
Structural Basis of Mutation-Dependent p53 Tetramerization Deficiency

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
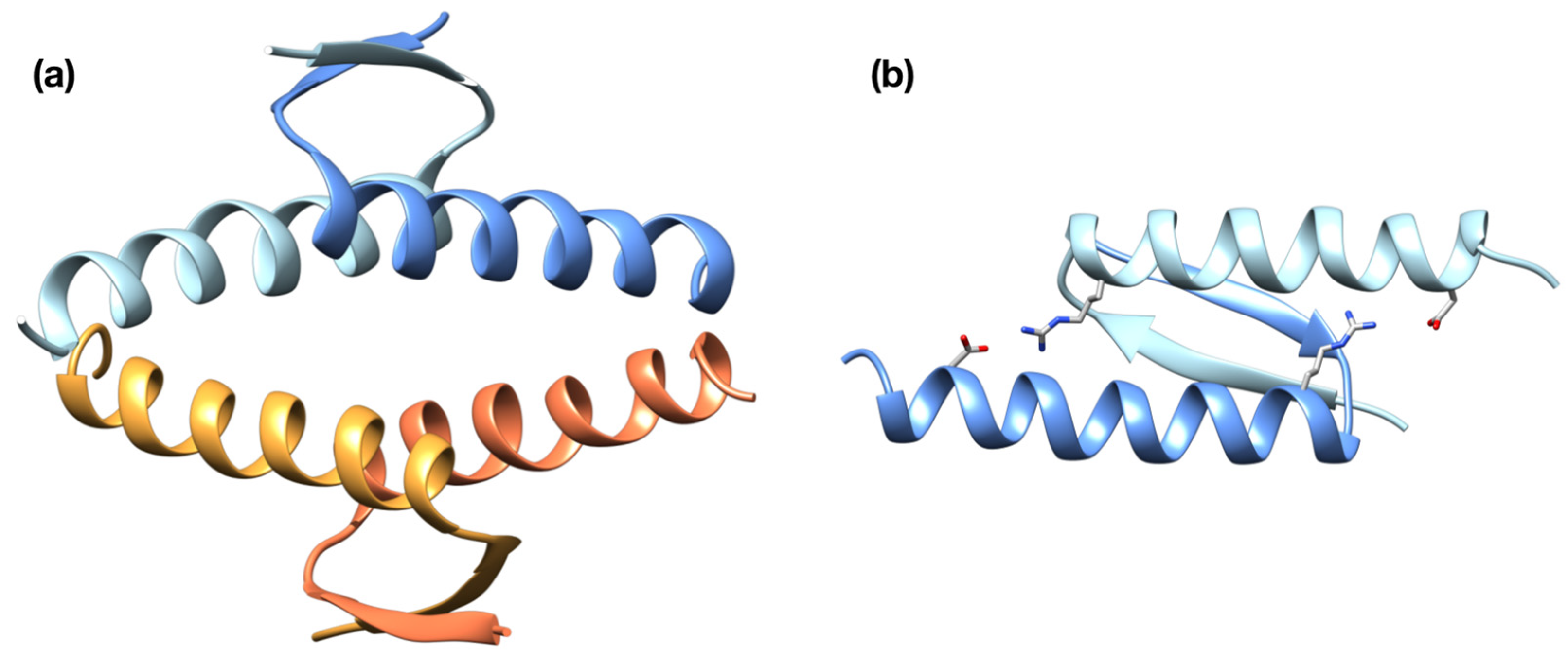
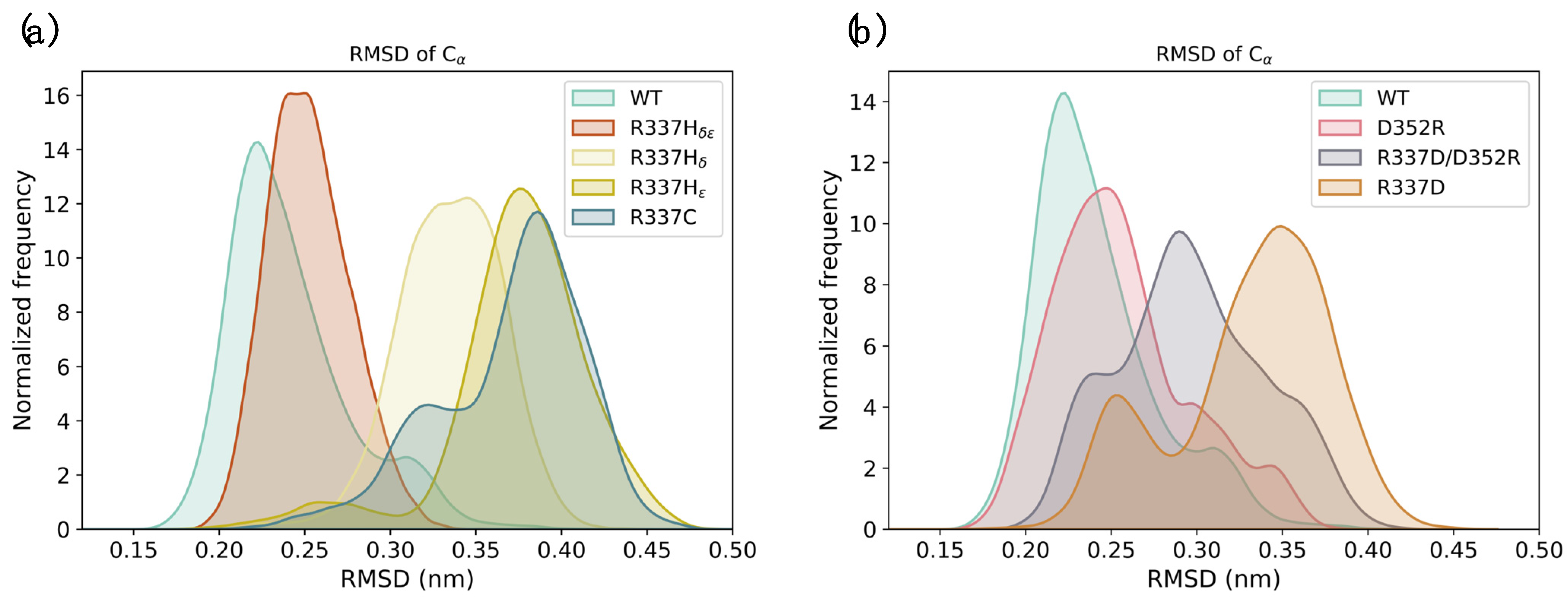
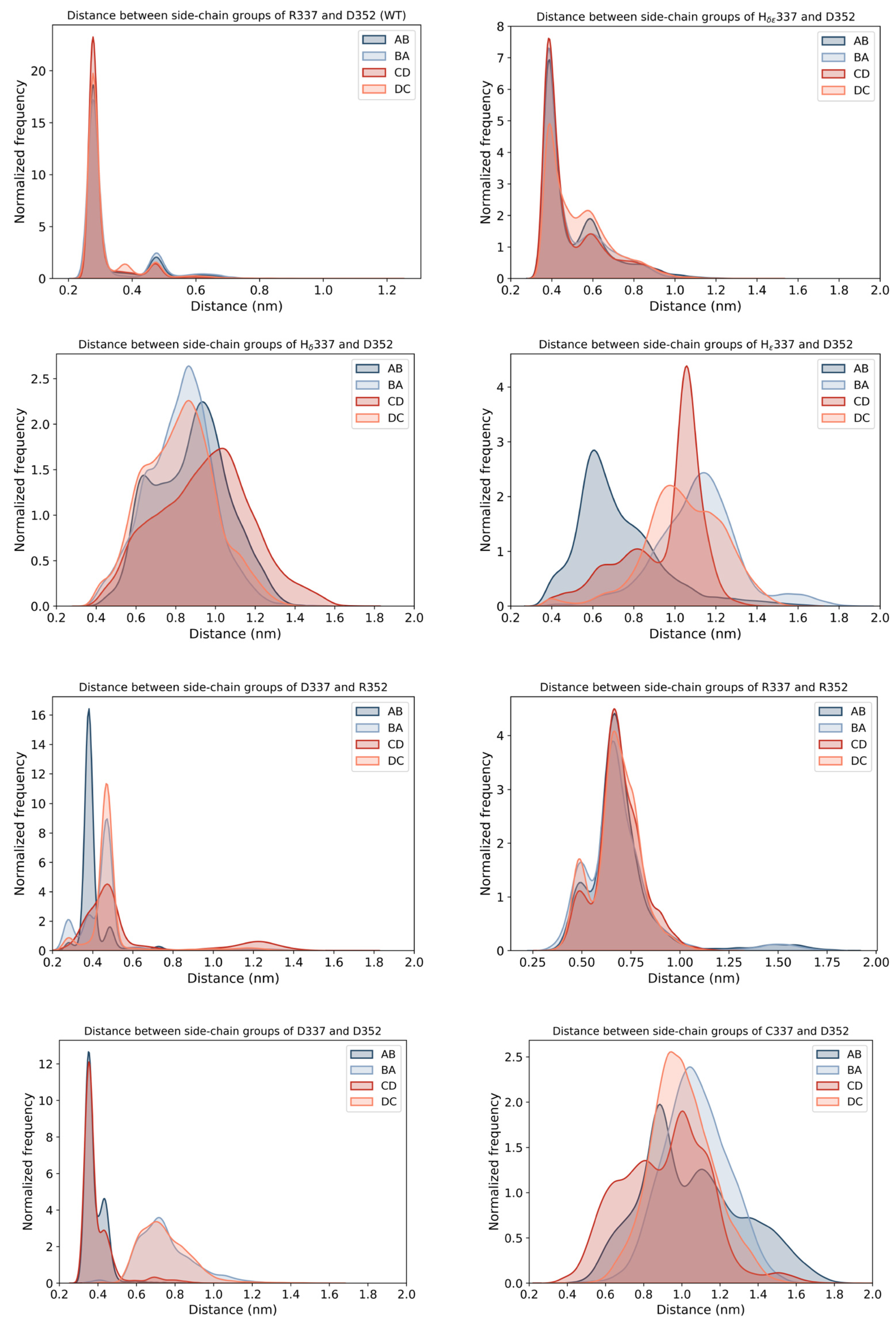
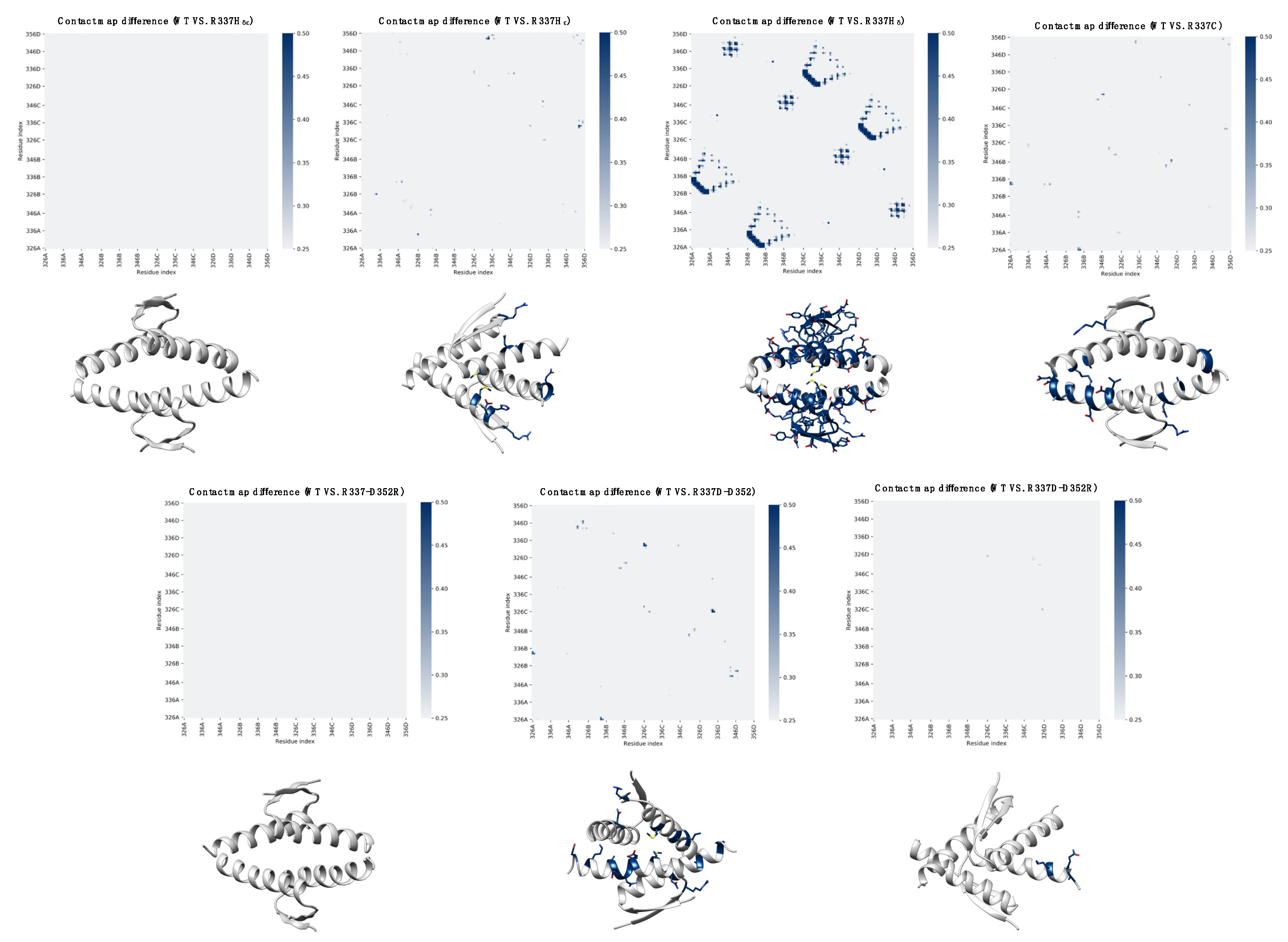
2. Results
2.1. Computational Prediction of the Effect of Naturally Occurring or Rationally Designed TP53 Mutation on the Stability of the p53 TET Domain
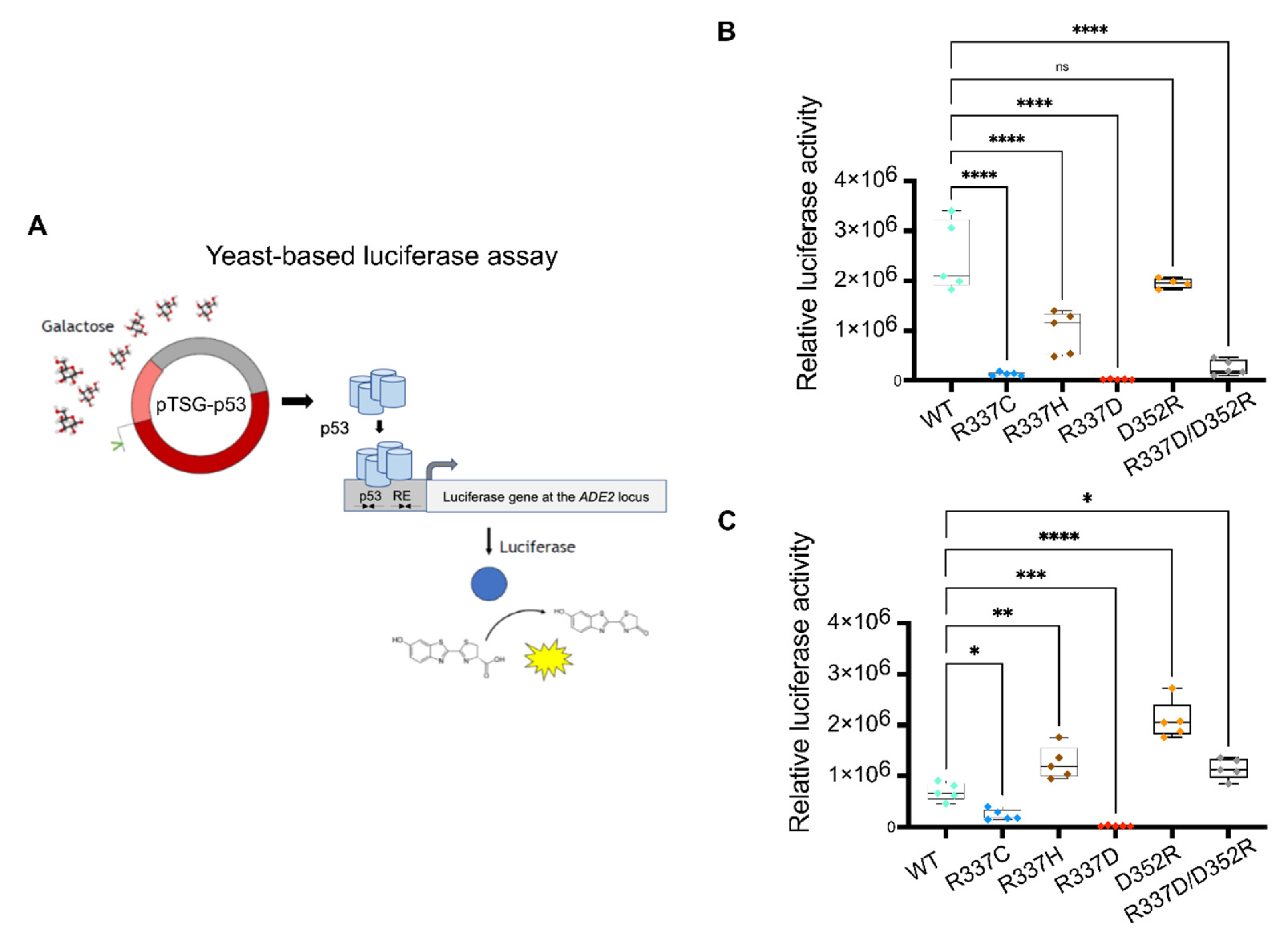
2.2. Experimental Analysis of Mutation-Dependent, p53-Driven Transcription
3. Discussion
4. Materials and Methods
4.1. Molecular Dynamics Simulations
4.2. Yeast Cultures
4.3. Luciferase Assay
4.4. Western Blotting
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kastenhuber, E.R.; Lowe, S.W. Putting P53 in Context. Cell 2017, 170, 1062–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sullivan, K.D.; Galbraith, M.D.; Andrysik, Z.; Espinosa, J.M. Mechanisms of Transcriptional Regulation by P53. Cell Death Differ. 2018, 25, 133–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrysik, Z.; Galbraith, M.D.; Guarnieri, A.L.; Zaccara, S.; Sullivan, K.D.; Pandey, A.; MacBeth, M.; Inga, A.; Espinosa, J.M. Identification of a Core TP53 Transcriptional Program with Highly Distributed Tumor Suppressive Activity. Genome Res. 2017, 27, 1645–1657. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H.; Prives, C. Blinded by the Light: The Growing Complexity of P53. Cell 2009, 137, 413–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; Kon, N.; Jiang, L.; Tan, M.; Ludwig, T.; Zhao, Y.; Baer, R.; Gu, W. Tumor Suppression in the Absence of P53-Mediated Cell-Cycle Arrest, Apoptosis, and Senescence. Cell 2012, 149, 1269–1283. [Google Scholar] [CrossRef] [Green Version]
- Kaiser, A.M.; Attardi, L.D. Deconstructing Networks of P53-Mediated Tumor Suppression in Vivo. Cell Death Differ. 2018, 25, 93–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizzotto, D.; Zaccara, S.; Rossi, A.; Galbraith, M.D.; Andrysik, Z.; Pandey, A.; Sullivan, K.D.; Quattrone, A.; Espinosa, J.M.; Dassi, E.; et al. Nutlin-Induced Apoptosis Is Specified by a Translation Program Regulated by PCBP2 and DHX30. Cell Rep. 2020, 30, 4355–4369. [Google Scholar] [CrossRef]
- McLure, K.G.; Lee, P.W. How P53 Binds DNA as a Tetramer. EMBO J. 1998, 17, 3342–3350. [Google Scholar] [CrossRef]
- Menendez, D.; Inga, A.; Resnick, M.A. The Expanding Universe of P53 Targets. Nat. Rev. Cancer 2009, 9, 724–737. [Google Scholar] [CrossRef]
- Kitayner, M.; Rozenberg, H.; Kessler, N.; Rabinovich, D.; Shaulov, L.; Haran, T.E.; Shakked, Z. Structural Basis of DNA Recognition by P53 Tetramers. Mol. Cell 2006, 22, 741–753. [Google Scholar] [CrossRef]
- Nicholls, C.D.; McLure, K.G.; Shields, M.A.; Lee, P.W. Biogenesis of P53 Involves Cotranslational Dimerization of Monomers and Posttranslational Dimerization of Dimers. Implications on the Dominant Negative Effect. J. Biol. Chem. 2002, 277, 12937–12945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vyas, P.; Beno, I.; Xi, Z.; Stein, Y.; Golovenko, D.; Kessler, N.; Rotter, V.; Shakked, Z.; Haran, T.E. Diverse P53/DNA Binding Modes Expand the Repertoire of P53 Response Elements. Proc. Natl. Acad. Sci. USA 2017, 114, 10624–10629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeffrey, P.D.; Gorina, S.; Pavletich, N.P. Crystal Structure of the Tetramerization Domain of the P53 Tumor Suppressor at 1.7 Angstroms. Science 1995, 267, 1498–1502. [Google Scholar] [CrossRef] [PubMed]
- Natan, E.; Baloglu, C.; Pagel, K.; Freund, S.M.; Morgner, N.; Robinson, C.V.; Fersht, A.R.; Joerger, A.C. Interaction of the P53 DNA-Binding Domain with Its n-Terminal Extension Modulates the Stability of the P53 Tetramer. J. Mol. Biol. 2011, 409, 358–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bista, M.; Freund, S.M.; Fersht, A.R. Domain-Domain Interactions in Full-Length P53 and a Specific DNA Complex Probed by Methyl NMR Spectroscopy. Proc. Natl. Acad. Sci. USA 2012, 109, 15752–15756. [Google Scholar] [CrossRef] [Green Version]
- D’Abramo, M.; Bešker, N.; Desideri, A.; Levine, A.J.; Melino, G.; Chillemi, G. The P53 Tetramer Shows an Induced-Fit Interaction of the C-Terminal Domain with the DNA-Binding Domain. Oncogene 2016, 35, 3272–3281. [Google Scholar] [CrossRef]
- Raj, N.; Attardi, L.D. The Transactivation Domains of the P53 Protein. Cold Spring Harb. Perspect. Med. 2017, 7, a026047. [Google Scholar] [CrossRef] [Green Version]
- Shin, J.S.; Ha, J.H.; Lee, D.H.; Ryu, K.S.; Bae, K.H.; Park, B.C.; Park, S.G.; Yi, G.S.; Chi, S.W. Structural Convergence of Unstructured P53 Family Transactivation Domains in Mdm2 Recognition. Cell Cycle 2015, 14, 533–543. [Google Scholar] [CrossRef] [Green Version]
- Ruan, H.; Yu, C.; Niu, X.; Zhang, W.; Liu, H.; Chen, L.; Xiong, R.; Sun, Q.; Jin, C.; Liu, Y.; et al. Computational Strategy for Intrinsically Disordered Protein Ligand Design Leads to the Discovery of P53 Transactivation Domain I Binding Compounds That Activate the P53 Pathway. Chem. Sci. 2021, 12, 3004–3016. [Google Scholar] [CrossRef]
- Krois, A.S.; Jane Dyson, H.; Wright, P.E. Long-Range Regulation of P53 DNA Binding by Its Intrinsically Disordered N-Terminal Transactivation Domain. Proc. Natl. Acad. Sci. USA 2018, 115, E11302–E11310. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Dyson, H.J.; Wright, P.E. A Phosphorylation-Dependent Switch in the Disordered P53 Transactivation Domain Regulates DNA Binding. Proc. Natl. Acad. Sci. USA 2020, 118, e2021456118. [Google Scholar] [CrossRef] [PubMed]
- Murray-Zmijewski, F.; Slee, E.A.; Lu, X. A Complex Barcode Underlies the Heterogeneous Response of P53 to Stress. Nat. Rev. Mol. Cell Biol. 2008, 9, 702–712. [Google Scholar] [CrossRef] [PubMed]
- Appella, E.; Anderson, C.W. Post-Translational Modifications and Activation of P53 by Genotoxic Stresses. Eur. J. Biochem. 2001, 268, 2764–2772. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.; Wang, D. Deciphering the PTM Codes of the Tumor Suppressor P53. J. Mol. Cell Biol. 2022, 13, 774–785. [Google Scholar] [CrossRef]
- Senitzki, A.; Safieh, J.; Sharma, V.; Golovenko, D.; Danin-Poleg, Y.; Inga, A.; Haran, T.E. The Complex Architecture of P53 Binding Sites. Nucleic Acids Res. 2021, 49, 1364–1382. [Google Scholar] [CrossRef]
- Farkas, M.; McMahon, S. Unlocking P53 Response Elements: DNA Shape Is the Key. Mol. Cell. Oncol. 2021, 8, 1905489. [Google Scholar] [CrossRef]
- Wang, B.; Xiao, Z.; Ren, E.C. Redefining the P53 Response Element. Proc. Natl. Acad. Sci. USA 2009, 106, 14373–14378. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.-A.T.; Grimm, S.A.; Bushel, P.R.; Li, J.; Li, Y.; Bennett, B.D.; Lavender, C.A.; Ward, J.M.; Fargo, D.C.; Anderson, C.W.; et al. Revealing a Human P53 Universe. Nucleic Acids Res. 2018, 46, 8153–8167. [Google Scholar] [CrossRef] [Green Version]
- Farkas, M.; Hashimoto, H.; Bi, Y.; Davuluri, R.V.; Resnick-Silverman, L.; Manfredi, J.J.; Debler, E.W.; McMahon, S.B. Distinct Mechanisms Control Genome Recognition by P53 at Its Target Genes Linked to Different Cell Fates. Nat. Commun. 2021, 12, 484. [Google Scholar] [CrossRef]
- Sammons, M.A.; Nguyen, T.A.T.; McDade, S.S.; Fischer, M. Tumor Suppressor P53: From Engaging DNA to Target Gene Regulation. Nucleic Acids Res. 2020, 48, 8848–8869. [Google Scholar] [CrossRef]
- Tebaldi, T.; Zaccara, S.; Alessandrini, F.; Bisio, A.; Ciribilli, Y.; Inga, A. Whole-Genome Cartography of P53 Response Elements Ranked on Transactivation Potential. BMC Genom. 2015, 16, 464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomso, D.J.; Inga, A.; Menendez, D.; Pittman, G.S.; Campbell, M.R.; Storici, F.; Bell, D.A.; Resnick, M.A. Functionally Distinct Polymorphic Sequences in the Human Genome That Are Targets for P53 Transactivation. Proc. Natl. Acad. Sci. USA 2005, 102, 6431–6436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bond, G.L.; Menin, C.; Bertorelle, R.; Alhorpuro, P.; Aaltonen, L.A.; Levine, A.J. MDM2 SNP309 Accelerates Colorectal Tumour Formation in Women. J. Med. Genet. 2006, 43, 950–952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeron-Medina, J.; Wang, X.; Repapi, E.; Campbell, M.R.; Su, D.; Castro-Giner, F.; Davies, B.; Peterse, E.F.; Sacilotto, N.; Walker, G.J.; et al. A Polymorphic P53 Response Element in KIT Ligand Influences Cancer Risk and Has Undergone Natural Selection. Cell 2013, 155, 410–422. [Google Scholar] [CrossRef] [Green Version]
- Kato, S.; Han, S.Y.; Liu, W.; Otsuka, K.; Shibata, H.; Kanamaru, R.; Ishioka, C. Understanding the Function-Structure and Function-Mutation Relationships of P53 Tumor Suppressor Protein by High-Resolution Missense Mutation Analysis. Proc. Natl. Acad. Sci. USA 2003, 100, 8424–8429. [Google Scholar] [CrossRef] [Green Version]
- Kawaguchi, T.; Kato, S.; Otsuka, K.; Watanabe, G.; Kumabe, T.; Tominaga, T.; Yoshimoto, T.; Ishioka, C. The Relationship among P53 Oligomer Formation, Structure and Transcriptional Activity Using a Comprehensive Missense Mutation Library. Oncogene 2005, 24, 6976–6981. [Google Scholar] [CrossRef] [Green Version]
- Fischer, M. Conservation and Divergence of the P53 Gene Regulatory Network between Mice and Humans. Oncogene 2019, 38, 4095–4109. [Google Scholar] [CrossRef] [Green Version]
- Joerger, A.C.; Wilcken, R.; Andreeva, A. Tracing the Evolution of the P53 Tetramerization Domain. Structure 2014, 22, 1301–1310. [Google Scholar] [CrossRef] [Green Version]
- Joerger, A.C.; Rajagopalan, S.; Natan, E.; Veprintsev, D.B.; Robinson, C.V.; Fersht, A.R. Structural Evolution of P53, P63, and P73: Implication for Heterotetramer Formation. Proc. Natl. Acad. Sci. USA 2009, 106, 17705–17710. [Google Scholar] [CrossRef] [Green Version]
- Chillemi, G.; Kehrloesser, S.; Bernassola, F.; Desideri, A.; Dötsch, V.; Levine, A.J.; Melino, G. Structural Evolution and Dynamics of the P53 Proteins. Cold Spring Harb. Perspect. Med. 2017, 7, a028308. [Google Scholar] [CrossRef] [Green Version]
- Mateu, M.G.; Fersht, A.R. Mutually Compensatory Mutations during Evolution of the Tetramerization Domain of Tumor Suppressor P53 Lead to Impaired Hetero-Oligomerization. Proc. Natl. Acad. Sci. USA 1999, 96, 3595–3599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belyi, V.A.; Ak, P.; Markert, E.; Wang, H.; Hu, W.; Puzio-Kuter, A.; Levine, A.J. The Origins and Evolution of the P53 Family of Genes. Cold Spring Harb. Perspect. Biol. 2010, 2, a001198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monti, P.; Bosco, B.; Gomes, S.; Saraiva, L.; Fronza, G.; Inga, A. Yeast as a Chassis for Developing Functional Assays to Study Human P53. J. Vis. Exp. 2019, 2019, e59071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, P.A.J.; Vousden, K.H. Mutant P53 in Cancer: New Functions and Therapeutic Opportunities. Cancer Cell 2014, 25, 304–317. [Google Scholar] [CrossRef] [Green Version]
- Malkin, D. Li-Fraumeni Syndrome. Genes Cancer 2001, 2, 474–484. [Google Scholar]
- Frebourg, T.; Bajalica Lagercrantz, S.; Oliveira, C.; Magenheim, R.; Evans, D.G.; Hoogerbrugge, N.; Ligtenberg, M.; Kets, M.; Oostenbrink, R.; Sijmons, R.; et al. Guidelines for the Li–Fraumeni and Heritable TP53-Related Cancer Syndromes. Eur. J. Hum. Genet. 2020, 28, 1379–1386. [Google Scholar] [CrossRef]
- Rana, H.Q.; Gelman, R.; LaDuca, H.; McFarland, R.; Dalton, E.; Thompson, J.; Speare, V.; Dolinsky, J.S.; Chao, E.C.; Garber, J.E. Differences in TP53 Mutation Carrier Phenotypes Emerge from Panel-Based Testing. J Natl. Cancer Inst. 2018, 110, 863–870. [Google Scholar] [CrossRef] [Green Version]
- Stein, Y.; Rotter, V.; Aloni-Grinstein, R. Gain-of-Function Mutant P53: All the Roads Lead to Tumorigenesis. Int. J. Mol. Sci. 2019, 20, 6197. [Google Scholar] [CrossRef] [Green Version]
- Bargonetti, J.; Prives, C. Gain-of-Function Mutant P53: History and Speculation. J. Mol. Cell Biol. 2019, 11, 605–609. [Google Scholar] [CrossRef]
- Gencel-Augusto, J.; Lozano, G. P53 Tetramerization: At the Center of the Dominant-Negative Effect of Mutant P53. Genes Dev. 2020, 34, 1128–1146. [Google Scholar] [CrossRef]
- Monti, P.; Perfumo, C.; Bisio, A.; Ciribilli, Y.; Menichini, P.; Russo, D.; Umbach, D.M.; Resnick, M.A.; Inga, A.; Fronza, G. Dominant-Negative Features of Mutant TP53 in Germline Carriers Have Limited Impact on Cancer Outcomes. Mol. Cancer Res. 2011, 9, 271–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamada, R.; Nomura, T.; Anderson, C.W.; Sakaguchi, K. Cancer-Associated P53 Tetramerization Domain Mutants: Quantitative Analysis Reveals a Low Threshold for Tumor Suppressor Inactivation. J. Biol. Chem. 2011, 286, 252–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathias, C.; Bortoletto, S.; Centa, A.; Komechen, H.; Lima, R.S.; Fonseca, A.S.; Sebastião, A.P.; Urban, C.A.; Soares, E.W.S.; Prando, C.; et al. Frequency of the TP53 R337H Variant in Sporadic Breast Cancer and Its Impact on Genomic Instability. Sci. Rep. 2020, 10, 16614. [Google Scholar] [CrossRef] [PubMed]
- Achatz, M.I.; Zambetti, G.P. The Inherited P53 Mutation in the Brazilian Population. Cold Spring Harb. Perspect. Med. 2016, 6, a026195. [Google Scholar] [CrossRef]
- Park, J.H.; Li, J.; Starost, M.F.; Liu, C.; Zhuang, J.; Chen, J.; Achatz, M.I.; Kang, J.G.; Wang, P.-Y.; Savage, S.A.; et al. Mouse Homolog of the Human TP53 R337H Mutation Reveals Its Role in Tumorigenesis. Cancer Res. 2018, 78, 5375–5383. [Google Scholar] [CrossRef] [Green Version]
- Pinto, E.M.; Billerbeck, A.E.C.; Villares, M.C.B.F.; Domenice, S.; Mendonça, B.B.; Latronico, A.C. Founder Effect for the Highly Prevalent R337H Mutation of Tumor Suppressor P53 in Brazilian Patients with Adrenocortical Tumors. Arq. Bras. Endocrinol. Metabol. 2004, 48, 647–650. [Google Scholar] [CrossRef] [Green Version]
- Pinto, E.M.; Zambetti, G.P. What 20 Years of Research Has Taught Us about the TP53 p.R337H Mutation. Cancer 2020, 126, 4678–4686. [Google Scholar] [CrossRef]
- Jansson, M.; Durant, S.T.; Cho, E.C.; Sheahan, S.; Edelmann, M.; Kessler, B.; la Thangue, N.B. Arginine Methylation Regulates the P53 Response. Nat. Cell Biol. 2008, 10, 1431–1439. [Google Scholar] [CrossRef]
- Khoo, K.H.; Verma, C.S.; Lane, D.P. Drugging the P53 Pathway: Understanding the Route to Clinical Efficacy. Nat. Rev. Drug Discov. 2014, 13, 217–236. [Google Scholar] [CrossRef]
- Brown, C.J.; Lain, S.; Verma, C.S.; Fersht, A.R.; Lane, D.P. Awakening Guardian Angels: Drugging the P53 Pathway. Nat. Rev. Cancer 2009, 9, 862–873. [Google Scholar] [CrossRef]
- Gomes, A.S.; Ramos, H.; Inga, A.; Sousa, E.; Saraiva, L. Structural and Drug Targeting Insights on Mutant P53. Cancers 2021, 13, 3344. [Google Scholar] [CrossRef] [PubMed]
- Bykov, V.J.; Issaeva, N.; Shilov, A.; Hultcrantz, M.; Pugacheva, E.; Chumakov, P.; Bergman, J.; Wiman, K.G.; Selivanova, G. Restoration of the Tumor Suppressor Function to Mutant P53 by a Low-Molecular-Weight Compound. Nat. Med. 2002, 8, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Mullard, A. P53 Programmes Plough On. Nat. Rev. Drug Discov. 2020, 19, 497–500. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wilcken, R.; Joerger, A.C.; Chuckowree, I.S.; Amin, J.; Spencer, J.; Fersht, A.R. Small Molecule Induced Reactivation of Mutant P53 in Cancer Cells. Nucleic Acids Res. 2013, 41, 6034–6044. [Google Scholar] [CrossRef] [PubMed]
- Bullock, A.N.; Henckel, J.; DeDecker, B.S.; Johnson, C.M.; Nikolova, P.V.; Proctor, M.R.; Lane, D.P.; Fersht, A.R. Thermodynamic Stability of Wild-Type and Mutant P53 Core Domain. Proc. Natl. Acad. Sci. USA 1997, 94, 14338–14342. [Google Scholar] [CrossRef] [Green Version]
- Brachmann, R.K.; Yu, K.; Eby, Y.; Pavletich, N.P.; Boeke, J.D. Genetic Selection of Intragenic Suppressor Mutations That Reverse the Effect of Common P53 Cancer Mutations. Embo. J. 1998, 17, 1847–1859. [Google Scholar] [CrossRef]
- Joerger, A.C.; Allen, M.D.; Fersht, A.R. Crystal Structure of a Superstable Mutant of Human P53 Core Domain. Insights into the Mechanism of Rescuing Oncogenic Mutations. J. Biol. Chem. 2004, 279, 1291–1296. [Google Scholar] [CrossRef] [Green Version]
- Gordo, S.; Martos, V.; Santos, E.; Menéndez, M.; Bo, C.; Giralt, E.; de Mendoza, J. Stability and Structural Recovery of the Tetramerization Domain of P53-R337H Mutant Induced by a Designed Templating Ligand. Proc. Natl. Acad. Sci. USA 2008, 105, 16426–16431. [Google Scholar] [CrossRef] [Green Version]
- Demir, Ö.; Barros, E.P.; Offutt, T.L.; Rosenfeld, M.; Amaro, R.E. An Integrated View of P53 Dynamics, Function, and Reactivation. Curr. Opin. Struct. Biol. 2021, 67, 187–194. [Google Scholar] [CrossRef]
- Lwin, T.Z.; Durant, J.J.; Bashford, D. A Fluid Salt-Bridging Cluster and the Stabilization of P53. J. Mol. Biol. 2007, 373, 1334–1347. [Google Scholar] [CrossRef] [Green Version]
- Blanden, A.R.; Yu, X.; Blayney, A.J.; Demas, C.; Ha, J.H.; Liu, Y.; Withers, T.; Carpizo, D.R.; Loh, S.N. Zinc Shapes the Folding Landscape of P53 and Establishes a Pathway for Reactivating Structurally Diverse Cancer Mutants. eLife 2020, 9, e61487. [Google Scholar] [CrossRef] [PubMed]
- Joerger, A.C.; Fersht, A.R. The Tumor Suppressor P53: From Structures to Drug Discovery. Cold Spring Harb. Perspect. Biol. 2010, 2, a000919. [Google Scholar] [CrossRef] [PubMed]
- Degtjarik, O.; Golovenko, D.; Diskin-Posner, Y.; Abrahmsén, L.; Rozenberg, H.; Shakked, Z. Structural Basis of Reactivation of Oncogenic P53 Mutants by a Small Molecule: Methylene Quinuclidinone (MQ). Nat. Commun. 2021, 12, 7057. [Google Scholar] [CrossRef] [PubMed]
- Golovenko, D.; Bräuning, B.; Vyas, P.; Haran, T.E.; Rozenberg, H.; Shakked, Z. New Insights into the Role of DNA Shape on Its Recognition by P53 Proteins. Structure 2018, 26, 1237–1250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitayner, M.; Rozenberg, H.; Rohs, R.; Suad, O.; Rabinovich, D.; Honig, B.; Shakked, Z. Diversity in DNA Recognition by P53 Revealed by Crystal Structures with Hoogsteen Base Pairs. Nat. Struct. Mol. Biol. 2010, 17, 423–429. [Google Scholar] [CrossRef] [PubMed]
- Wells, M.; Tidow, H.; Rutherford, T.J.; Markwick, P.; Jensen, M.R.; Mylonas, E.; Svergun, D.I.; Blackledge, M.; Fersht, A.R. Structure of Tumor Suppressor P53 and Its Intrinsically Disordered N-Terminal Transactivation Domain. Proc. Natl. Acad. Sci. USA 2008, 105, 5762–5767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canadillas, J.M.; Tidow, H.; Freund, S.M.; Rutherford, T.J.; Ang, H.C.; Fersht, A.R. Solution Structure of P53 Core Domain: Structural Basis for Its Instability. Proc. Natl. Acad. Sci. USA 2006, 103, 2109–2114. [Google Scholar] [CrossRef] [Green Version]
- Emamzadah, S.; Tropia, L.; Halazonetis, T.D. Crystal Structure of a Multidomain Human P53 Tetramer Bound to the Natural CDKN1A (P21) P53-Response Element. Mol. Cancer Res. 2011, 9, 1493–1499. [Google Scholar] [CrossRef] [Green Version]
- Wieczorek, A.M.; Waterman, J.L.; Waterman, M.J.; Halazonetis, T.D. Structure-Based Rescue of Common Tumor-Derived P53 Mutants. Nat. Med. 1996, 2, 1143–1146. [Google Scholar] [CrossRef]
- Cho, Y.; Gorina, S.; Jeffrey, P.D.; Pavletich, N.P. Crystal Structure of a P53 Tumor Suppressor-DNA Complex: Understanding Tumorigenic Mutations [See Comments]. Science 1994, 265, 346–355. [Google Scholar] [CrossRef]
- Tidow, H.; Melero, R.; Mylonas, E.; Freund, S.M.; Grossmann, J.G.; Carazo, J.M.; Svergun, D.I.; Valle, M.; Fersht, A.R. Quaternary Structures of Tumor Suppressor P53 and a Specific P53 DNA Complex. Proc. Natl. Acad. Sci. USA 2007, 104, 12324–12329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okorokov, A.L.; Sherman, M.B.; Plisson, C.; Grinkevich, V.; Sigmundsson, K.; Selivanova, G.; Milner, J.; Orlova, E.V. The Structure of P53 Tumour Suppressor Protein Reveals the Basis for Its Functional Plasticity. EMBO J. 2006, 25, 5191–5200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikolova, P.V.; Henckel, J.; Lane, D.P.; Fersht, A.R. Semirational Design of Active Tumor Suppressor P53 DNA Binding Domain with Enhanced Stability. Proc. Natl. Acad. Sci. USA 1998, 95, 14675–14680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baroni, T.E.; Wang, T.; Qian, H.; Dearth, L.R.; Truong, L.N.; Zeng, J.; Denes, A.E.; Chen, S.W.; Brachmann, R.K. A Global Suppressor Motif for P53 Cancer Mutants. Proc. Natl. Acad. Sci. USA 2004, 101, 4930–4935. [Google Scholar] [CrossRef] [Green Version]
- Inga, A.; Monti, P.; Fronza, G.; Darden, T.; Resnick, M.A. P53 Mutants Exhibiting Enhanced Transcriptional Activation and Altered Promoter Selectivity Are Revealed Using a Sensitive, Yeast-Based Functional Assay. Oncogene 2001, 20, 501–513. [Google Scholar] [CrossRef] [Green Version]
- Roth, J.; Lenz-Bauer, C.; Contente, A.; Löhr, K.; Koch, P.; Bernard, S.; Dobbelstein, M. Reactivation of Mutant P53 by a One-Hybrid Adaptor Protein. Cancer Res. 2003, 63, 3904–3908. [Google Scholar]
- Monti, P.; Menichini, P.; Speciale, A.; Cutrona, G.; Fais, F.; Taiana, E.; Neri, A.; Bomben, R.; Gentile, M.; Gattei, V.; et al. Heterogeneity of TP53 Mutations and P53 Protein Residual Function in Cancer: Does It Matter? Front. Oncol. 2020, 10, 593383. [Google Scholar] [CrossRef]
- Wiman, K.G. Pharmacological Reactivation of Mutant P53: From Protein Structure to the Cancer Patient. Oncogene 2010, 29, 4245–4252. [Google Scholar] [CrossRef] [Green Version]
- Terakawa, T.; Takada, S. P53 Dynamics upon Response Element Recognition Explored by Molecular Simulations. Sci. Rep. 2015, 5, 17107. [Google Scholar] [CrossRef] [Green Version]
- Barros, E.P.; Demir, Ö.; Soto, J.; Cocco, M.J.; Amaro, R.E. Markov State Models and NMR Uncover an Overlooked Allosteric Loop in P53. Chem. Sci. 2021, 12, 1891–1900. [Google Scholar] [CrossRef]
- Tan, Y.S.; Mhoumadi, Y.; Verma, C.S. Roles of Computational Modelling in Understanding P53 Structure, Biology, and Its Therapeutic Targeting. J. Mol. Cell Biol. 2019, 11, 306–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pradhan, M.R.; Siau, J.W.; Kannan, S.; Nguyen, M.N.; Ouaray, Z.; Kwoh, C.K.; Lane, D.P.; Ghadessy, F.; Verma, C.S. Simulations of Mutant P53 DNA Binding Domains Reveal a Novel Druggable Pocket. Nucleic Acids Res. 2019, 47, 1637–1652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demir; Ieong, P.U.; Amaro, R.E. Full-Length P53 Tetramer Bound to DNA and Its Quaternary Dynamics. Oncogene 2017, 36, 1451–1460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jorgensen, W.; Chandrasekhar, J.; Madura, J.; Impey, R.; Klein, M.L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical Sampling through Velocity Rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parrinello, M.; Rahman, A. Polymorphic Transitions in Single Crystals: A New Molecular Dynamics Method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A Linear Constraint Solver for Molecular Simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. Ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from Ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Bekker, H.; Berendsen, H.; Dijkstra, E.; Achterop, S.; van Drunen, R.; van der Spoel, D.; Sijbers, A.; Keegstra, H.; Reitsma, B.; Renardus, M. Gromacs: A Parallel Computer for Molecular Dynamics Simulations. Phys. Comput. 1993, 92. [Google Scholar]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindah, E. Gromacs: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Bonomi, M.; Branduardi, D.; Gervasio, F.L.; Parrinello, M. The Unfolded Ensemble and Folding Mechanism of the C-Terminal GB1 β-Hairpin. J. Am. Chem. Soc. 2008, 130, 13938–13944. [Google Scholar] [CrossRef]
- Storici, F.; Resnick, M.A. The Delitto Perfetto Approach to in Vivo Site-Directed Mutagenesis and Chromosome Rearrangements with Synthetic Oligonucleotides in Yeast. Methods Enzymol. 2006, 409, 329–345. [Google Scholar]
- Inga, A.; Storici, F.; Darden, T.A.; Resnick, M.A. Differential Transactivation by the P53 Transcription Factor Is Highly Dependent on P53 Level and Promoter Target Sequence. Mol. Cell. Biol. 2002, 22, 8612–8625. [Google Scholar] [CrossRef] [Green Version]
- Andreotti, V.; Ciribilli, Y.; Monti, P.; Bisio, A.; Lion, M.; Jordan, J.; Fronza, G.; Menichini, P.; Resnick, M.A.; Inga, A. P53 Transactivation and the Impact of Mutations, Cofactors and Small Molecules Using a Simplified Yeast-Based Screening System. PLoS ONE 2011, 6, e20643. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rigoli, M.; Spagnolli, G.; Lorengo, G.; Monti, P.; Potestio, R.; Biasini, E.; Inga, A. Structural Basis of Mutation-Dependent p53 Tetramerization Deficiency. Int. J. Mol. Sci. 2022, 23, 7960. https://doi.org/10.3390/ijms23147960
Rigoli M, Spagnolli G, Lorengo G, Monti P, Potestio R, Biasini E, Inga A. Structural Basis of Mutation-Dependent p53 Tetramerization Deficiency. International Journal of Molecular Sciences. 2022; 23(14):7960. https://doi.org/10.3390/ijms23147960
Chicago/Turabian StyleRigoli, Marta, Giovanni Spagnolli, Giulia Lorengo, Paola Monti, Raffaello Potestio, Emiliano Biasini, and Alberto Inga. 2022. "Structural Basis of Mutation-Dependent p53 Tetramerization Deficiency" International Journal of Molecular Sciences 23, no. 14: 7960. https://doi.org/10.3390/ijms23147960