
Transcriptome and Metabolome Analyses Revealed the Response Mechanism of Sugar Beet to Salt Stress of Different Durations


Abstract
:1. Introduction
2. Results
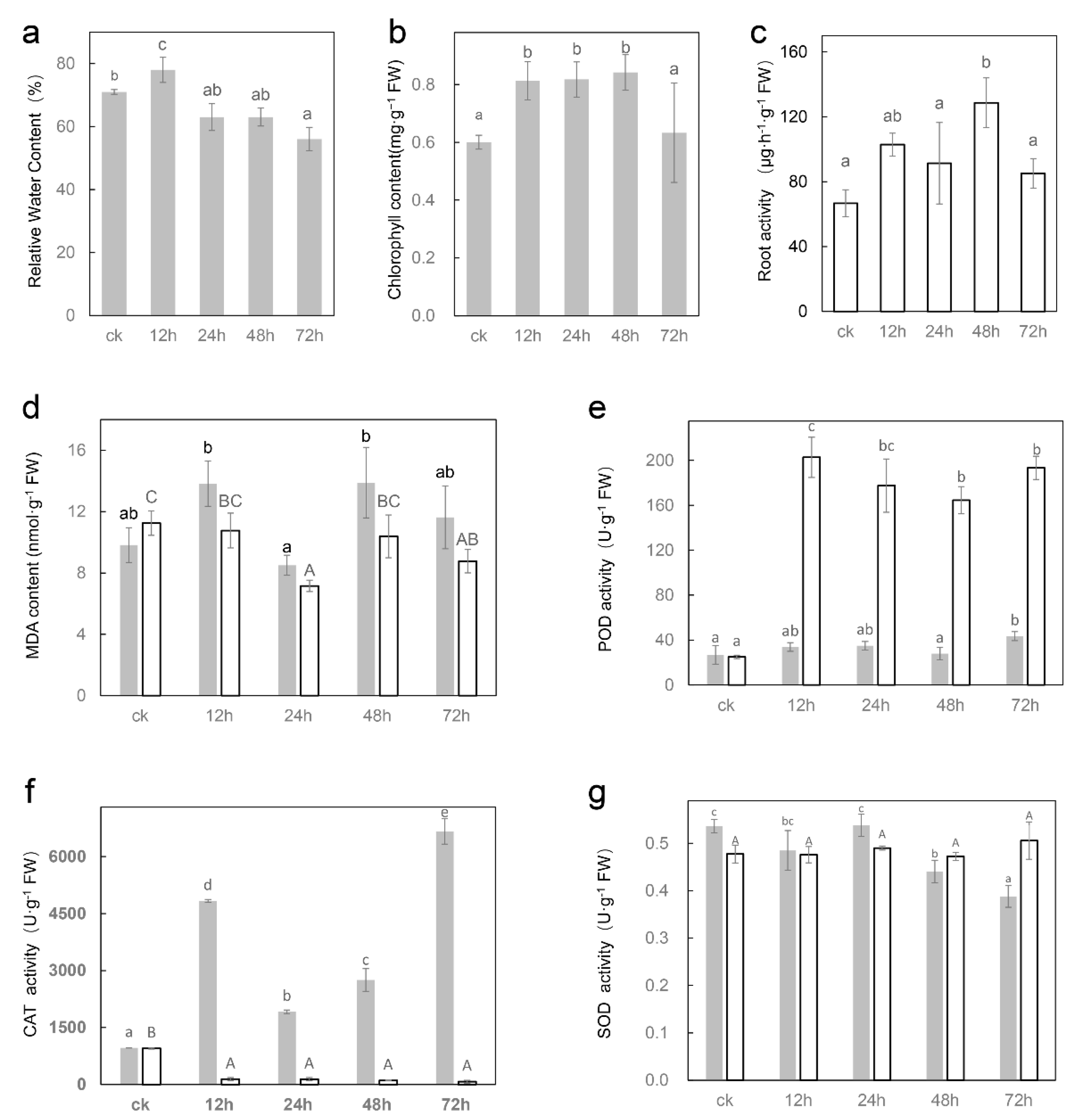
2.1. Salt Affects the Morphology and Physiological Indexes of Sugar Beet
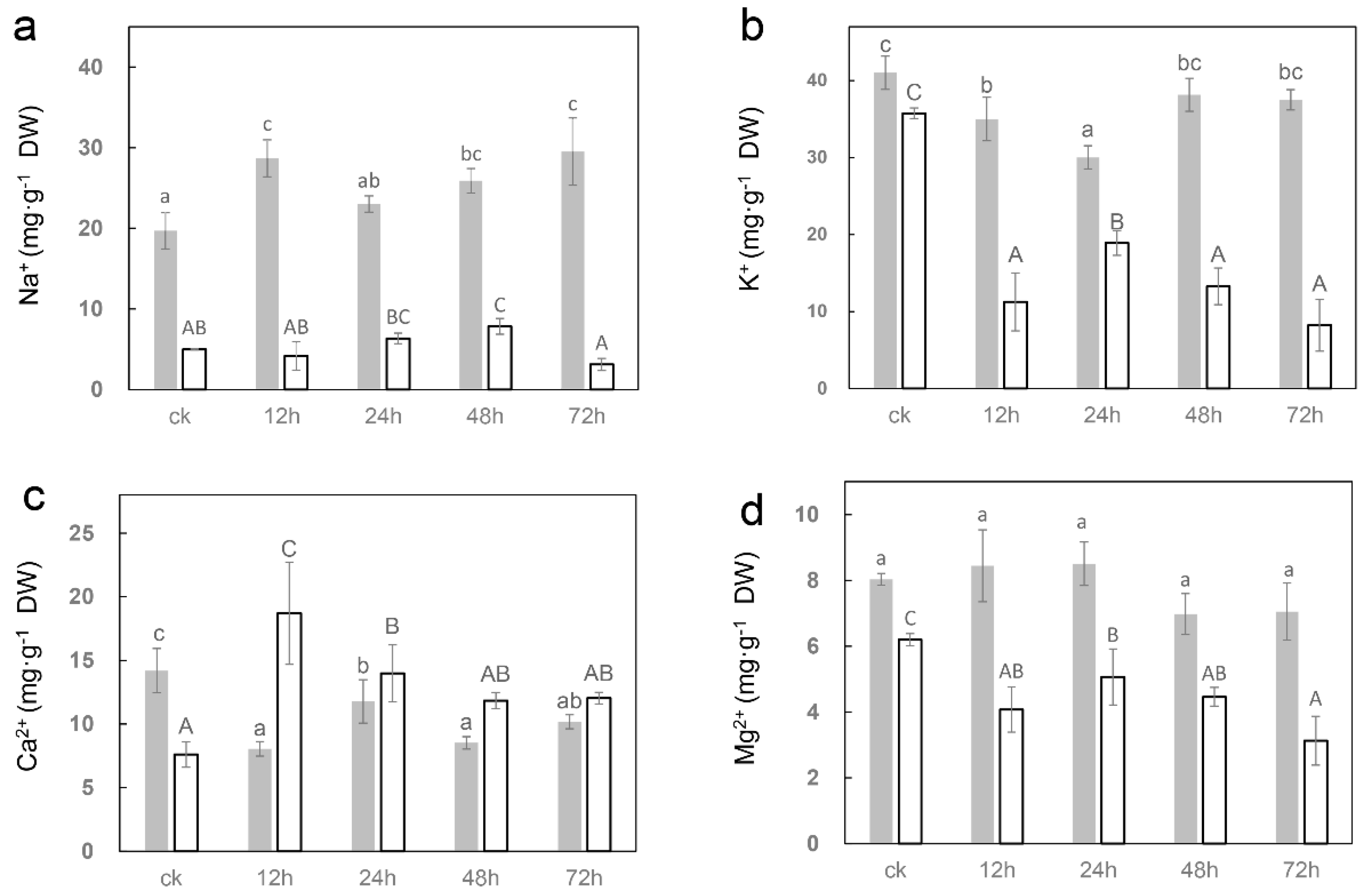
2.2. Salt Affects the Ion Content of Sugar Beet
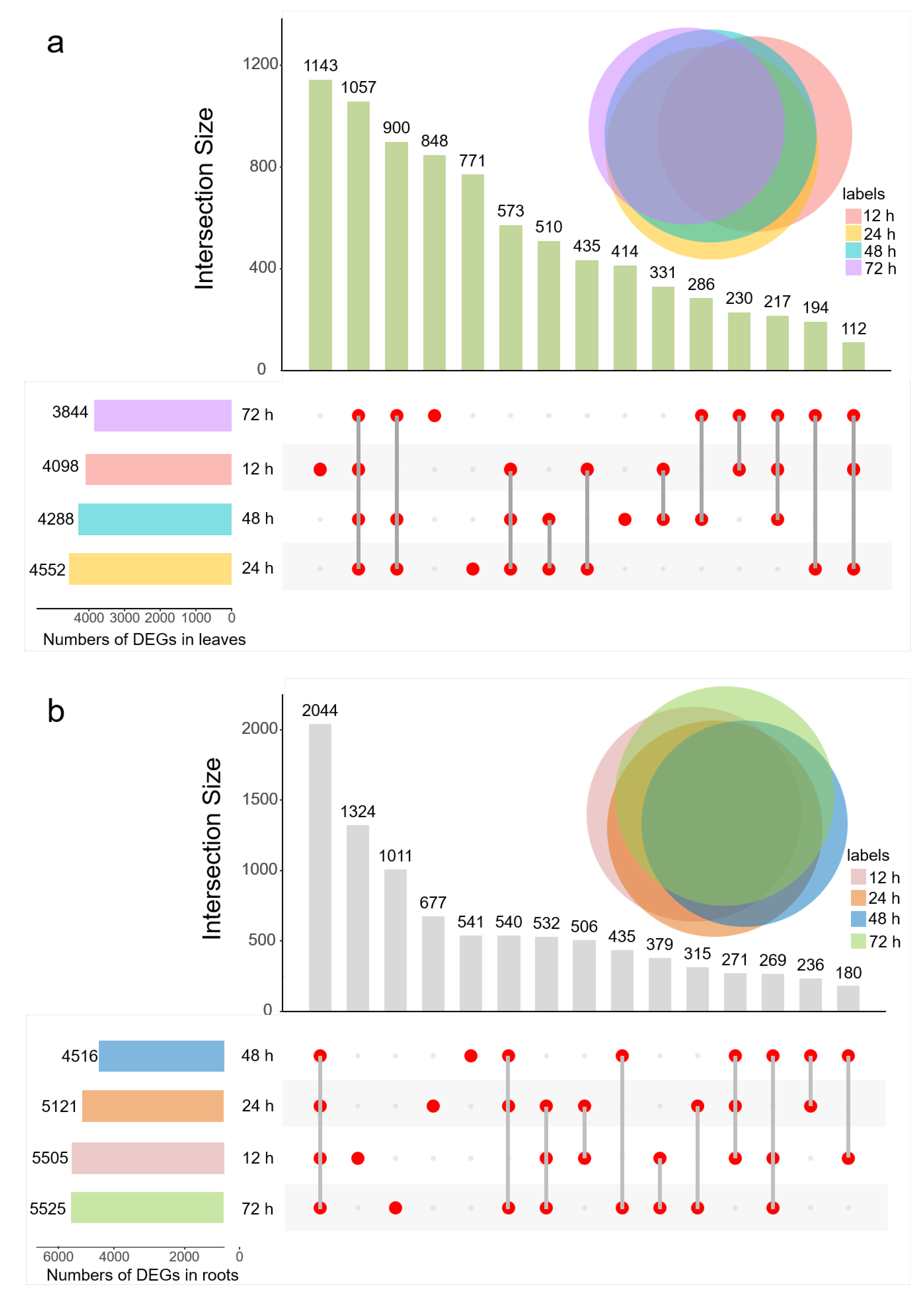
2.3. Salt Affects the Gene Expression of Sugar Beet
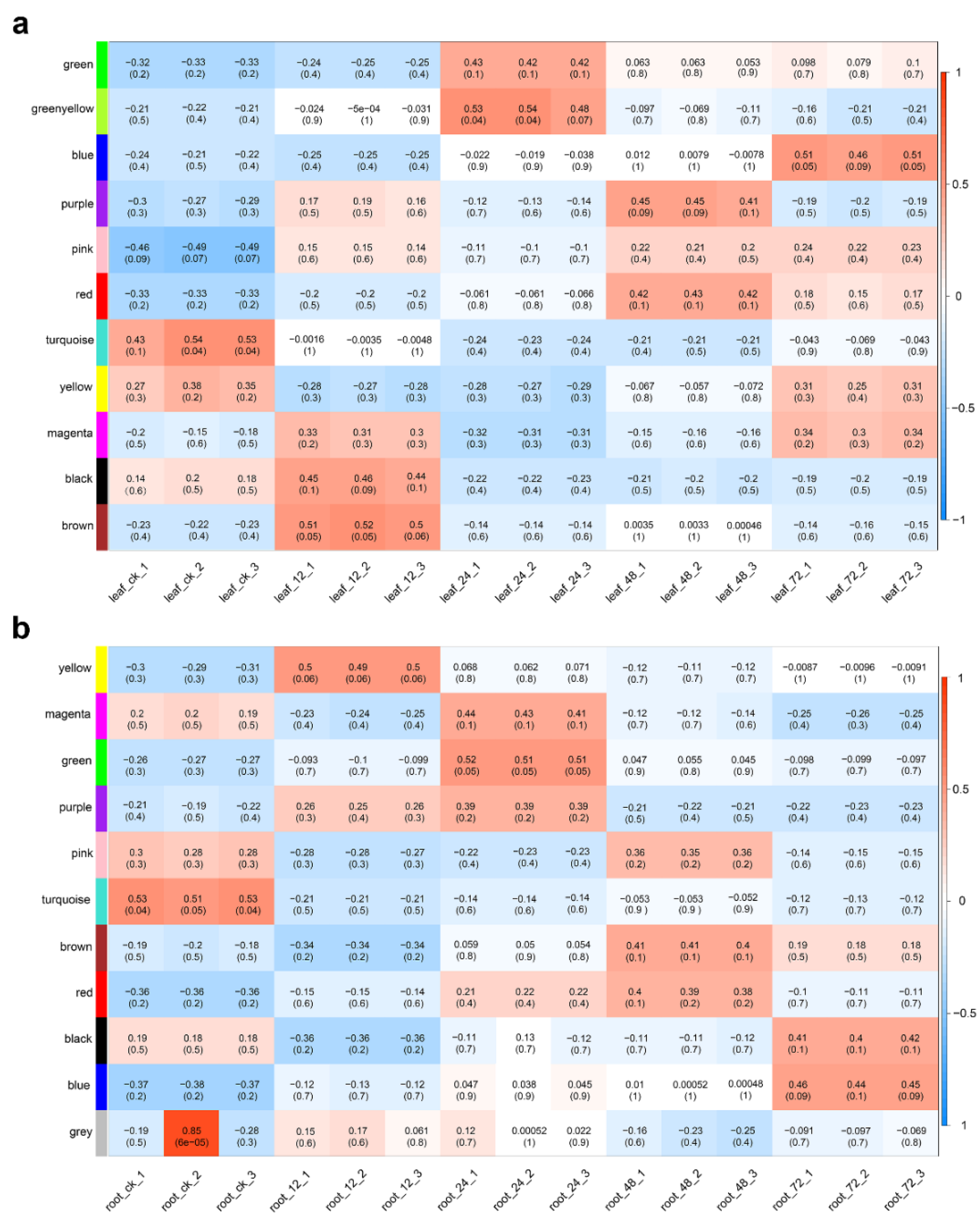
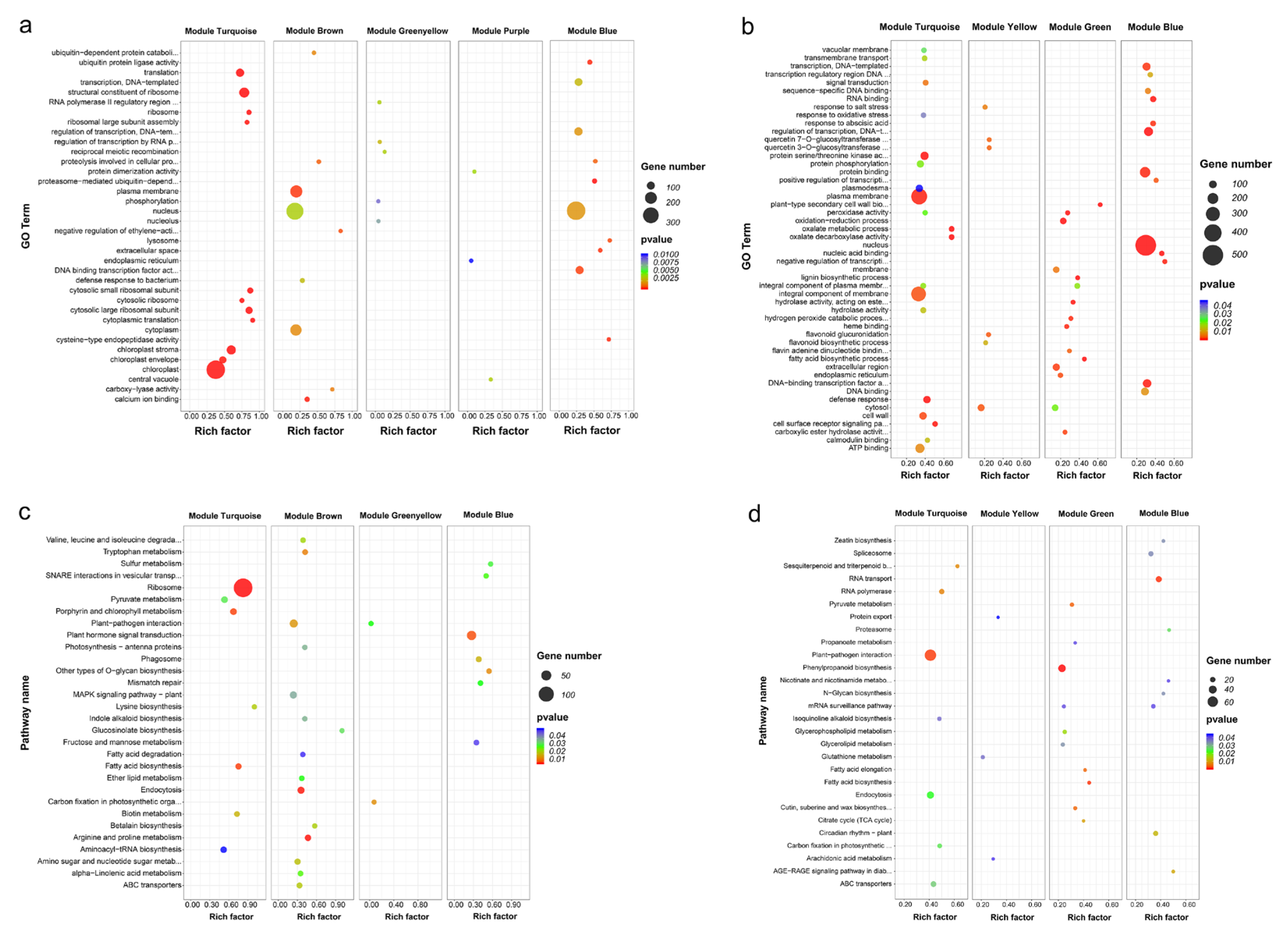
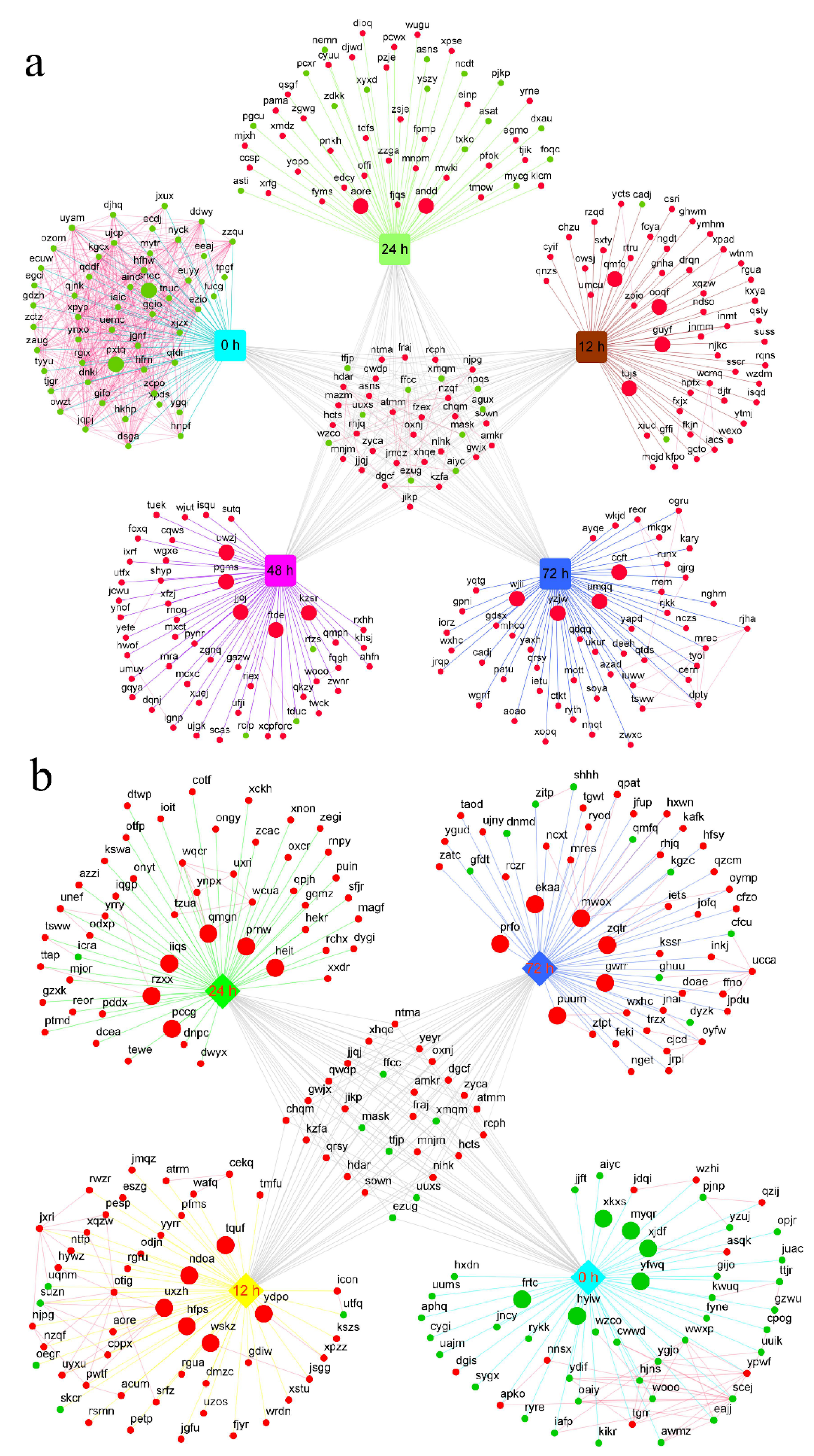
2.4. Co-Expression Network Analysis of Differentially Expressed Genes
2.5. Hub Gene Identification and Expression Pattern Analysis
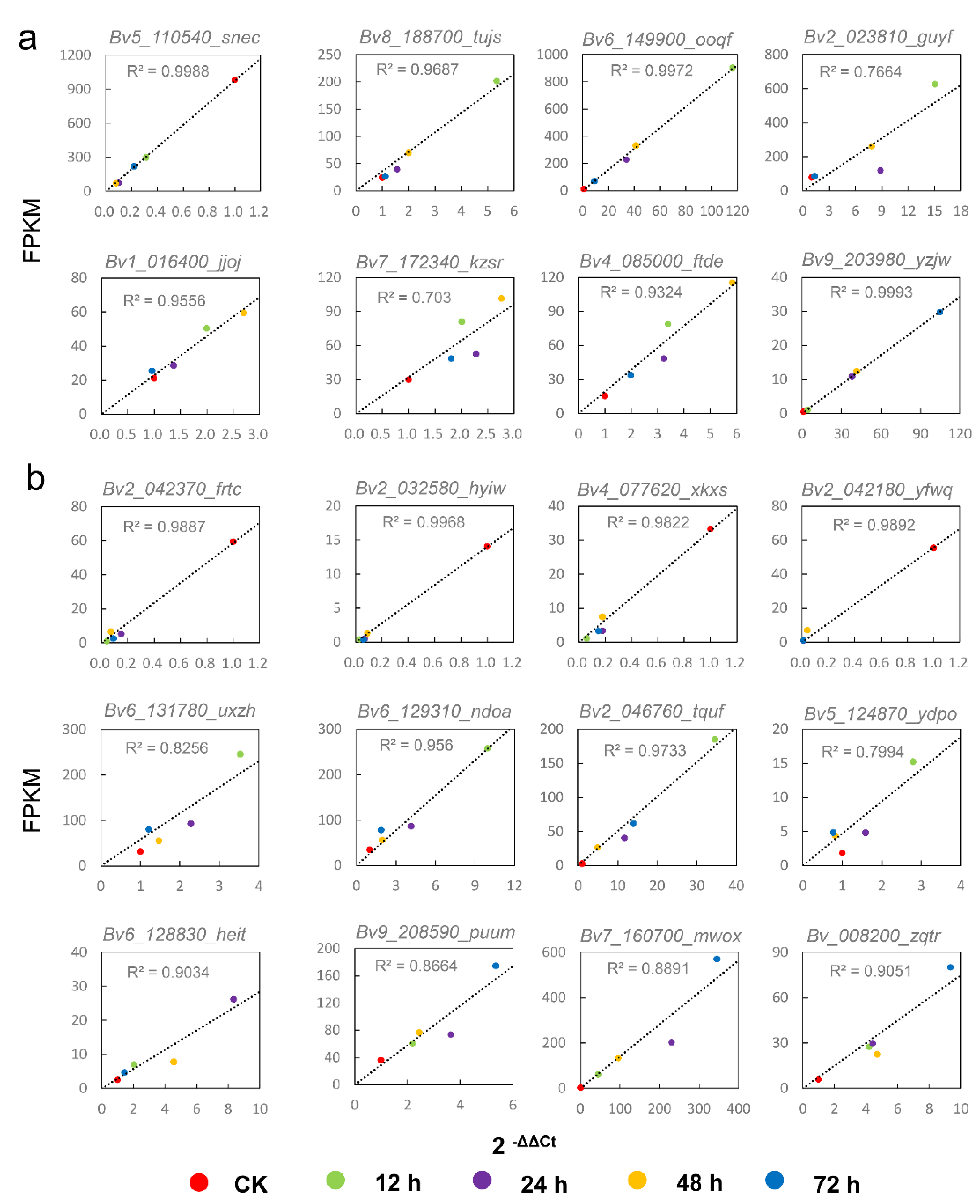
2.6. Validation of DEGs by qRT-PCR
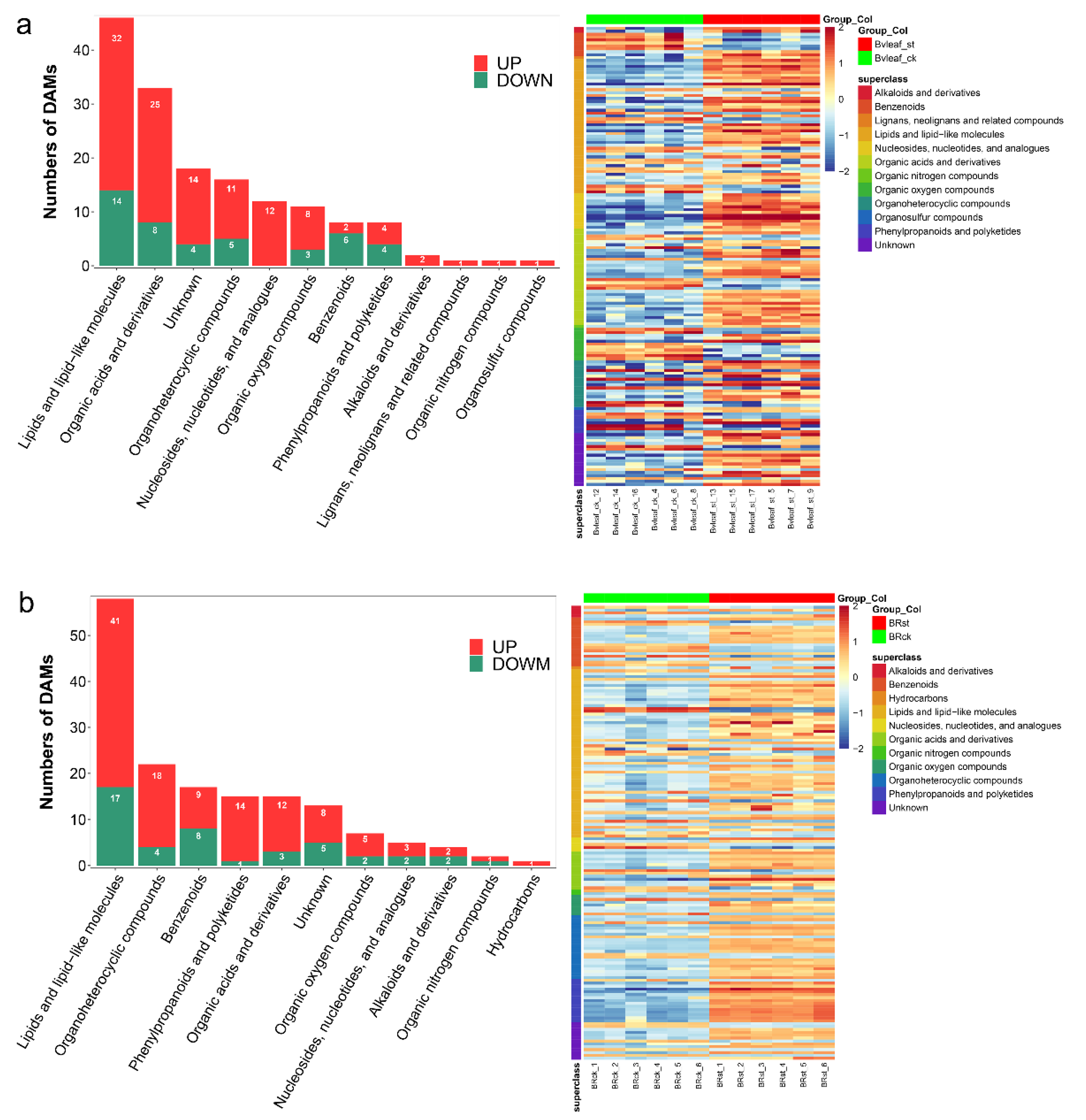
2.7. Salt Affects the Metabolomic of Sugar Beet
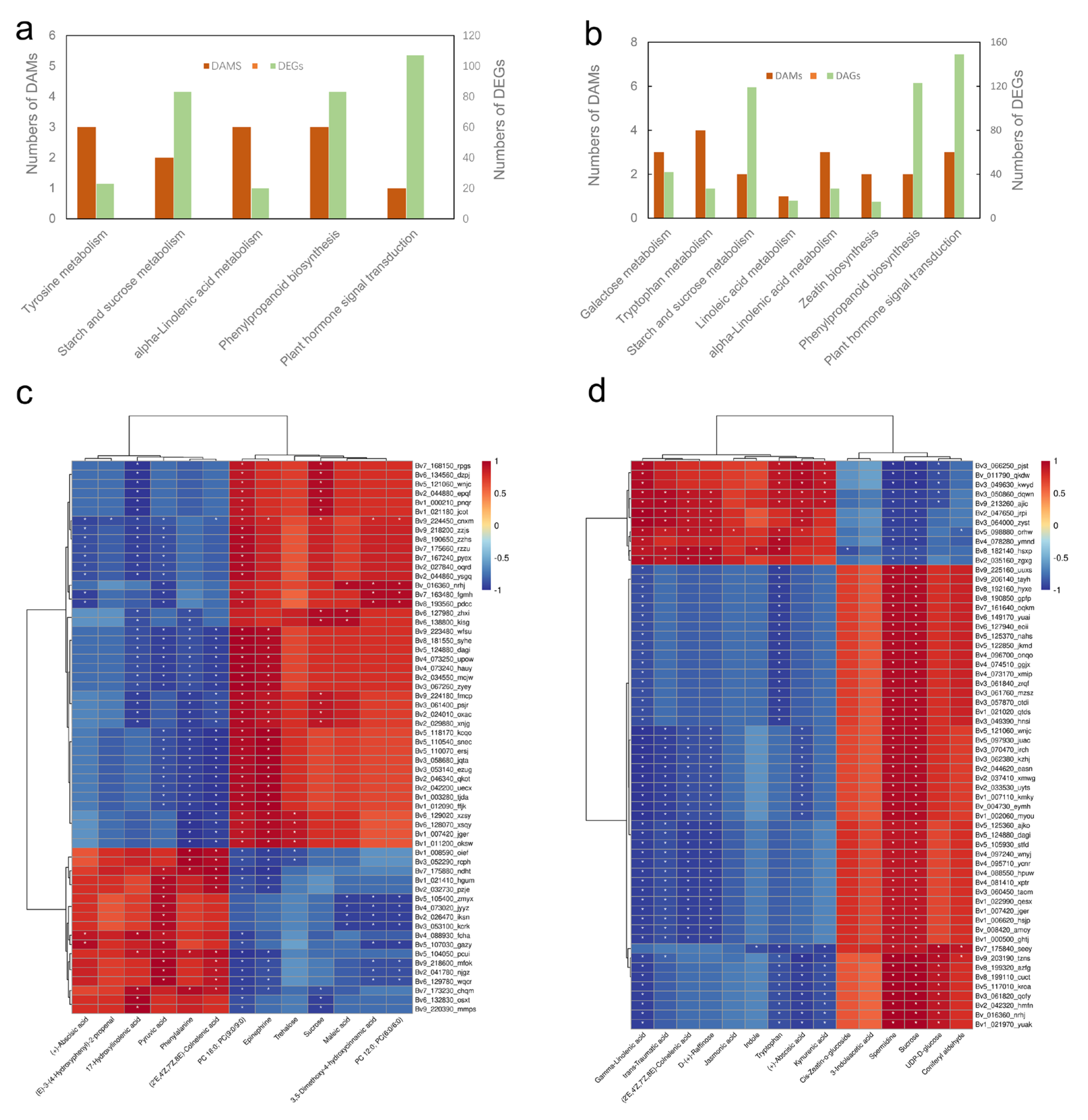
2.8. Profiles of DEGs and DAMs under Salt Stress in Sugar Beet
3. Discussion
4. Materials and Methods
4.1. Plant Growth and Treatment
4.2. Physiologic Indexes Detection and Ion Content Detection
4.3. RNA Extraction and Transcriptome Sequencing
4.4. Analysis of Sequencing Data
4.5. Construction of Gene Co-Expression Network Analysis
4.6. Establishment of Candidate Hub Genes and Construction of the Gene Networks
4.7. Real-Time Quantitative Reverse Transcription PCR (RT-qPCR) Analysis
4.8. Metabolite Analysis
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| DEG | Differentially expressed genes |
| DAM | Differentially accumulated metabolites |
| FPKM | Fragments per kilo-base per million reads |
| GO | Gene ontology |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| qRT-PCR | Quantitative real-time polymerase chain reaction |
| MDA | Malonaldehyde |
| POD | Peroxidase |
| SOD | Superoxide dismutase |
| CAT | Catalase |
| ABA | Abscisic acid |
| ROS | Reactive oxygen species |
References
- Rengasamy, P. Soil processes affecting crop production in salt-affected soils. Funct. Plant Biol. 2010, 37, 613–620. [Google Scholar] [CrossRef]
- Munns, R.; Gilliham, M. Salinity tolerance of crops—What is the cost? New Phytol. 2015, 208, 668–673. [Google Scholar] [CrossRef] [PubMed]
- Tester, M.; Davenport, R. Na+ Tolerance and Na+ Transport in Higher Plants. Ann. Bot. 2003, 91, 503–527. [Google Scholar] [CrossRef] [PubMed]
- Flowers, T.J.; Colmer, T.D. Plant salt tolerance: Adaptations in halophytes. Ann. Bot. 2015, 115, 327–331. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Wang, Q.; Yu, M.; Zhang, Y.; Wu, Y.; Zhang, H. Transgenic salt-tolerant sugar beet (Beta vulgaris L.) constitutively expressing anArabidopsis thalianavacuolar Na+/H+antiporter gene, AtNHX3, accumulates more soluble sugar but less salt in storage roots. Plant Cell Environ. 2008, 31, 1325–1334. [Google Scholar] [CrossRef] [PubMed]
- Skorupa, M.; Gołębiewski, M.; Kurnik, K.; Niedojadło, J.; Kęsy, J.; Klamkowski, K.; Wójcik, K.; Treder, W.; Tretyn, A.; Tyburski, J. Salt stress vs. salt shock - the case of sugar beet and its halophytic ancestor. BMC Plant Biol. 2019, 19, 57. [Google Scholar] [CrossRef] [PubMed]
- Khavari-Ne, R.; Najafi, F.; Khavari-Ne, S. Growth and Some Physiological Parameters of Four Sugar Beet (Beta vulgaris L.) Cultivars as Affected by Salinity. Pak. J. Biol. Sci. PJBS 2008, 11, 1390–1393. [Google Scholar] [CrossRef] [PubMed]
- Munns, R. Tester M. Mechanisms of Salinity Tolerance. Annu. Rev. Plant Biol. 2008, 59, 651–681. [Google Scholar] [CrossRef] [PubMed]
- Munns, R. Physiological processes limiting plant growth in saline soils: Some dogmas and hypotheses. Plant Cell Environ. 1993, 16, 15–24. [Google Scholar] [CrossRef]
- Garthwaite, A.J.; Von Bothmer, R.; Colmer, T.D. Salt tolerance in wild Hordeum species is associated with restricted entry of Na+ and Cl− into the shoots. J. Exp. Bot. 2005, 56, 2365–2378. [Google Scholar] [CrossRef]
- Rausell, A.; Kanhonou, R.; Yenush, L.; Serrano, R.; Ros, R. The translation initiation factor eIF1A is an important determinant in the tolerance to NaCl stress in yeast and plants. Plant J. 2003, 34, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Parida, A.K.; Das, A.B. Salt tolerance and salinity effects on plants: A review. Ecotoxicol. Environ. Saf. 2005, 60, 324–349. [Google Scholar] [CrossRef]
- Subbarao, G.V.; Wheeler, R.M.; Stutte, G.W.; Levine, L.H. How far can sodium substitute for potassium in red beet? J. Plant Nutr. 1999, 22, 1745–1761. [Google Scholar] [CrossRef] [PubMed]
- Subbarao, G.V.; Wheeler, R.M.; Stutte, G.W.; Levine, L.H. Low potassium enhances sodium uptake in red-beet under moderate saline conditions. J. Plant Nutr. 2000, 23, 1449–1470. [Google Scholar] [CrossRef]
- Pi, Z.; Stevanato, P.; Sun, F.; Yang, Y.; Sun, X.; Zhao, H.; Geng, G.; Yu, L. Proteomic changes induced by potassium deficiency and potassium substitution by sodium in sugar beet. J. Plant Res. 2016, 129, 527–538. [Google Scholar] [CrossRef] [PubMed]
- D’Onofrio, C.; Kader, A.; Lindberg, S. Uptake of sodium in quince, sugar beet, and wheat protoplasts determined by the fluorescent sodium-binding dye benzofuran isophthalate. J. Plant Physiol. 2005, 162, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Skorupa, M.; Goł˛ebiewski, M.; Domagalski, K.; Kurnika, K.; Nahia, K.A.; Złoch, M.; Tyburski, J. Transcriptomic profiling of the salt stress response in excised leaves of the halophyte Beta vulgaris ssp. maritima. Plant Sci. 2016, 243, 56–70. [Google Scholar] [CrossRef]
- Hossain, M.S.; Persicke, M.; ElSayed, A.I.; Kalinowski, J.; Dietz, K.-J. Metabolite profiling at the cellular and subcellular level reveals metabolites associated with salinity tolerance in sugar beet. J. Exp. Bot. 2017, 68, 5961–5976. [Google Scholar] [CrossRef]
- Lv, X.; Jin, Y.; Wang, Y. De novo transcriptome assembly and identification of salt-responsive genes in sugar beet M14. Comput. Biol. Chem. 2018, 75, 1–10. [Google Scholar] [CrossRef]
- Zhu, M.; Xie, H.; Wei, X.; Dossa, K.; Yu, Y.; Hui, S.; Tang, G.; Zeng, X.; Yu, Y.; Hu, P.; et al. WGCNA Analysis of Salt-Responsive Core Transcriptome Identifies Novel Hub Genes in Rice. Genes 2019, 10, 719. [Google Scholar] [CrossRef] [Green Version]
- Zhou, P.; Li, Q.; Liu, G.L.; Xu, N.; Yang, Y.J.; Wang, Y.; Zeng, W.L.; Wang, S.S.; Chen, A.G. Transcriptomic Analysis of Chilling-treated Tobacco (Nicotiana tabacum) Leaves Reveals Chilling-Induced Lignin Biosynthetic Pathways. Curr. Sci. 2019, 117, 1885–1892. [Google Scholar] [CrossRef]
- Zhang, X.; Huang, N.; Mo, L.; Lv, M.; Gao, Y.; Wang, J.; Liu, C.; Yin, S.; Zhou, J.; Xiao, N.; et al. Global Transcriptome and Co-Expression Network Analysis Reveal Contrasting Response of Japonica and Indica Rice Cultivar to γ Radiation. Int. J. Mol. Sci. 2019, 20, 4358. [Google Scholar] [CrossRef] [PubMed]
- Sahu, J.; Panda, D.; Baruah, G.; Patar, L.; Sen, P.; Borah, B.K.; Modi, M.K. Revealing shared differential co-expression profiles in rice infected by virus from reoviridae and sequiviridae group. Gene 2019, 698, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Amrine, K.C.H.; Blanco-Ulate, B.; Cantu, D. Discovery of Core Biotic Stress Responsive Genes in Arabidopsis by Weighted Gene Co-Expression Network Analysis. Plos ONE 2015, 10, e0118731. [Google Scholar]
- Ghatak, A.; Chaturvedi, P.; Weckwerth, W. Metabolomics in Plant Stress Physiology. Adv. Biochem. Eng. Biotechnol. 2018, 164, 187–236. [Google Scholar]
- Shi, S.; Cui, J.; Lu, Z.; Cheng, D.; Luo, C. Screening of tolerance to NaCl in sugar beet germplasms. China Beet Sugar 2008, 4, 7–9. [Google Scholar]
- Karlova, R.; Boer, D.; Hayes, S.; Testerink, C. Root plasticity under abiotic stress. Plant Physiol. 2021, 187, 1057–1070. [Google Scholar] [CrossRef]
- Liu, S.; Ye, T.; Li, Z.; Li, J.; Jamil, A.M.; Zhou, Y.; Hua, G.; Liang, A.; Deng, T.; Yang, L. Identifying Hub Genes for Heat Tolerance in Water Buffalo (Bubalus bubalis) Using Transcriptome Data. Front. Genet. 2019, 10, 209. [Google Scholar] [CrossRef]
- Julkowska, M.M.; Testerink, C. Tuning plant signaling and growth to survive salt. Trends Plant Sci. 2015, 20, 586–594. [Google Scholar] [CrossRef]
- Joseph, S.; Murphy, D.; Bhave, M. Glycine betaine biosynthesis in saltbushes (Atriplex spp.) under salinity stress. Biologia 2013, 68, 879–895. [Google Scholar] [CrossRef]
- Sharma, A.; Shahzad, B.; Rehman, A.; Bhardwaj, R.; Landi, M.; Zheng, B. Response of Phenylpropanoid Pathway and the Role of Polyphenols in Plants under Abiotic Stress. Molecules 2019, 24, 2452. [Google Scholar] [CrossRef] [PubMed]
- Pan, Z.; Zhao, Y.; Zheng, Y.; Liu, J.; Jiang, X.; Guo, Y. A High-Throughput Method for Screening Arabidopsis Mutants with Disordered Abiotic Stress-Induced Calcium Signal. J. Genet. Genom. 2012, 39, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Yuan, F.; Yang, H.; Xue, Y.; Kong, D.; Ye, R.; Li, C.; Zhang, J.; Theprungsirikul, L.; Shrift, T.; Krichilsky, B.; et al. OSCA1 mediates osmotic-stress-evoked Ca2+ increases vital for osmosensing in Arabidopsis. Nature 2014, 514, 367–371. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Ye, J.; Yang, Y.; Lin, H.; Yue, L.; Luo, J.; Long, Y.; Fu, H.; Liu, X.; Zhang, Y.; et al. The SOS2-SCaBP8 Complex Generates and Fine-Tunes an AtANN4-Dependent Calcium Signature under Salt Stress. Dev. Cell 2019, 48, 697–709.e695. [Google Scholar] [CrossRef]
- Osakabe, Y.; Yamaguchi-Shinozaki, K.; Shinozaki, K.; Tran, L.-S.P. Sensing the environment: Key roles of membrane-localized kinases in plant perception and response to abiotic stress. J. Exp. Bot. 2013, 64, 445–458. [Google Scholar] [CrossRef]
- Sun, X.-L.; Yu, Q.-Y.; Tang, L.-L.; Ji, W.; Bai, X.; Cai, H.; Liu, X.-F.; Ding, X.-D.; Zhu, Y.-M. GsSRK, a G-type lectin S-receptor-like serine/threonine protein kinase, is a positive regulator of plant tolerance to salt stress. J. Plant Physiol. 2013, 170, 505–515. [Google Scholar] [CrossRef]
- Dubiella, U.; Seybold, H.; Durian, G.; Komander, E.; Lassig, R.; Witte, C.-P.; Schulze, W.X.; Romeis, T. Calcium-dependent protein kinase/NADPH oxidase activation circuit is required for rapid defense signal propagation. Proc. Natl. Acad. Sci. USA 2013, 110, 8744–8749. [Google Scholar] [CrossRef]
- Huang, Y.; Jiao, Y.; Xie, N.; Guo, Y.; Zhang, F.; Xiang, Z.; Wang, R.; Wang, F.; Gao, Q.; Tian, L.; et al. OsNCED5, a 9-cis-epoxycarotenoid dioxygenase gene, regulates salt and water stress tolerance and leaf senescence in rice. Plant Sci. 2019, 287, 110188. [Google Scholar] [CrossRef]
- Ismail, A.; Takeda, S.; Nick, P. Life and death under salt stress: Same players, different timing? J. Exp. Bot. 2014, 65, 2963–2979. [Google Scholar] [CrossRef]
- Li, L.; Li, N.; Qi, X.; Bai, Y.; Chen, Q.; Fang, H.; Yu, X.; Liu, D.; Liang, C.; Zhou, Y. Characterization of the Glehnia littoralis Non-specific Phospholipase C Gene GlNPC3 and Its Involvement in the Salt Stress Response. Front. Plant Sci. 2021, 12, 769599. [Google Scholar] [CrossRef]
- Sagar, S.; Singh, A. Emerging role of phospholipase C mediated lipid signaling in abiotic stress tolerance and development in plants. Plant Cell Rep. 2021, 40, 2123–2133. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-C.; Gunawan, F.; Yunus, I.S.; Nakamura, Y. Arabidopsis Serine Decarboxylase 1 (SDC1) in Phospholipid and Amino Acid Metabolism. Front. Plant Sci. 2018, 9, 972. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Kanehara, K.; Nakamura, Y. Arabidopsis CHOLINE/ETHANOLAMINE KINASE 1 (CEK1) is a primary choline kinase localized at the endoplasmic reticulum (ER) and involved in ER stress tolerance. New Phytol. 2019, 223, 1904–1917. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Taylor, M.C.; Barrow, R.A.; Croyal, M.; Masle, J. Loss of Phosphoethanolamine N-Methyltransferases Abolishes Phosphatidylcholine Synthesis and Is Lethal. Plant Physiol. 2019, 179, 124–142. [Google Scholar] [CrossRef]
- Naeem, M.; Shahzad, K.; Saqib, S.; Shahzad, A.; Nasrullah; Younas, M.; Afridi, M.I. The Solanum melongena COP1LIKE manipulates fruit ripening and flowering time in tomato (Solanum lycopersicum). Plant Growth Regul. 2022, 96, 369–382. [Google Scholar] [CrossRef]
- Gao, J. Plant Physiology Experiment Guide; Higher Education Press: Beijing, China, 2006. [Google Scholar]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.-C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2009, 26, 139–140. [Google Scholar] [CrossRef]
- Conway, J.R.; Lex, A.; Gehlenborg, N. UpSetR: An R package for the visualization of intersecting sets and their properties. Bioinformatics 2017, 33, 2938–2940. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2007, 36, D480–D484. [Google Scholar] [CrossRef] [PubMed]
- Han, J.-D.J.; Bertin, N.; Hao, T.; Goldberg, D.S.; Berriz, G.F.; Zhang, L.V.; Dupuy, D.; Walhout, M.; Cusick, M.E.; Roth, F.; et al. Evidence for dynamically organized modularity in the yeast protein–protein interaction network. Nature 2004, 430, 88–93. [Google Scholar] [CrossRef]
- Mering, C.; Jensen, L.J.; Snel, B.; Hooper, S.D.; Krupp, M.; Foglierini, M.; Jouffre, N.; Huynen, M.A.; Bork, P. STRING: Known and predicted protein-protein associations, integrated and transferred across organisms. Nucleic Acids Res. 2005, 33, D433–D437. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Li, J.; Cui, J.; Dai, C.; Liu, T.; Cheng, D.; Luo, C. Whole-Transcriptome RNA Sequencing Reveals the Global Molecular Responses and CeRNA Regulatory Network of mRNAs, lncRNAs, miRNAs and circRNAs in Response to Salt Stress in Sugar Beet (Beta vulgaris). Int. J. Mol. Sci. 2020, 22, 289. [Google Scholar] [CrossRef]
- Li, Y.; Fang, J.; Qi, X.; Lin, M.; Zhong, Y.; Sun, L.; Cui, W. Combined Analysis of the Fruit Metabolome and Transcriptome Reveals Candidate Genes Involved in Flavonoid Biosynthesis in Actinidia arguta. Int. J. Mol. Sci. 2018, 19, 1471. [Google Scholar] [CrossRef]
- Smith, C.A.; Want, E.J.; O’Maille, G.; Abagyan, R.; Siuzdak, G. XCMS: Processing Mass Spectrometry Data for Metabolite Profiling Using Nonlinear Peak Alignment, Matching, and Identification. Anal. Chem. 2006, 78, 779–787. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Module | Gene ID | Annotation |
|---|---|---|
| Leaves | ||
| Turquoise | Bv2_029170_pxtq | cycloartenol-C-24-methyltransferase |
| Bv5_110540_snec | beta-glucosidase 13 | |
| Brown | Bv8_188700_tujs | miraculin |
| Bv6_149930_qmfq | shikimate kinase, chloroplastic | |
| Bv2_023810_guyf | DExH-box ATP-dependent RNA helicase DExH17 | |
| Bv6_149900_ooqf | GEM-like protein 5 | |
| Greenyellow | Bv4_074970_andd | thioredoxin F-type, chloroplastic |
| Bv4_095830_aore | probable glutathione S-transferase | |
| Purple | Bv1_016400_jjoj | chaperone protein dnaJ 11, chloroplastic |
| Bv4_086990_pgms | probable ubiquitin-conjugating enzyme E2 26 | |
| Bv_007910_uwzj | transcription factor bHLH35 | |
| Bv7_172340_kzsr | non-specific phospholipase C1 | |
| Bv4_085000_ftde | B-box zinc finger protein 22 | |
| Blue | Bv9_203980_yzjw | two-pore potassium channel 3 |
| Bv2_033120_umqq | vacuolar-processing enzyme | |
| Bv2_042060_ccft | probable choline kinase 2 | |
| Bv6_139660_wjii | homeobox-leucine zipper protein ATHB-40 | |
| Roots | ||
| Turquoise | Bv4_090510_myqr | L-type lectin-domain containing receptor kinase S.7 |
| Bv2_042370_frtc | LRR receptor-like serine/threonine-protein kinase | |
| Bv_016430_xjdf | heavy metal-associated isoprenylated plant protein 47 | |
| Bv2_032580_hyiw | ankyrin repeat-containing protein NPR4 | |
| Bv4_077620_xkxs | 7-deoxyloganetic acid glucosyltransferase-like | |
| Bv2_042180_yfwq | cytosolic sulfotransferase 5 | |
| Yellow | Bv1_009510_hfps | WD repeat-containing protein C2A9.03-like |
| Bv6_131780_uxzh | glutamate decarboxylase 4 | |
| Bv6_129310_ndoa | AAA-ATPase At3g50940 | |
| Bv3_067190_wskz | peroxisomal fatty acid beta-oxidation multifunctional protein MFP2 | |
| Bv2_046760_tquf | vacuolar amino acid transporter 1 | |
| Bv5_124870_ydpo | protein DETOXIFICATION 48 | |
| Green | Bv2_044780_rzxx | 3-ketoacyl-CoA synthase 10 |
| Bv1_016480_prnw | protein IN2-1 homolog B | |
| Bv4_083640_iiqs | slit homolog 3 protein | |
| Bv2_043550_pccg | ABC transporter G family member 32 | |
| Bv6_128830_heit | epoxide hydrolase 4 | |
| Bv3_066080_qmgn | peroxidase 66 | |
| Blue | Bv4_091130_ekaa | hypothetical protein BVRB_4g091130 |
| Bv6_135390_gwrr | ethylene-responsive transcription factor RAP2-4 | |
| Bv9_208590_puum | ethylene-responsive transcription factor ERF054 | |
| Bv7_160700_mwox | AF4/FMR2 family member 4 | |
| Bv6_133000_prfo | uncharacterized protein LOC104895442 | |
| Bv_008200_zqtr | calmodulin-like protein 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cui, J.; Li, J.; Dai, C.; Li, L. Transcriptome and Metabolome Analyses Revealed the Response Mechanism of Sugar Beet to Salt Stress of Different Durations. Int. J. Mol. Sci. 2022, 23, 9599. https://doi.org/10.3390/ijms23179599
Cui J, Li J, Dai C, Li L. Transcriptome and Metabolome Analyses Revealed the Response Mechanism of Sugar Beet to Salt Stress of Different Durations. International Journal of Molecular Sciences. 2022; 23(17):9599. https://doi.org/10.3390/ijms23179599
Chicago/Turabian StyleCui, Jie, Junliang Li, Cuihong Dai, and Liping Li. 2022. "Transcriptome and Metabolome Analyses Revealed the Response Mechanism of Sugar Beet to Salt Stress of Different Durations" International Journal of Molecular Sciences 23, no. 17: 9599. https://doi.org/10.3390/ijms23179599