
Chronic Alcohol Exposure Promotes Cancer Stemness and Glycolysis in Oral/Oropharyngeal Squamous Cell Carcinoma Cell Lines by Activating NFAT Signaling
 and
and 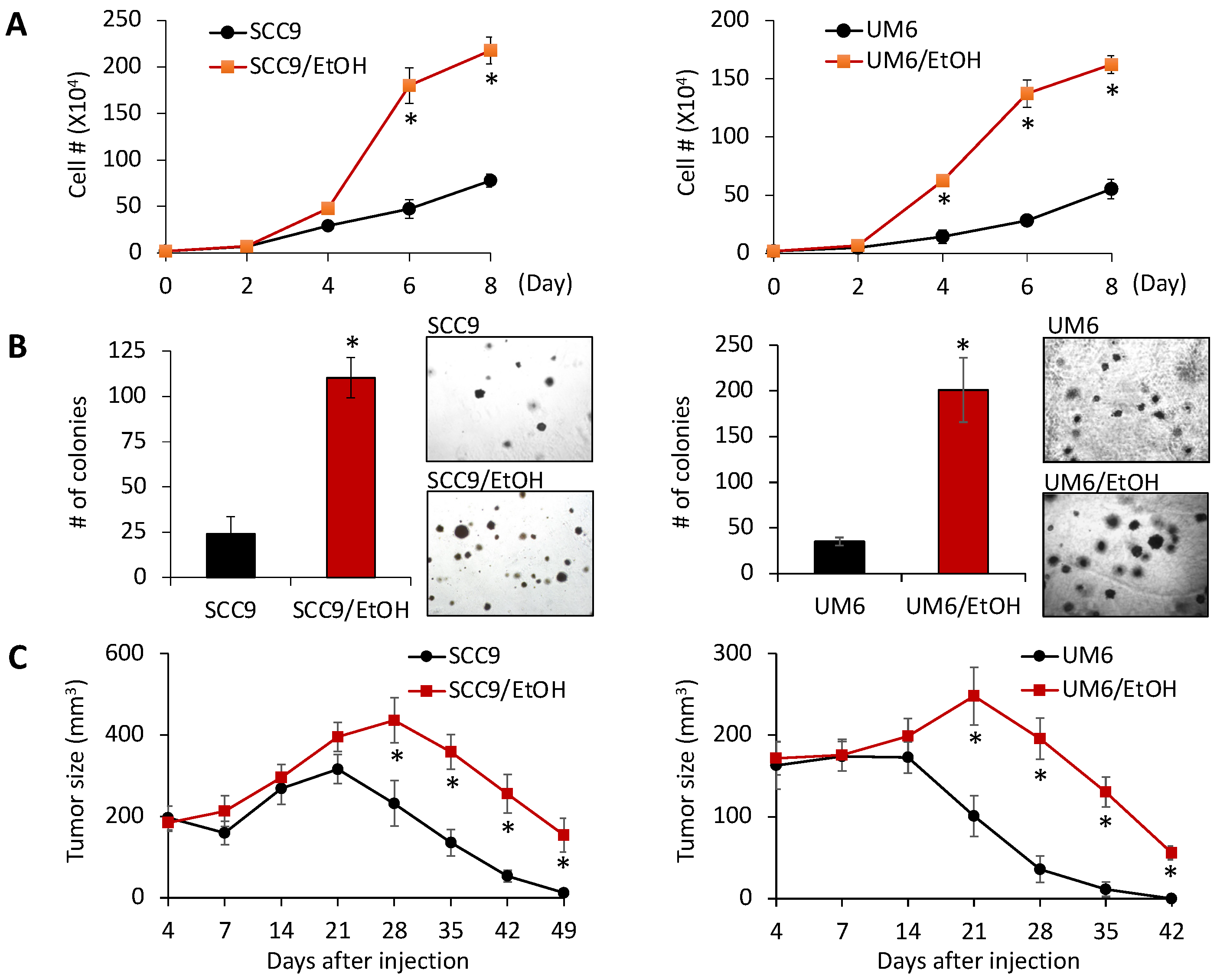{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Chronic EtOH Exposure Increases OSCC Growth In Vitro and In Vivo
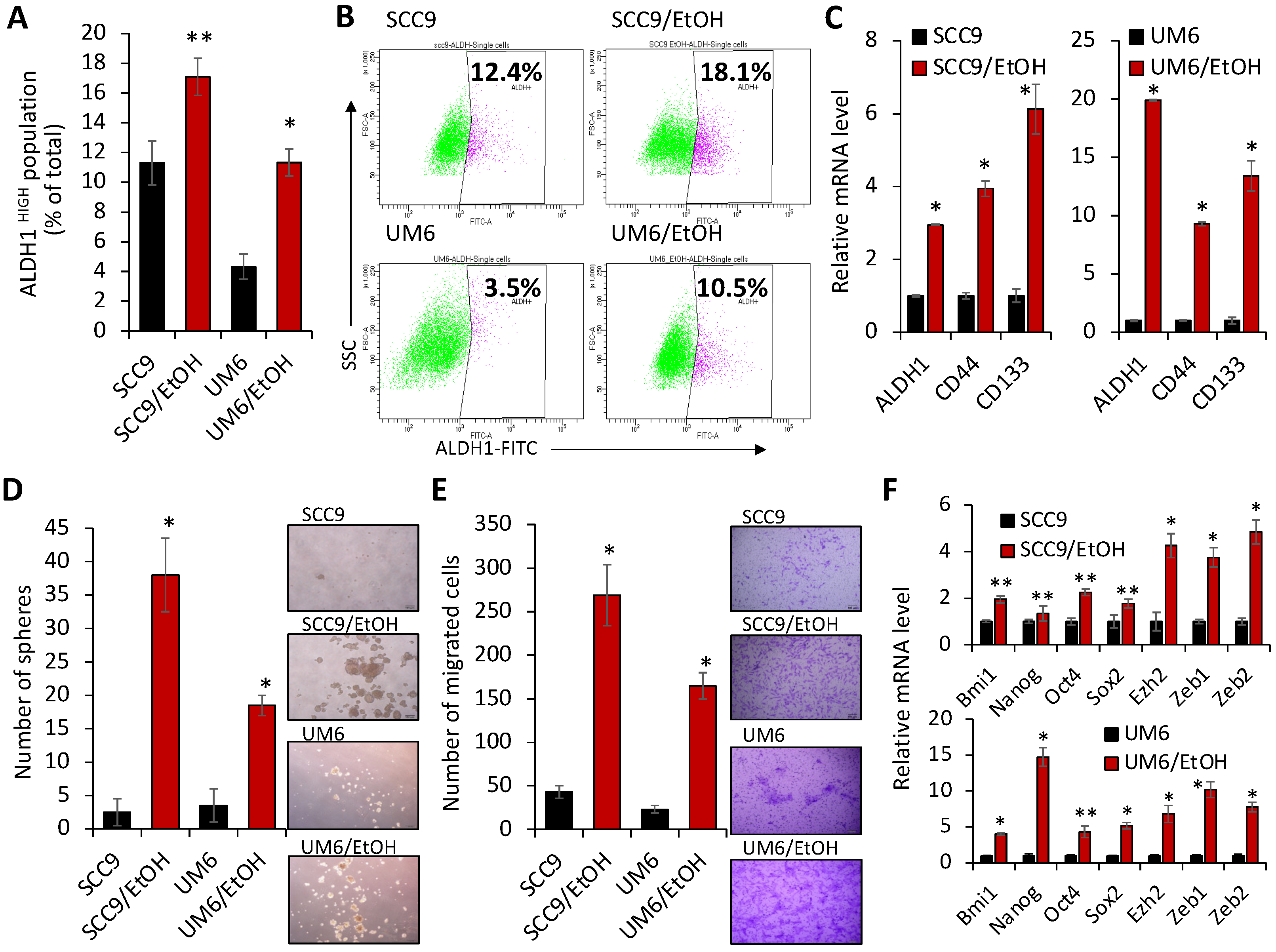
2.2. Chronic EtOH Exposure Increases the Number of ALDH1HIGH Stem-like Cell Populations and Characteristics Associated with Stemness in OSCC
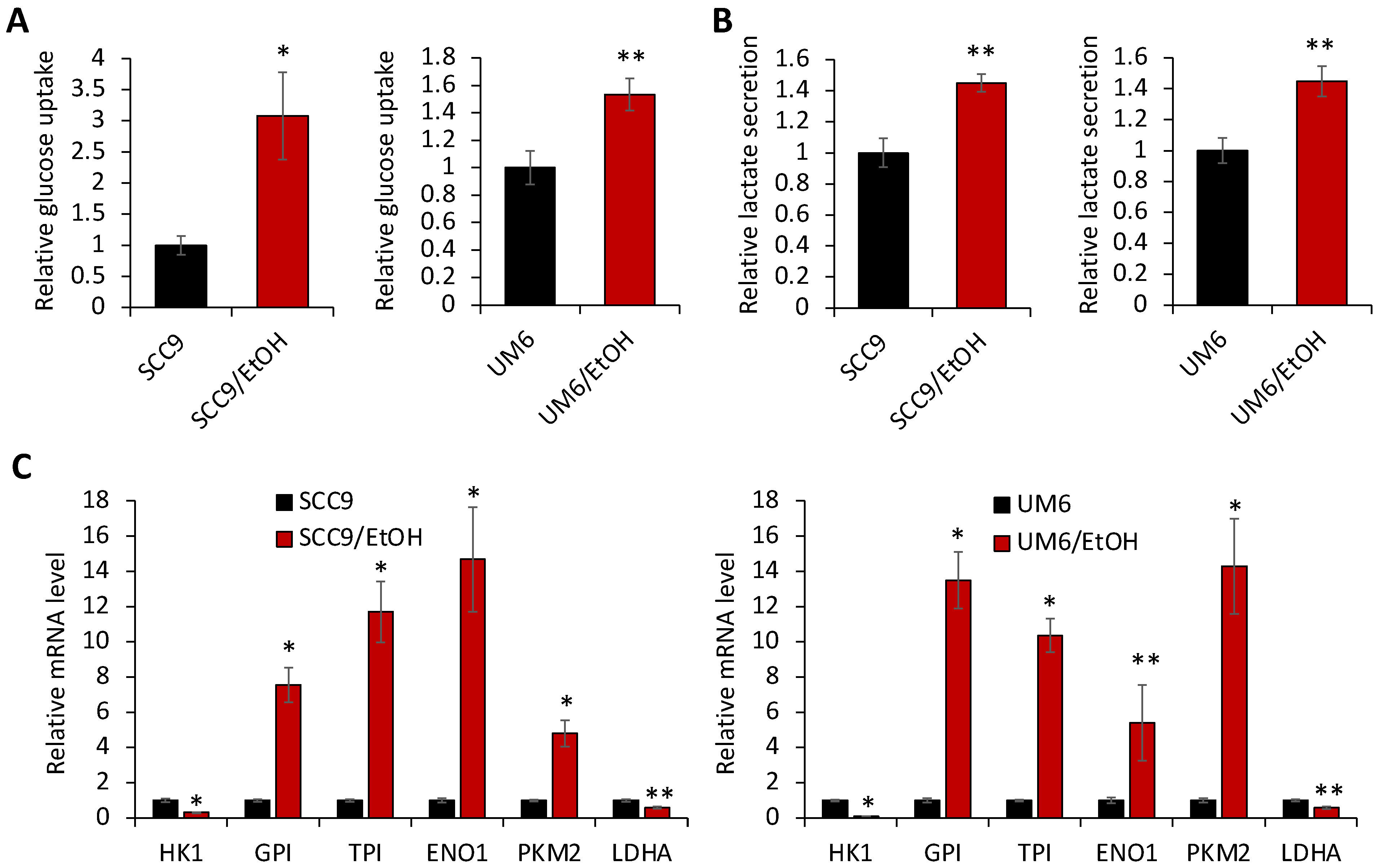
2.3. Chronic EtOH Exposure Promotes Aerobic Glycolysis in OSCC
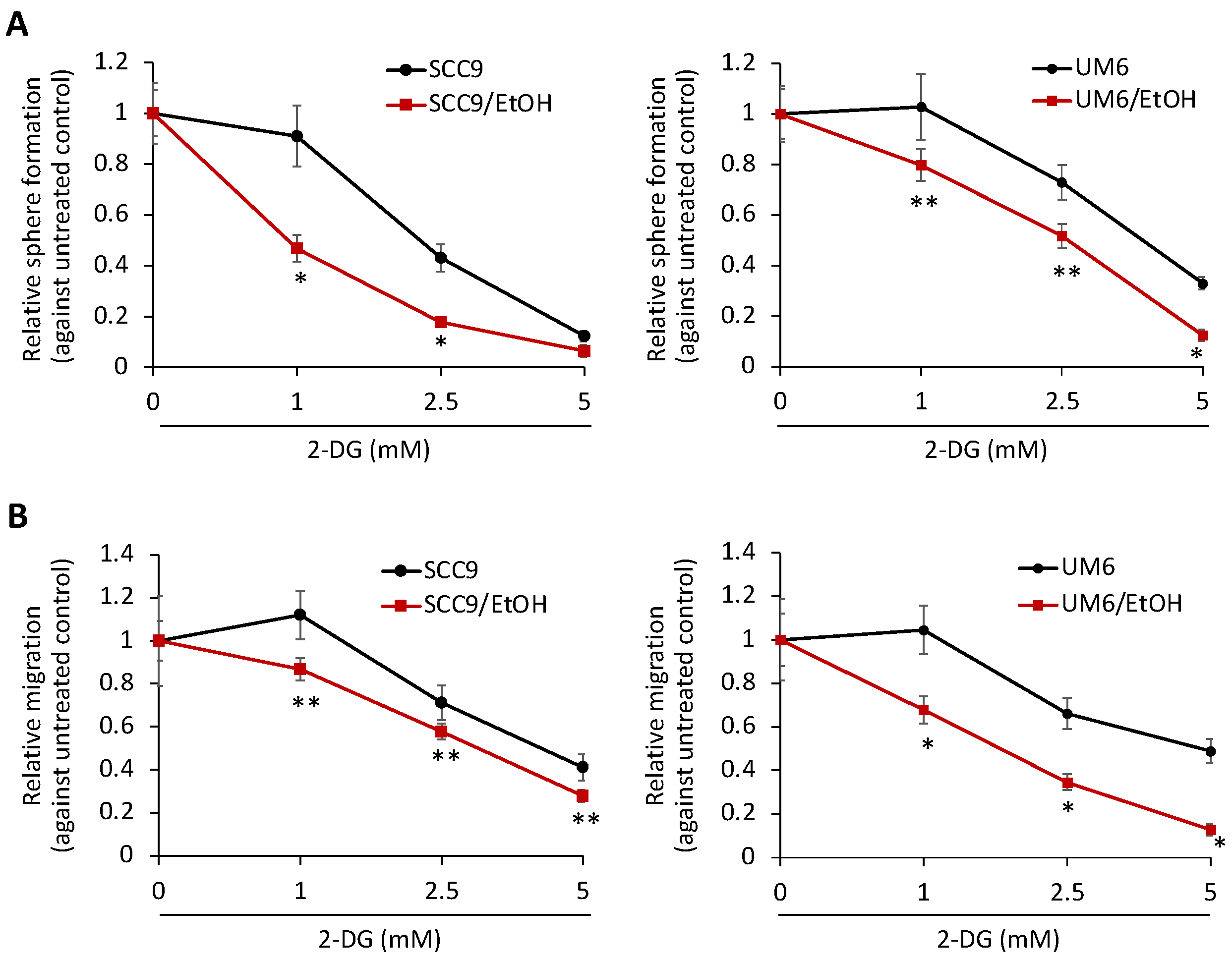
2.4. Increased Aerobic Glycolysis Is Required to Maintain the Stemness Characteristics of EtOH-Exposed OSCC Cells
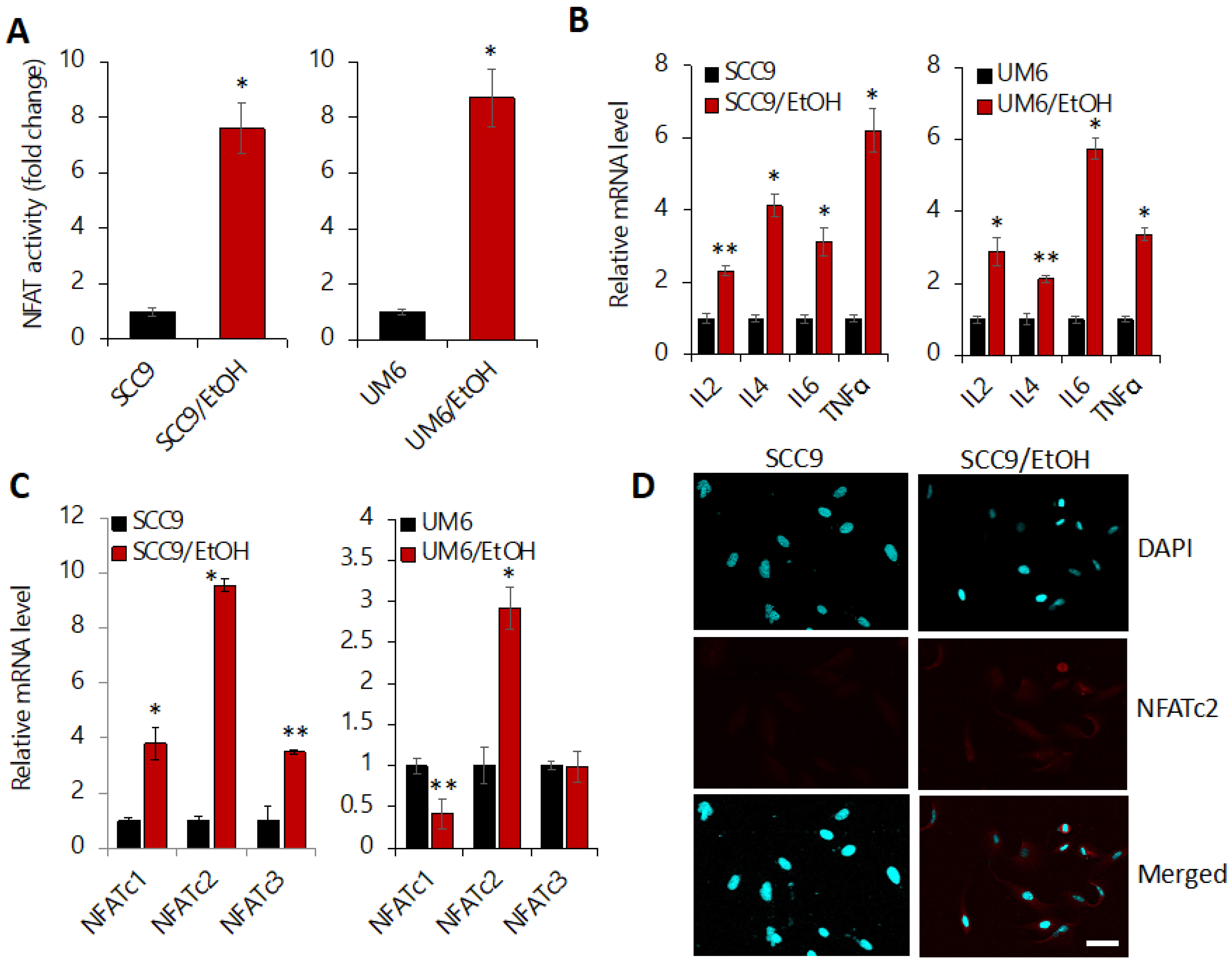
2.5. Chronic EtOH Exposure Activates NFAT Signaling
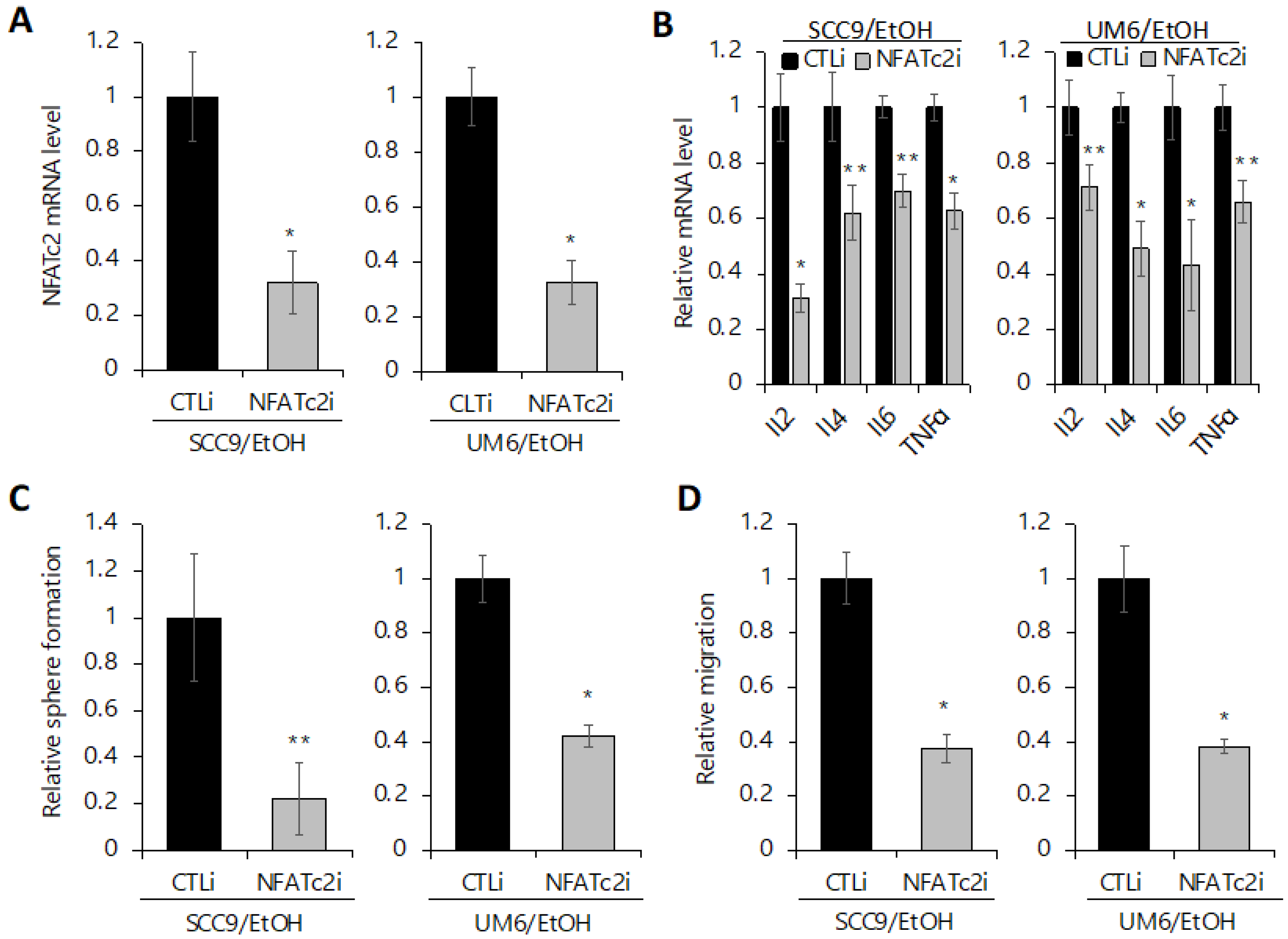
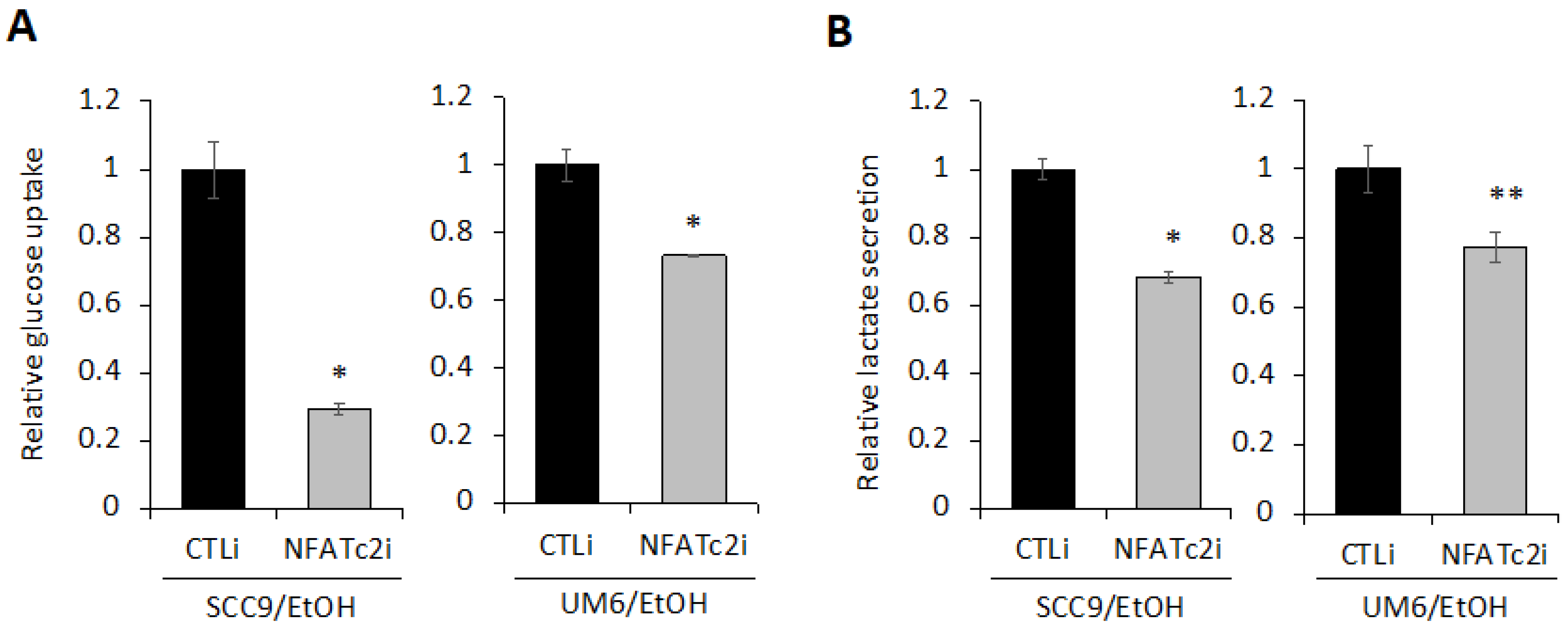
2.6. Silencing NFATc2 Inhibits Cancer Stemness and Aerobic Glycolysis in EtOH-Treated OSCC
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Reagent
4.2. Cell Proliferation Assay
4.3. Anchorage-Independent Growth
4.4. In Vivo Xenograft Tumor Assay
4.5. Glucose Uptake and Lactate Secretion
4.6. Quantitative Real-Time PCR (qPCR)
4.7. ALDH1 Assay
4.8. Tumor Sphere Formation Assay
4.9. Migration Assay
4.10. Luciferase Reporter Assay
4.11. Confocal Laser Scanning Microscopy
4.12. Small Interfering RNA (siRNA) Transfection
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Al-Swiahb, J.N.; Chen, C.H.; Chuang, H.C.; Fang, F.M.; Tasi, H.T.; Chien, C.Y. Clinical, pathological and molecular determinants in squamous cell carcinoma of the oral cavity. Future Oncol. 2010, 6, 837–850. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Shiboski, C.H.; Schmidt, B.L.; Jordan, R.C.K. Tongue and tonsil carcinoma—Increasing trends in the US population ages 20–44 years. Cancer-Am. Cancer Soc. 2005, 103, 1843–1849. [Google Scholar] [CrossRef]
- Chaturvedi, A.K.; Engels, E.A.; Pfeiffer, R.M.; Hernandez, B.Y.; Xiao, W.H.; Kim, E.; Jiang, B.; Goodman, M.T.; Sibug-Saber, M.; Cozen, W.; et al. Human Papillomavirus and Rising Oropharyngeal Cancer Incidence in the United States. J. Clin. Oncol. 2011, 29, 4294–4301. [Google Scholar] [CrossRef]
- Baan, R.; Straif, K.; Grosse, Y.; Secretan, B.; El Ghissassi, F.; Bouvard, V.; Altieri, A.; Cogliano, V.; WHO International Agency for Research on Cancer Monograph Working Group. Carcinogenicity of alcoholic beverages. Lancet Oncol. 2007, 8, 292–293. [Google Scholar] [CrossRef]
- Nelson, D.E.; Jarman, D.W.; Rehm, J.; Greenfield, T.K.; Rey, G.; Kerr, W.C.; Miller, P.; Shield, K.D.; Ye, Y.; Naimi, T.S. Alcohol-attributable cancer deaths and years of potential life lost in the United States. Am. J. Public Health 2013, 103, 641–648. [Google Scholar] [CrossRef]
- Garro, A.J.; Lieber, C.S. Alcohol and cancer. Annu. Rev. Pharmacol. Toxicol. 1990, 30, 219–249. [Google Scholar] [CrossRef]
- Seitz, H.K.; Homann, N. The role of acetaldehyde in alcohol-associated cancer of the gastrointestinal tract. Novartis Found. Symp. 2007, 285, 110–119. [Google Scholar]
- Pelucchi, C.; Tramacere, I.; Boffetta, P.; Negri, E.; La Vecchia, C. Alcohol consumption and cancer risk. Nutr. Cancer 2011, 63, 983–990. [Google Scholar] [CrossRef]
- Lieber, C.S. Mechanism of ethanol induced hepatic injury. Pharmacol. Ther. 1990, 46, 1–41. [Google Scholar] [CrossRef]
- Seitz, H.K.; Stickel, F.; Homann, N. Pathogenetic mechanisms of upper aerodigestive tract cancer in alcoholics. Int. J. Cancer 2004, 108, 483–487. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Wang, X.; Zhang, X.; Sun, Z.; Chen, X. Ethanol promotes chemically induced oral cancer in mice through activation of the 5-lipoxygenase pathway of arachidonic acid metabolism. Cancer Prev. Res. (Phila.) 2011, 4, 1863–1872. [Google Scholar] [CrossRef] [PubMed]
- Osei-Sarfo, K.; Tang, X.H.; Urvalek, A.M.; Scognamiglio, T.; Gudas, L.J. The molecular features of tongue epithelium treated with the carcinogen 4-nitroquinoline-1-oxide and alcohol as a model for HNSCC. Carcinogenesis 2013, 34, 2673–2681. [Google Scholar] [CrossRef] [PubMed]
- Shin, K.-H.; Kim, R.H. An Updated Review of Oral Cancer Stem Cells and Their Stemness Regulation. Crit. Rev. Oncog. 2018, 23, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Dean, M.; Fojo, T.; Bates, S. Tumour stem cells and drug resistance. Nat. Rev. Cancer 2005, 5, 275–284. [Google Scholar] [CrossRef]
- Guo, W.; Lasky, J.L., 3rd; Wu, H. Cancer stem cells. Pediatr. Res. 2006, 59, 59R–64R. [Google Scholar] [CrossRef]
- Crea, F.; Danesi, R.; Farrar, W.L. Cancer stem cell epigenetics and chemoresistance. Epigenomics 2009, 1, 63–79. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef]
- Zhu, X.; Chen, H.H.; Gao, C.Y.; Zhang, X.X.; Jiang, J.X.; Zhang, Y.; Fang, J.; Zhao, F.; Chen, Z.G. Energy metabolism in cancer stem cells. World J. Stem Cells 2020, 12, 448–461. [Google Scholar] [CrossRef]
- Ciavardelli, D.; Rossi, C.; Barcaroli, D.; Volpe, S.; Consalvo, A.; Zucchelli, M.; De Cola, A.; Scavo, E.; Carollo, R.; D’Agostino, D.; et al. Breast cancer stem cells rely on fermentative glycolysis and are sensitive to 2-deoxyglucose treatment. Cell Death Dis. 2014, 5, e1336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, J.; Qian, F.; Tchabo, N.; Mhawech-Fauceglia, P.; Beck, A.; Qian, Z.; Wang, X.; Huss, W.J.; Lele, S.B.; Morrison, C.D.; et al. Ovarian cancer spheroid cells with stem cell-like properties contribute to tumor generation, metastasis and chemotherapy resistance through hypoxia-resistant metabolism. PLoS ONE 2014, 9, e84941. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhou, Y.; Shingu, T.; Feng, L.; Chen, Z.; Ogasawara, M.; Keating, M.J.; Kondo, S.; Huang, P. Metabolic alterations in highly tumorigenic glioblastoma cells: Preference for hypoxia and high dependency on glycolysis. J. Biol. Chem. 2011, 286, 32843–32853. [Google Scholar] [CrossRef]
- Song, K.; Kwon, H.; Han, C.; Zhang, J.; Dash, S.; Lim, K.; Wu, T. Active glycolytic metabolism in CD133(+) hepatocellular cancer stem cells: Regulation by MIR-122. Oncotarget 2015, 6, 40822–40835. [Google Scholar] [CrossRef]
- Feng, W.; Gentles, A.; Nair, R.V.; Huang, M.; Lin, Y.; Lee, C.Y.; Cai, S.; Scheeren, F.A.; Kuo, A.H.; Diehn, M. Targeting unique metabolic properties of breast tumor initiating cells. Stem Cells 2014, 32, 1734–1745. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Duan, Q.; Zhang, Z.; Li, H.; Wu, H.; Shen, Q.; Wang, C.; Yin, T. Up-regulation of glycolysis promotes the stemness and EMT phenotypes in gemcitabine-resistant pancreatic cancer cells. J. Cell Mol. Med. 2017, 21, 2055–2067. [Google Scholar] [CrossRef]
- Zhu, T.; Zheng, J.; Zhuo, W.; Pan, P.; Li, M.; Zhang, W.; Zhou, H.; Gao, Y.; Li, X.; Liu, Z. ETV4 promotes breast cancer cell stemness by activating glycolysis and CXCR4-mediated sonic Hedgehog signaling. Cell Death Discov. 2021, 7, 126. [Google Scholar] [CrossRef]
- Chen, C.; Bai, L.; Cao, F.; Wang, S.; He, H.; Song, M.; Chen, H.; Liu, Y.; Guo, J.; Si, Q.; et al. Targeting LIN28B reprograms tumor glucose metabolism and acidic microenvironment to suppress cancer stemness and metastasis. Oncogene 2019, 38, 4527–4539. [Google Scholar] [CrossRef]
- O’Neill, S.; Porter, R.K.; McNamee, N.; Martinez, V.G.; O’Driscoll, L. 2-Deoxy-D-Glucose inhibits aggressive triple-negative breast cancer cells by targeting glycolysis and the cancer stem cell phenotype. Sci. Rep. 2019, 9, 3788. [Google Scholar] [CrossRef]
- Chae, Y.C.; Kim, J.H. Cancer stem cell metabolism: Target for cancer therapy. BMB Rep. 2018, 51, 319–326. [Google Scholar] [CrossRef]
- Lee, K.M.; Giltnane, J.M.; Balko, J.M.; Schwarz, L.J.; Guerrero-Zotano, A.L.; Hutchinson, K.E.; Nixon, M.J.; Estrada, M.V.; Sanchez, V.; Sanders, M.E.; et al. MYC and MCL1 Cooperatively Promote Chemotherapy-Resistant Breast Cancer Stem Cells via Regulation of Mitochondrial Oxidative Phosphorylation. Cell Metab. 2017, 26, 633–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanford, N.N.; Sher, D.J.; Xu, X.H.; Ahn, C.; D’Amico, A.V.; Aizer, A.A.; Mahal, B.A. Alcohol Use Among Patients With Cancer and Survivors in the United States, 2000–2017. J. Natl. Compr. Cancer Netw. 2020, 18, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Holm, M.; Olsen, A.; Christensen, J.; Kroman, N.T.; Bidstrup, P.E.; Johansen, C.; Overvad, K.; Tjonneland, A. Pre-diagnostic alcohol consumption and breast cancer recurrence and mortality: Results from a prospective cohort with a wide range of variation in alcohol intake. Int. J. Cancer 2013, 132, 686–694. [Google Scholar] [CrossRef] [PubMed]
- Kwan, M.L.; Kushi, L.H.; Weltzien, E.; Tam, E.K.; Castillo, A.; Sweeney, C.; Caan, B.J. Alcohol Consumption and Breast Cancer Recurrence and Survival Among Women With Early-Stage Breast Cancer: The Life After Cancer Epidemiology Study. J. Clin. Oncol. 2010, 28, 4410–4416. [Google Scholar] [CrossRef]
- Li, Y.J.; Mao, Y.Y.; Zhang, Y.; Cai, S.F.; Chen, G.D.; Ding, Y.; Guo, J.; Chen, K.; Jin, M.J. Alcohol drinking and upper aerodigestive tract cancer mortality: A systematic review and meta-analysis. Oral Oncol. 2014, 50, 269–275. [Google Scholar] [CrossRef]
- Schwedhelm, C.; Boeing, H.; Hoffmann, G.; Aleksandrova, K.; Schwingshackl, L. Effect of diet on mortality and cancer recurrence among cancer survivors: A systematic review and meta-analysis of cohort studies. Nutr. Rev. 2016, 74, 737–748. [Google Scholar] [CrossRef]
- Lee, S.H.; Hong, H.S.; Liu, Z.X.; Kim, R.H.; Kang, M.K.; Park, N.H.; Shin, K.H. TNFalpha enhances cancer stem cell-like phenotype via Notch-Hes1 activation in oral squamous cell carcinoma cells. Biochem. Biophys. Res. Commun. 2012, 424, 58–64. [Google Scholar] [CrossRef]
- Lee, S.H.; Lee, C.R.; Rigas, N.K.; Kim, R.H.; Kang, M.K.; Park, N.H.; Shin, K.H. Human papillomavirus 16 (HPV16) enhances tumor growth and cancer stemness of HPV-negative oral/oropharyngeal squamous cell carcinoma cells via miR-181 regulation. Papillomavirus Res. 2015, 1, 116–125. [Google Scholar] [CrossRef]
- Clay, M.R.; Tabor, M.; Owen, J.H.; Carey, T.E.; Bradford, C.R.; Wolf, G.T.; Wicha, M.S.; Prince, M.E. Single-marker identification of head and neck squamous cell carcinoma cancer stem cells with aldehyde dehydrogenase. Head Neck 2010, 32, 1195–1201. [Google Scholar] [CrossRef]
- Prince, M.E.; Sivanandan, R.; Kaczorowski, A.; Wolf, G.T.; Kaplan, M.J.; Dalerba, P.; Weissman, I.L.; Clarke, M.F.; Ailles, L.E. Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proc. Natl. Acad. Sci. USA 2007, 104, 973–978. [Google Scholar] [CrossRef]
- Prince, M.E.P.; Zhou, L.; Moyer, J.S.; Tao, H.M.; Lu, L.; Owen, J.; Egenti, M.; Zheng, F.; Chang, A.E.; Xia, J.C.; et al. Evaluation of the immunogenicity of ALDH (high) human head and neck squamous cell carcinoma cancer stem cells in vitro. Oral Oncol. 2016, 59, 30–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, Y.; Zuo, X.; Wei, D. Concise Review: Emerging Role of CD44 in Cancer Stem Cells: A Promising Biomarker and Therapeutic Target. Stem Cells Transl. Med. 2015, 4, 1033–1043. [Google Scholar] [CrossRef] [PubMed]
- Aghajani, M.; Mansoori, B.; Mohammadi, A.; Asadzadeh, Z.; Baradaran, B. New emerging roles of CD133 in cancer stem cell: Signaling pathway and miRNA regulation. J. Cell Physiol. 2019, 234, 21642–21661. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.S.; He, R.Z.; Jiang, Y.H.; Liu, D.J.; Tao, L.Y.; Yang, M.W.; Lin, C.Y.; Shen, Y.; Fu, X.L.; Yang, J.Y.; et al. Transcription factor NFAT5 contributes to the glycolytic phenotype rewiring and pancreatic cancer progression via transcription of PGK1. Cell Death Dis. 2019, 10, 948. [Google Scholar] [CrossRef]
- Liu, Y.Y.; Liang, T.; Qiu, X.K.; Ye, X.X.; Li, Z.W.; Tian, B.Q.; Yan, D.L. Down-Regulation of Nfatc1 Suppresses Proliferation, Migration, Invasion, and Warburg Effect in Prostate Cancer Cells. Med. Sci. Monitor 2019, 25, 1572–1581. [Google Scholar] [CrossRef]
- Clipstone, N.A.; Crabtree, G.R. Identification of Calcineurin as a Key Signaling Enzyme in Lymphocyte-T Activation. Nature 1992, 357, 695–697. [Google Scholar] [CrossRef]
- Daniel, C.; Gerlach, K.; Väth, M.; Neurath, M.F.; Weigmann, B. Nuclear factor of activated T cells—A transcription factor family as critical regulator in lung and colon cancer. Int. J. Cancer 2014, 134, 1767–1775. [Google Scholar] [CrossRef]
- Ogawa, T.; Washio, J.; Takahashi, T.; Echigo, S.; Takahashi, N. Glucose and glutamine metabolism in oral squamous cell carcinoma: Insight from a quantitative metabolomic approach. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2014, 118, 218–225. [Google Scholar] [CrossRef]
- Lelevich, V.V. Effect of Ethanol and Metronidazole on Glycolysis in Some Parts of Rat-Brain. Vopr. Meditsinskoi Khimii 1987, 33, 139–142. [Google Scholar]
- Kang, H.; Choi, S.J.; Park, K.H.; Lee, C.K.; Moon, J.S. Impaired Glycolysis Promotes Alcohol Exposure-Induced Apoptosis in HEI-OC1 Cells via Inhibition of EGFR Signaling. Int. J. Mol. Sci. 2020, 21, 476. [Google Scholar] [CrossRef]
- Valkenburg, K.C.; Graveel, C.R.; Zylstra-Diegel, C.R.; Zhong, Z.; Williams, B.O. Wnt/beta-catenin Signaling in Normal and Cancer Stem Cells. Cancers 2011, 3, 2050–2079. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Wang, S.; Ren, Z.; Frank, J.A.; Yang, X.H.; Zhang, Z.; Ke, Z.J.; Shi, X.; Luo, J. Chronic ethanol exposure enhances the aggressiveness of breast cancer: The role of p38gamma. Oncotarget 2016, 7, 3489–3505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, M.; Ren, Z.H.; Wang, X.; Comer, A.; Frank, J.A.; Ke, Z.J.; Huang, Y.; Zhang, Z.; Shi, X.L.; Wang, S.Y.; et al. ErbB2 and p38 gamma MAPK mediate alcohol-induced increase in breast cancer stem cells and metastasis. Mol. Cancer 2016, 15, 52. [Google Scholar] [CrossRef]
- Machida, K.; Tsukamoto, H.; Mkrtchyan, H.; Duan, L.; Dynnyk, A.; Liu, H.M.; Asahina, K.; Govindarajan, S.; Raye, R.; Ou, J.J.; et al. Toll-like receptor 4 mediates synergism between alcohol and HCV in hepatic oncogenesis involving stem cell marker Nanog. Proc. Natl. Acad. Sci. USA 2009, 106, 1548–1553. [Google Scholar] [CrossRef] [PubMed]
- Ambade, A.; Satishchandran, A.; Szabo, G. Alcoholic hepatitis accelerates early hepatobiliary cancer by increasing stemness and miR-122-mediated HIF-1 alpha activation. Sci. Rep.-UK 2016, 6, 21340. [Google Scholar] [CrossRef]
- Liu, P.P.; Liao, J.; Tang, Z.J.; Wu, W.J.; Yang, J.; Zeng, Z.L.; Hu, Y.; Wang, P.; Ju, H.Q.; Xu, R.H.; et al. Metabolic regulation of cancer cell side population by glucose through activation of the Akt pathway. Cell Death Differ. 2014, 21, 124–135. [Google Scholar] [CrossRef]
- Xu, Q.; Zhang, Q.; Ishida, Y.; Hajjar, S.; Tang, X.; Shi, H.; Dang, C.V.; Le, A.D. EGF induces epithelial-mesenchymal transition and cancer stem-like cell properties in human oral cancer cells via promoting Warburg effect. Oncotarget 2017, 8, 9557–9571. [Google Scholar] [CrossRef]
- Zhao, Z.; Song, Z.J.; Liao, Z.J.; Liu, Z.G.; Sun, H.F.; Lei, B.X.; Chen, W.J.; Dang, C.X. PKM2 promotes stemness of breast cancer cell by through Wnt/beta-catenin pathway. Tumor Biol. 2016, 37, 4223–4234. [Google Scholar] [CrossRef]
- Yang, T.; Shu, X.; Zhang, H.W.; Sun, L.X.; Yu, L.; Liu, J.; Sun, L.C.; Yang, Z.H.; Ran, Y.L. Enolase 1 regulates stem cell-like properties in gastric cancer cells by stimulating glycolysis. Cell Death Dis. 2020, 11, 870. [Google Scholar] [CrossRef]
- Hong, H.S.; Akhavan, J.; Lee, S.H.; Kim, R.H.; Kang, M.K.; Park, N.H.; Shin, K.H. Proinflammatory cytokine TNFalpha promotes HPV-associated oral carcinogenesis by increasing cancer stemness. Int. J. Oral Sci. 2020, 12, 3. [Google Scholar] [CrossRef]
- Lang, T.Y.; Ding, X.J.; Kong, L.S.; Zhou, X.Y.; Zhang, Z.Q.; Ju, H.X.; Ding, S. NFATC2 is a novel therapeutic target for colorectal cancer stem cells. Oncotargets Ther. 2018, 11, 6911–6924. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Z.J.; Liu, J.; Wang, S.Q.; Zhu, Y.; Gao, X.Y.; Tin, V.P.; Qin, J.; Wang, J.W.; Wong, M.P. NFATc2 enhances tumor-initiating phenotypes through the NFATc2/SOX2/ALDH axis in lung adenocarcinoma. Elife 2017, 6, e26733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walczak-Drzewiecka, A.; Ratajewski, M.; Wagner, W.; Dastych, J. HIF-1 alpha is up-regulated in activated mast cells by a process that involves calcineurin and NFAT. J. Immunol. 2008, 181, 1665–1672. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Kieu, C.; Martin, C.E.; Han, J.; Chen, W.; Kim, J.S.; Kang, M.K.; Kim, R.H.; Park, N.H.; Kim, Y.; et al. NFATc3 plays an oncogenic role in oral/oropharyngeal squamous cell carcinomas by promoting cancer stemness via expression of OCT4. Oncotarget 2019, 10, 2306–2319. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.R.; Lee, S.H.; Rigas, N.K.; Kim, R.H.; Kang, M.K.; Park, N.H.; Shin, K.H. Elevated expression of JMJD6 is associated with oral carcinogenesis and maintains cancer stemness properties. Carcinogenesis 2016, 37, 119–128. [Google Scholar] [CrossRef]
- Shin, K.H.; Kim, R.H.; Kim, R.H.; Kang, M.K.; Park, N.H. hnRNP G elicits tumor-suppressive activity in part by upregulating the expression of Txnip. Biochem. Biophys. Res. Commun. 2008, 372, 880–885. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, A.; Kim, A.H.; Kang, M.K.; Park, N.-H.; Kim, R.H.; Kim, Y.; Shin, K.-H. Chronic Alcohol Exposure Promotes Cancer Stemness and Glycolysis in Oral/Oropharyngeal Squamous Cell Carcinoma Cell Lines by Activating NFAT Signaling. Int. J. Mol. Sci. 2022, 23, 9779. https://doi.org/10.3390/ijms23179779
Nguyen A, Kim AH, Kang MK, Park N-H, Kim RH, Kim Y, Shin K-H. Chronic Alcohol Exposure Promotes Cancer Stemness and Glycolysis in Oral/Oropharyngeal Squamous Cell Carcinoma Cell Lines by Activating NFAT Signaling. International Journal of Molecular Sciences. 2022; 23(17):9779. https://doi.org/10.3390/ijms23179779
Chicago/Turabian StyleNguyen, Anthony, Anna H. Kim, Mo K. Kang, No-Hee Park, Reuben H. Kim, Yong Kim, and Ki-Hyuk Shin. 2022. "Chronic Alcohol Exposure Promotes Cancer Stemness and Glycolysis in Oral/Oropharyngeal Squamous Cell Carcinoma Cell Lines by Activating NFAT Signaling" International Journal of Molecular Sciences 23, no. 17: 9779. https://doi.org/10.3390/ijms23179779