and-Management-Network (accessed on 14 July 2022).
- Klarin, D.; Lynch, J.; Aragam, K.; Chaffin, M.; Assimes, T.L.; Huang, J.; Lee, K.M.; Shao, Q.; Huffman, J.E.; Natarajan, P.; et al. VA Million Veteran Program. Genome-wide association study of peripheral artery disease in the Million Veteran Program. Nat. Med. 2019, 25, 1274–1279. [Google Scholar] [CrossRef]
- Abrantes, P.; Rosa, A.; Francisco, V.; Sousa, I.; Xavier, J.M.; Oliveira, S.A. Mitochondrial genome association study with peripheral arterial disease and venous thromboembolism. Atherosclerosis 2016, 252, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Panagiotou, O.A.; Ioannidis, J.P. Genome-Wide Significance Project. What should the genome-wide significance threshold be? Empirical replication of borderline genetic associations. Int. J. Epidemiol. 2012, 41, 273–286. [Google Scholar] [CrossRef] [PubMed]
- Safarova, M.S.; Fan, X.; Austin, E.E.; van Zuydam, N.; Hopewell, J.; Schaid, D.J.; Kullo, I.J. Targeted Sequencing Study to Uncover Shared Genetic Susceptibility Between Peripheral Artery Disease and Coronary Heart Disease-Brief Report. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1227–1233. [Google Scholar] [CrossRef] [PubMed]
- Badimon, L.; Vilahur, G. Thrombosis formation on atherosclerotic lesions and plaque rupture. J. Intern. Med. 2014, 276, 618–632. [Google Scholar] [CrossRef]
- Ladich, E.R.; Virmani, R. What Is the Role of Thrombosis in the Pathophysiology of Atherosclerosis; Burke, A.P., Ed.; Available online: https://www.medscape.com/answers/1612610-193803/what-is-the-role-of-thrombosis-in-the-pathophysiology-of-atherosclerosis (accessed on 10 July 2022).
- Mueller, T.; Marschon, R.; Dieplinger, B.; Haidinger, D.; Gegenhuber, A.; Poelz, W.; Webersinke, G.; Haltmayer, M. Factor V Leiden, prothrombin G20210A, and methylenetetrahydrofolate reductase C677T mutations are not associated with chronic limb ischemia: The Linz Peripheral Arterial Disease (LIPAD) study. J. Vasc. Surg. 2005, 41, 808–815. [Google Scholar] [CrossRef]
- Chen, Q.; Smith, C.Y.; Bailey, K.R.; Wennberg, P.W.; Kullo, I.J. Disease location is associated with survival in patients with peripheral arterial disease. J. Am. Heart Assoc. 2013, 2, e000304. [Google Scholar] [CrossRef]
- Wang, W.; Zhao, T.; Geng, K.; Yuan, G.; Chen, Y.; Xu, Y. Smoking and the Pathophysiology of Peripheral Artery Disease. Front. Cardiovasc. Med. 2021, 8, 704106. [Google Scholar] [CrossRef]
- Justice, A.E.; Winkler, T.W.; Feitosa, M.F.; Graff, M.; Fisher, V.A.; Young, K.; Barata, L.; Deng, X.; Czajkowski, J.; Hadley, D.; et al. Genome-wide meta-analysis of 241,258 adults accounting for smoking behaviour identifies novel loci for obesity traits. Nat. Commun. 2017, 8, 14977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khera, A.V.; Chaffin, M.; Aragam, K.G.; Haas, M.E.; Roselli, C.; Choi, S.H.; Natarajan, P.; Lander, E.S.; Lubitz, S.A.; Ellinor, P.T.; et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat. Genet. 2018, 50, 1219–1224. [Google Scholar] [CrossRef]
- Ntalla, I.; Kanoni, S.; Zeng, L.; Giannakopoulou, O.; Daneshm, J.; Watkins, H.; Samani, N.J.; Deloukas, P.; Schunkert, H.; UK Biobank CardioMetabolic Consortium CHD Working Group. Genetic Risk Score for Coronary Disease Identifies Predispositions to Cardiovascular and Noncardiovascular Diseases. J. Am. Coll. Cardiol. 2019, 73, 2932–2942. [Google Scholar] [CrossRef]
- Tam, V.; Patel, N.; Turcotte, M.; Bossé, Y.; Paré, G.; Meyre, D. Benefits and limitations of genome-wide association studies. Nat. Rev. Genet. 2019, 20, 467–484. [Google Scholar] [CrossRef] [PubMed]
- Huyghe, J.R.; Jackson, A.U.; Fogarty, M.P.; Buchkovich, M.L.; Stančáková, A.; Stringham, H.M.; Sim, X.; Yang, L.; Fuchsberger, C.; Cederberg, H.; et al. Exome array analysis identifies new loci and low-frequency variants influencing insulin processing and secretion. Nat. Genet. 2013, 45, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Shameer, K.; Klee, E.W.; Dalenberg, A.K.; Kullo, I.J. Whole exome sequencing implicates an INO80D mutation in a syndrome of aortic hypoplasia, premature atherosclerosis, and arterial stiffness. Circ. Cardiovasc. Genet. 2014, 7, 607–614. [Google Scholar] [CrossRef] [PubMed]
- St Hilaire, C.; Ziegler, S.G.; Markello, T.C.; Brusco, A.; Groden, C.; Gill, F.; Carlson-Donohoe, H.; Lederman, R.J.; Chen, M.Y.; Yang, D.; et al. NT5E mutations and arterial calcifications. N. Engl. J. Med. 2011, 364, 432–442. [Google Scholar] [CrossRef]
- Lai, W.K.; Kan, M.Y. Homocysteine-Induced Endothelial Dysfunction. Ann. Nutr. Metab. 2015, 67, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Guo, X.; Xia, Y.; Mao, L. An update on the phenotypic switching of vascular smooth muscle cells in the pathogenesis of atherosclerosis. Cell. Mol. Life Sci. 2021, 79, 6. [Google Scholar] [CrossRef]
- Lee, D.Y.; Chiu, J.J. Atherosclerosis and flow: Roles of epigenetic modulation in vascular endothelium. J. Biomed. Sci. 2019, 26, 56. [Google Scholar] [CrossRef]
- Xu, H.; Li, S.; Liu, Y.S. Roles and Mechanisms of DNA Methylation in Vascular Aging and Related Diseases. Front. Cell Dev. Biol. 2021, 9, 699374. [Google Scholar] [CrossRef]
- Dabravolski, S.A.; Sukhorukov, V.N.; Kalmykov, V.A.; Grechko, A.V.; Shakhpazyan, N.K.; Orekhov, A.N. The Role of KLF2 in the Regulation of Atherosclerosis Development and Potential Use of KLF2-Targeted Therapy. Biomedicines 2022, 10, 254. [Google Scholar] [CrossRef]
- Alhayaza, R.; Haque, E.; Karbasiafshar, C.; Sellke, F.W.; Abid, M.R. The Relationship Between Reactive Oxygen Species and Endothelial Cell Metabolism. Front. Chem. 2020, 8, 592688. [Google Scholar] [CrossRef]
- Liu, S.; Lin, Z. Vascular Smooth Muscle Cells Mechanosensitive Regulators and Vascular Remodeling. J. Vasc. Res. 2022, 59, 90–113. [Google Scholar] [CrossRef] [PubMed]
- Liu, O.H.; Kiema, M.; Beter, M.; Ylä-Herttuala, S.; Laakkonen, J.P.; Kaikkonen, M.U. Hypoxia-Mediated Regulation of Histone Demethylases Affects Angiogenesis-Associated Functions in Endothelial Cells. Arterioscler. Thromb Vasc. Biol. 2020, 40, 2665–2677. [Google Scholar] [CrossRef] [PubMed]
- Churov, A.; Summerhill, V.; Grechko, A.; Orekhova, V.; Orekhov, A. MicroRNAs as Potential Biomarkers in Atherosclerosis. Int. J. Mol. Sci. 2019, 20, 5547. [Google Scholar] [CrossRef] [PubMed]
- de Gonzalo-Calvo, D.; Iglesias-Gutiérrez, E.; Llorente-Cortés, V. Epigenetic Biomarkers and Cardiovascular Disease: Circulating MicroRNAs. Rev. Esp. Cardiol. 2017, 70, 763–769. [Google Scholar] [CrossRef] [PubMed]
- Kazmi, N.; Elliott, H.R.; Burrows, K.; Tillin, T.; Hughes, A.D.; Chaturvedi, N.; Gaunt, T.R.; Relton, C.L. Associations between high blood pressure and DNA methylation. PLoS ONE 2020, 15, e0227728. [Google Scholar] [CrossRef]
- Gonzalez-Jaramillo, V.; Portilla-Fernandez, E.; Glisic, M.; Voortman, T.; Bramer, W.; Chowdhury, R.; Roks, A.J.M.; Danser, A.H.J.; Muka, T.; Nano, J.; et al. Correction to: The role of DNA methylation and histone modifications in blood pressure: A systematic review. J. Hum. Hypertens. 2020, 34, 193, Erratum in J. Hum. Hypertens. 2019, 33, 703–715. [Google Scholar] [CrossRef]
- Stoll, S.; Wang, C.; Qiu, H. DNA Methylation and Histone Modification in Hypertension. Int. J. Mol. Sci. 2018, 19, 1174. [Google Scholar] [CrossRef]
- Yamunadevi, A.; Pratibha, R.; Rajmohan, M.; Mahendraperumal, S.; Ganapathy, N. Basics of Epigenetics and Role of Epigenetics in Diabetic Complications. J. Pharm. Bioallied. Sci. 2021, 13, S336–S343. [Google Scholar] [CrossRef]
- Maas, S.C.E.; Mens, M.M.J.; Kühnel, B.; van Meurs, J.B.J.; Uitterlinden, A.G.; Peters, A.; Prokisch, H.; Herder, C.; Grallert, H.; Kunze, S.; et al. Smoking-related changes in DNA methylation and gene expression are associated with cardio-metabolic traits. Clin. Epigenet. 2020, 12, 157. [Google Scholar] [CrossRef]
- Siemelink, M.A.; van der Laan, S.W.; Haitjema, S.; van Koeverden, I.D.; Schaap, J.; Wesseling, M.; de Jager, S.C.A.; Mokry, M.; van Iterson, M.; Dekkers, K.F.; et al. Smoking is Associated to DNA Methylation in Atherosclerotic Carotid Lesions. Circ. Genom. Precis. Med. 2018, 11, e002030. [Google Scholar] [CrossRef]
- Kinser, H.E.; Pincus, Z. MicroRNAs as modulators of longevity and the aging process. Hum. Genet. 2020, 139, 291–308. [Google Scholar] [CrossRef] [PubMed]
- Światowy, W.J.; Drzewiecka, H.; Kliber, M.; Sąsiadek, M.; Karpiński, P.; Pławski, A.; Jagodziński, P.P. Physical Activity and DNA Methylation in Humans. Int. J. Mol. Sci. 2021, 22, 12989. [Google Scholar] [CrossRef] [PubMed]
- Plaza-Diaz, J.; Izquierdo, D.; Torres-Martos, Á.; Baig, A.T.; Aguilera, C.M.; Ruiz-Ojeda, F.J. Impact of Physical Activity and Exercise on the Epigenome in Skeletal Muscle and Effects on Systemic Metabolism. Biomedicines 2022, 10, 126. [Google Scholar] [CrossRef] [PubMed]
- Gorabi, A.M.; Penson, P.E.; Banach, M.; Motallebnezhad, M.; Jamialahmadi, T.; Sahebkar, A. Epigenetic control of atherosclerosis via DNA methylation: A new therapeutic target? Life Sci. 2020, 253, 117682. [Google Scholar] [CrossRef] [PubMed]
- Masud, R.; Shameer, K.; Dhar, A.; Ding, K.; Kullo, I.J. Gene expression profiling of peripheral blood mononuclear cells in the setting of peripheral arterial disease. J. Clin. Bioinform. 2012, 2, 6. [Google Scholar] [CrossRef]
- Li, X.; Meng, X.; Spiliopoulou, A.; Timofeeva, M.; Wei, W.Q.; Gifford, A.; Shen, X.; He, Y.; Varley, T.; McKeigue, P.; et al. MR-PheWAS: Exploring the causal effect of SUA level on multiple disease outcomes by using genetic instruments in UK Biobank. Ann. Rheum. Dis. 2018, 77, 1039–1047. [Google Scholar] [CrossRef]
- Available online: https://www.feinberg.northwestern.edu/researchday/showItem.php?id=989493 (accessed on 28 July 2022).
- Available online: https://www.genome.gov/Health/Genomics-and-Medicine/Polygenic-risk-scores (accessed on 28 July 2022).
- Available online: https://www.genome.gov/Funded-Programs-Projects/PRIMED-Consortium (accessed on 28 July 2022).
- Wang, F.; Ghanzouri, I.; Leeper, N.J.; Tsao, P.S.; Ross, E.G. Development of a polygenic risk score to improve detection of peripheral artery disease. Vasc. Med. 2022, 27, 219–227. [Google Scholar] [CrossRef]
- Kullo, I.J. Polygenic risk score for peripheral artery disease: A tool to refine risk stratification. Vasc. Med. 2022, 27, 228–229. [Google Scholar] [CrossRef]
- Small, A.M.; Huffman, J.E.; Klarin, D.; Sabater-Lleal, M.; Lynch, J.A.; Assimes, T.L.; Sun, Y.V.; Miller, D.; Freiberg, M.S.; Morrison, A.C.; et al. Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) Hemostasis Working Group and the VA Million Veteran Program. Mendelian Randomization Analysis of Hemostatic Factors and Their Contribution to Peripheral Artery Disease-Brief Report. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 380–386. [Google Scholar] [CrossRef] [PubMed]
- Dikilitas, O.; Satterfield, B.A.; Kullo, I.J. Risk factors for polyvascular involvement in patients with peripheral artery disease: A Mendelian Randomization Study. J. Am. Heart Assoc. 2020, 9, e017740. [Google Scholar] [CrossRef]
- Levin, M.G.; Zuber, V.; Walker, V.M.; Klarin, D.; Lynch, J.; Malik, R.; Aday, A.W.; Bottolo, L.; Pradhan, A.D.; Dichgans, M. Prioritizing the Role of Major Lipoproteins and Subfractions as Risk Factors for Peripheral Artery Disease. Circulation 2021, 144, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Levin, M.G.; Klarin, D.; Assimes, T.L.; Freiberg, M.S.; Ingelsson, E.; Lynch, J.; Natarajan, P.; O’Donnell, C.; Rader, D.J.; Tsao, P.S.; et al. VA Million Veteran Program. Genetics of Smoking and Risk of Atherosclerotic Cardiovascular Diseases: A Mendelian Randomization Study. JAMA Netw. Open. 2021, 4, e2034461. [Google Scholar] [CrossRef] [PubMed]
- Wootton, R.E.; Richmond, R.C.; Stuijfzand, B.G.; Lawn, R.B.; Sallis, H.M.; Taylor, G.M.J.; Hemani, G.; Jones, H.J.; Zammit, S.; Davey Smith, G.; et al. Evidence for causal effects of lifetime smoking on risk for depression and schizophrenia: A Mendelian randomisation study. Psychol. Med. 2020, 50, 2435–2443. [Google Scholar] [CrossRef] [PubMed]
- Nikpay, M.; Goel, A.; Won, H.H.; Hall, L.M.; Willenborg, C.; Kanoni, S.; Saleheen, D.; Kyriakou, T.; Nelson, C.P.; Hopewell, J.C.; et al. A comprehensive 1000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Nat. Genet. 2015, 47, 1121–1130. [Google Scholar] [CrossRef]
- Levin, M.G.; Klarin, D.; Walker, V.M.; Gill, D.; Lynch, J.; Hellwege, J.N.; Keaton, J.M.; Lee, K.M.; Assimes, T.L.; Natarajan, P.; et al. Association between Genetic Variation in Blood Pressure and Increased Lifetime Risk of Peripheral Artery Disease. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 2027–2034. [Google Scholar] [CrossRef]
- Koren, O.; Spor, A.; Felin, J.; Fåk, F.; Stombaugh, J.; Tremaroli, V.; Behre, C.J.; Knight, R.; Fagerberg, B.; Ley, R.E.; et al. Human oral, gut, and plaque microbiota in patients with atherosclerosis. Proc. Natl. Acad. Sci. USA 2011, 108, 4592–4598. [Google Scholar] [CrossRef]
- Wang, Z.; Klipfell, E.; Bennett, B.J.; Koeth, R.; Levison, B.S.; Dugar, B.; Feldstein, A.E.; Britt, E.B.; Fu, X.; Chung, Y.M.; et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 2011, 472, 57–63. [Google Scholar] [CrossRef]
- Tran, B. Assessment and management of peripheral arterial disease: What every cardiologist should know. Heart 2021, 107, 1835–1843. [Google Scholar] [CrossRef]
- Ruiz-Canela, M.; Martínez-González, M.A. Lifestyle and dietary risk factors for peripheral artery disease. Circ. J. 2014, 78, 553–559. [Google Scholar] [CrossRef]
- Tierney, S.; Fennessy, F.; Hayes, D.B. ABC of arterial and vascular disease. Secondary prevention of peripheral vascular disease. BMJ 2000, 320, 1262–1265. [Google Scholar] [CrossRef]
- Keller, S.; Del Giorno, R.; Buso, G.; Deslarzes, C.; Calanca, L.; Lanzi, S.; Mazzolai, L. Passeport vasculaire: Un outil pour la prévention secondaire des patients avec maladie artérielle périphérique [Vascular passport : A tool for secondary prevention among patients with peripheral artery disease]. Rev. Med. Suisse. 2021, 17, 2128–2131. (In French) [Google Scholar] [PubMed]
- Signorelli, S.S.; Fiore, V.; Malaponte, G. Inflammation and peripheral arterial disease: The value of circulating biomarkers (Review). Int. J. Mol. Med. 2014, 33, 777–783. [Google Scholar] [CrossRef]
- Wildman, R.P.; Muntner, P.; Chen, J.; Sutton-Tyrrell, K.; He, J. Relation of inflammation to peripheral arterial disease in the national health and nutrition examination survey, 1999–2002. Am. J. Cardiol. 2005, 96, 1579–1583. [Google Scholar] [CrossRef] [PubMed]
- Conte, M.S.; Bradbury, A.W.; Kolh, P.; White, J.V.; Dick, F.; Fitridge, R.; Mills, J.L.; Ricco, J.B.; Suresh, K.R.; Murad, M.H.; et al. Global vascular guidelines on the management of chronic limb-threatening ischemia. J. Vasc. Surg. 2019, 69, 3S–125S.e40, Erratum in J. Vasc. Surg. 2019, 70, 662. [Google Scholar] [CrossRef] [PubMed]
- Poredos, P. Secondary Prevention of Peripheral Arterial Disease. ESC, European Society of Cardiology, E-Journal of the ESC Council for Cardiology Practice. 2004, 3. Available online: https://www.escardio.org/Journals/E-Journal-of-Cardiology-Practice/Volume-3/Secondary-prevention-of-peripheral-arterial-disease-Title-Secondary-preventio (accessed on 15 July 2022).
- American Heart Association. Prevention and Treatment of PAD. Last Reviewed: 2 June 2021. Available online: https://www.heart.org/en/health-topics/peripheral-artery-disease/prevention-and-treatment-of-pad (accessed on 15 July 2022).
- Forster, R.; Liew, A.; Bhattacharya, V.; Shaw, J.; Stansby, G. Gene therapy for peripheral arterial disease. Cochrane Database Syst. Rev. 2018, 10, CD012058. Available online: https://www.cochrane.org/CD012058/PVD_gene-therapy-peripheral-arterial-disease (accessed on 15 July 2022). [CrossRef]
- Salybekov, A.A.; Wolfien, M.; Kobayashi, S.; Steinhoff, G.; Asahara, T. Personalized Cell Therapy for Patients with Peripheral Arterial Diseases in the Context of Genetic Alterations: Artificial Intelligence-Based Responder and Non-Responder Prediction. Cells 2021, 10, 3266. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Polymorphism | Type of Study | Diagnosis | Location/ Chromosome | The Nearest Gene (s) | References |
|---|---|---|---|---|---|
| LA studies/SNPs | |||||
| LA/SNPs | LEAD/POAD | 1p31 | Unknown | [23] | |
| LA/SNPs | LEAD | 1p | HTR6 5, PLA2G2E, ECE1, IL22RA1, ECE1, IL28RA, LDLRAP1, PTAFR, FABP3, COL16A1, FNDC5, COL8A | [24] | |
| LA/SNPs | LEAD | 3p,3q | IL1RAP, FGF12 | [24,25] | |
| LA/SNPs | LEAD | 6q | LPA, PLG | [24] | |
| LA/SNPs | LEAD | 7q | Unidentified | [24] | |
| LA/SNPs | LEAD | 10p | IL15RA, ITGA8 | [24] | |
| LA/SNPs | LEAD | 16q | IL4R, IL21R | [24] | |
| CGS/SNPs studies | |||||
| rs2171209 | CGS/SNPs | LEAD/ABI | 6q25.3 | SYTL3 | [25,26] |
| rs290481 | CGS/SNPs | LEAD/ABI | 10q25.2–q25.3 | TCF7L2 | [25,26] |
| rs3745274 | CGS/SNPs | LEAD/ABI | 19q13.2 | CYP2B6 | [24,25] |
| rs891512, rs1808593 | CG/SNPs | LEAD | 7q36.1 | NOS3 | [25,27] |
| rs1042713 | CGS/SNPs | LEAD/Lp(a) | 5q32 | ADRB2 | [24,25,27] |
| rs828853, rs1299142 | CGS/SNPs | LEAD/DM | 2p13.1 | SLC4A5 | [25,27] |
| rs3917187/rs2284791, rs668871 | CGS/SNPs | LEAD/TG | 14q24.3, 6q25.3 | TGFB3, SLC22A3 | [24,25,27] |
| rs2110981, rs2270042 | CGS/SNPs | LEAD CRP | 2p13.3 | ADD2 | [25,27] |
| rs22394, rs10247, rs11681 | CGS/SNPs | LEAD/FB | 2p13.3 | ATP6B1 | [25,27] |
| rs207129, rs154027, rs257376 | CGS/SNPs | LEAD/Hyc | 6p22.2, 7q22.3 | SLC17A2, PKRAR2B | [24,25,27] |
| Relevant LEAD-related GWAS studies | |||||
| rs10757269 | GWAS1 | CAD LEAD/ABI | 9p21 | CDKN2B | [25,28,29,30] |
| rs13290547 | GWAS1 | LEAD | 9q33 | DAB21P | [25,28] |
| rs3794624 | GWAS3 | LEAD | 16q24.2 | CYBA | [25,28] |
| rs1122608 | GWAS3 | LEAD | 19p13.2 | LDLR | [24,25,28] |
| rs10757278 | GWAS1 | AAA/CAD/ LEAD/ICA | 9p21 | CDKN2A/CDKN2B | [29] |
| rs1333049 | GWAS1 | LEAD/MI | 9p21 | CDKN2A/CDKN2B | [28,30] |
| rs1051730 | GWAS3 | No. of CS | 15q25.1 | CHRNA5/A3/B4 | [25,31] |
| rs7025486 | GWAS3 | AAA/LEAD/MI | 9q33 | DAB2IP | [25,28,32] |
| rs3794624 | GWAS1 | LEAD | 16q24.2 | CYBA | [25,28] |
| rs376511 | GWAS3 | TAO | 3p25.3 | IL17RC | [25,33,34] |
| rs7632505 | GWAS3 | TAO | 3q21.1 | SEMA5B | [25,33,35] |
| rs10178082 | GWAS3 | TAO | 7p21.3 | RPA3 | [25,33] |
| rs1902341 | GWAS1 | LEAD | 3p23–p22.3 | OSBPL10 | [25,36] |
| rs9584669 | GWAS1 | LEAD | 13q32.2 | IPO5/RAP2A | [17,25,37,38,39] |
| rs6842241 | GWAS1 | LEAD | 4q31.22–q31.23 | EDNRA | [25,37] |
| rs2074633 | GWAS2 | LEAD | 7p21.1 | HDAC9 | [25,37] |
| rs653178/rs3184504 | EMR GWAS2 | LEAD/MI | 12q24.12 | ATXN2-SH2B3 | [25,40] |
| rs2554503 | GWAS3 | LEAD | 8p23 | CSMD1 | [25,36] |
| rs235243 | GWAS3 | LEAD | 1p36.22–p36.21 | VSP13D | [25,36] |
| Other Genetic Polymorphisms Associated with the Pathophysiological Mechanisms of LEAD | |||||
| Genes involved in lipid metabolism | LEAD | 2p24.1 19q13.32 | APOB APOE | [25,41] [25,42] | |
| Genes involved in the mechanism of inflammation | LEAD | 7p15.3 1q24.2 19p13.2 17q12 11q22.2 | IL6 SELE ICAM1 MCP1/CCL2 MMP1, MP3 | [13,43,44] [13,45] [25] [13,25,45] | |
| Genes involved in blood pressure regulation | LEAD | 17q23.3 3q23 1q42.2 | ACE AGT1R AGT | [13,25,46,47,48,49,50] | |
| Genes involved in the function of VSMc | LEAD | 7q36.1 | NOS3 | [25,51,52,53,54,55] | |
| Genes involved in vascular homeostasis | LEAD/DM/AT | 20q13.12 | SLC2A10 | [13,25,56] | |
| Genes involved in atherothrombosis | LEAD | 11p11.2 1q24.2 3q21.5 4q31.3 | F2 F5 P2YR12 FGB | [25,57,58] [25,58,59] [25,60,61] [25,62,63] | |
| HHcy | LEAD | 1q36.22 | MTHFR | [25,64,65] | |
| Clinical Features | Fountain Classification | Rutherford Classification | |
|---|---|---|---|
| Stage | Grade | Category | |
| Asymptomatic | I | 0 | 0 |
| Mild claudication | IIa | I | 1 |
| Moderate claudication Severe claudication | IIb | I | 2 |
| I | 3 | ||
| Ischemic rest pain | III | II | 4 |
| Minor tissue loss (Ischemic ulcers of the digits of the foot) | IV | III | 5 |
| Major tissue loss (severe ischemic ulcers or gangrene) | IV | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Butnariu, L.I.; Gorduza, E.V.; Florea, L.; Țarcă, E.; Moisă, Ș.M.; Tradafir, L.M.; Cojocaru, E.; Luca, A.-C.; Stătescu, L.; Bădescu, M.C. The Genetic Architecture of the Etiology of Lower Extremity Peripheral Artery Disease: Current Knowledge and Future Challenges in the Era of Genomic Medicine. Int. J. Mol. Sci. 2022, 23, 10481. https://doi.org/10.3390/ijms231810481
Butnariu LI, Gorduza EV, Florea L, Țarcă E, Moisă ȘM, Tradafir LM, Cojocaru E, Luca A-C, Stătescu L, Bădescu MC. The Genetic Architecture of the Etiology of Lower Extremity Peripheral Artery Disease: Current Knowledge and Future Challenges in the Era of Genomic Medicine. International Journal of Molecular Sciences. 2022; 23(18):10481. https://doi.org/10.3390/ijms231810481
Chicago/Turabian StyleButnariu, Lăcrămioara Ionela, Eusebiu Vlad Gorduza, Laura Florea, Elena Țarcă, Ștefana Maria Moisă, Laura Mihaela Tradafir, Elena Cojocaru, Alina-Costina Luca, Laura Stătescu, and Minerva Codruța Bădescu. 2022. "The Genetic Architecture of the Etiology of Lower Extremity Peripheral Artery Disease: Current Knowledge and Future Challenges in the Era of Genomic Medicine" International Journal of Molecular Sciences 23, no. 18: 10481. https://doi.org/10.3390/ijms231810481