
Efficient Identification of the MYC Regulator with the Use of the CRISPR Library and Context-Matched Database Screenings

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
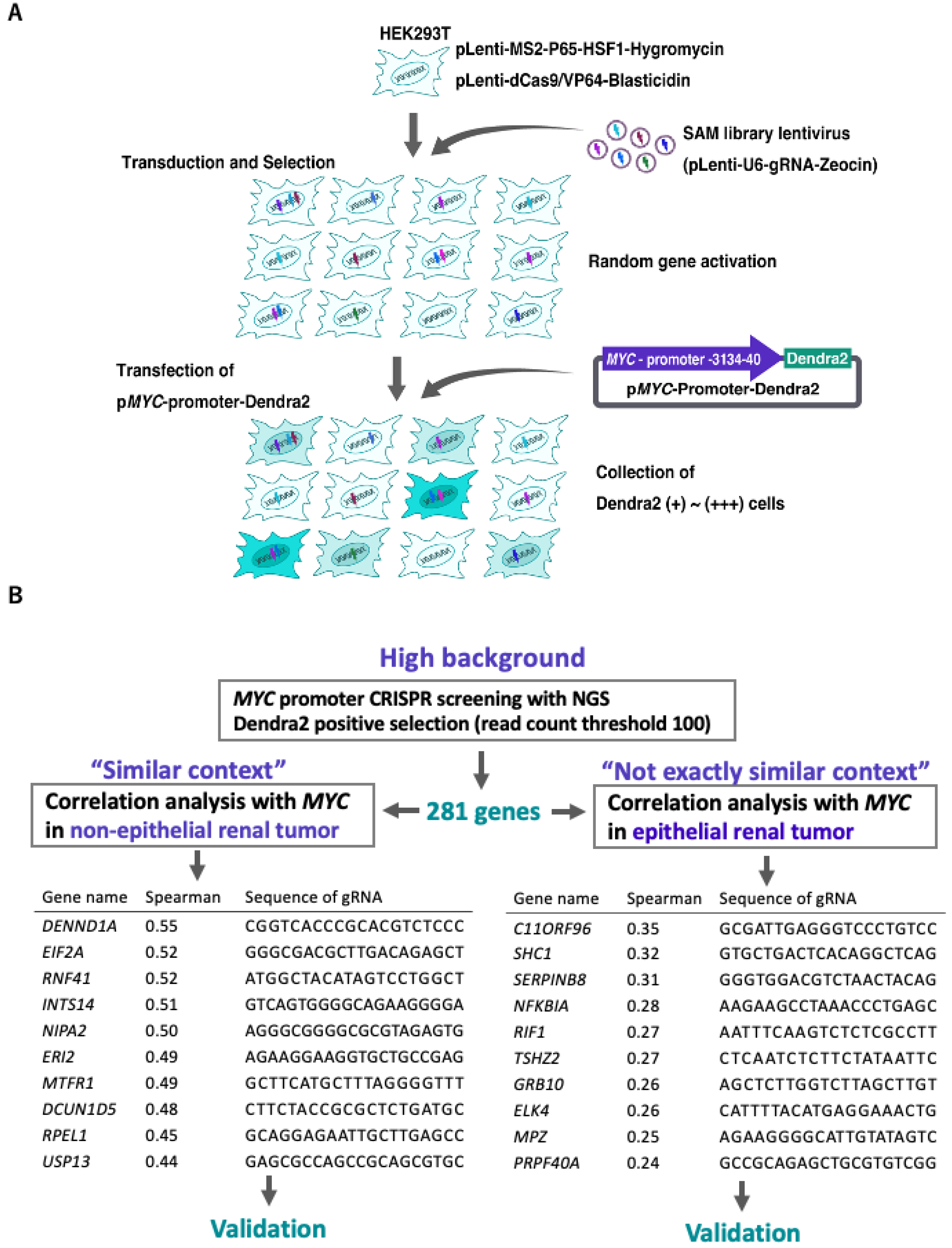
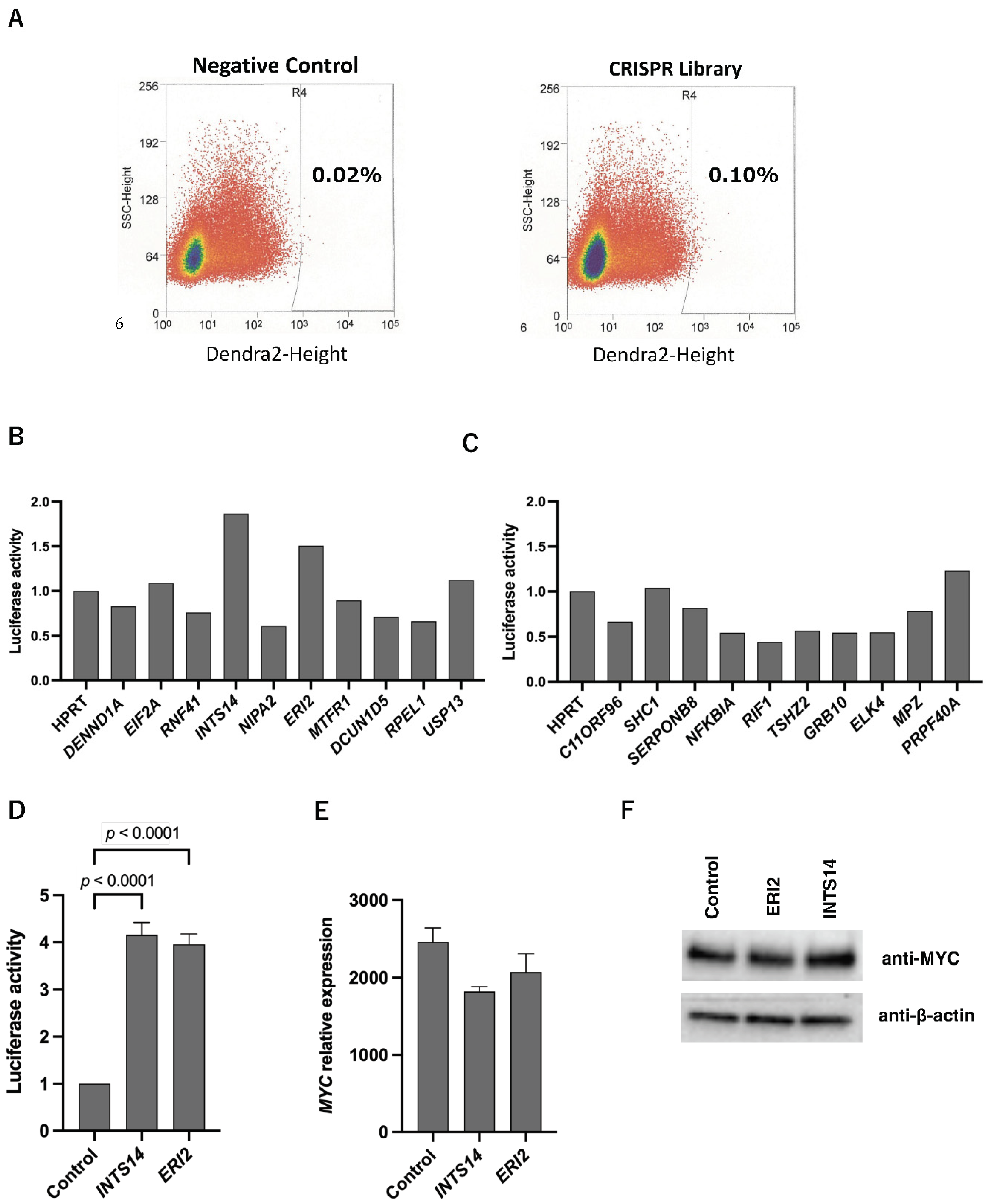
2.1. High-Throughput Gene Purification to Narrow down Candidate Genes
2.2. INTS14 and ERI2 Identified as Candidates for the MYC Regulator with Context-Matched Database Screening
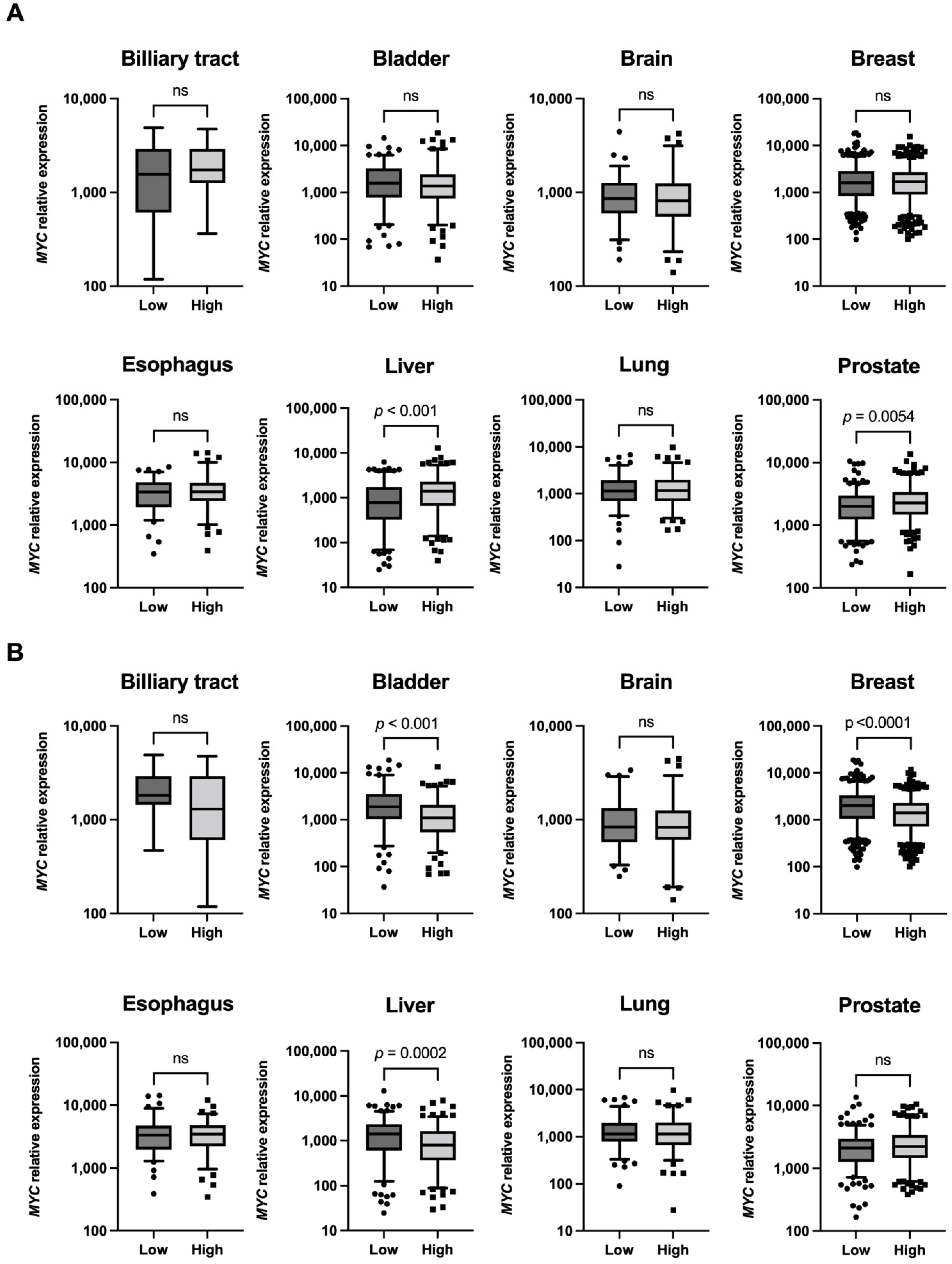
2.3. High MYC Expression in Tumors Associated with the High Expression of INTS14
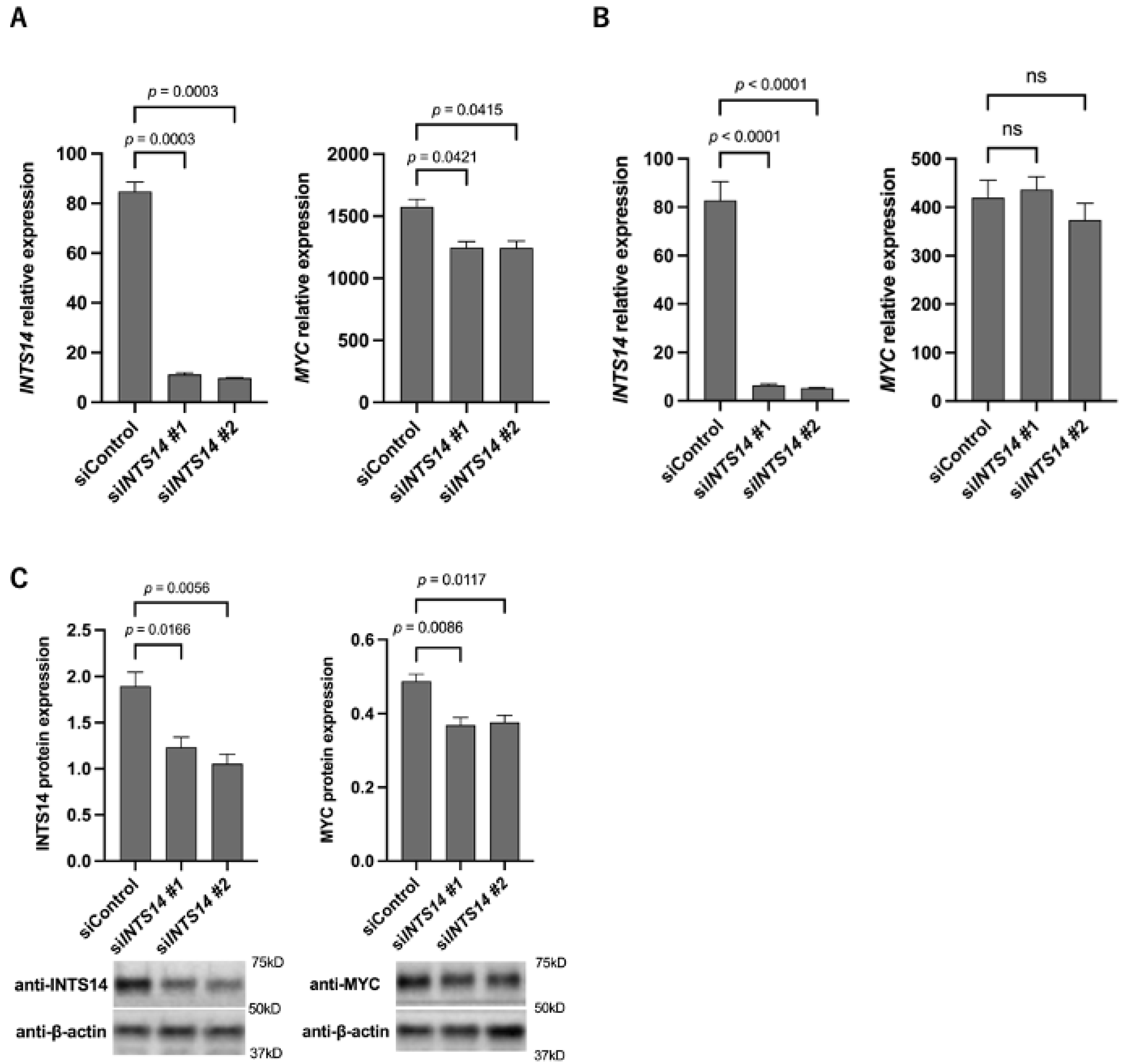
2.4. The Knockdown of INTS14 Decreases MYC Expression in the Prostate Cancer Cell Line
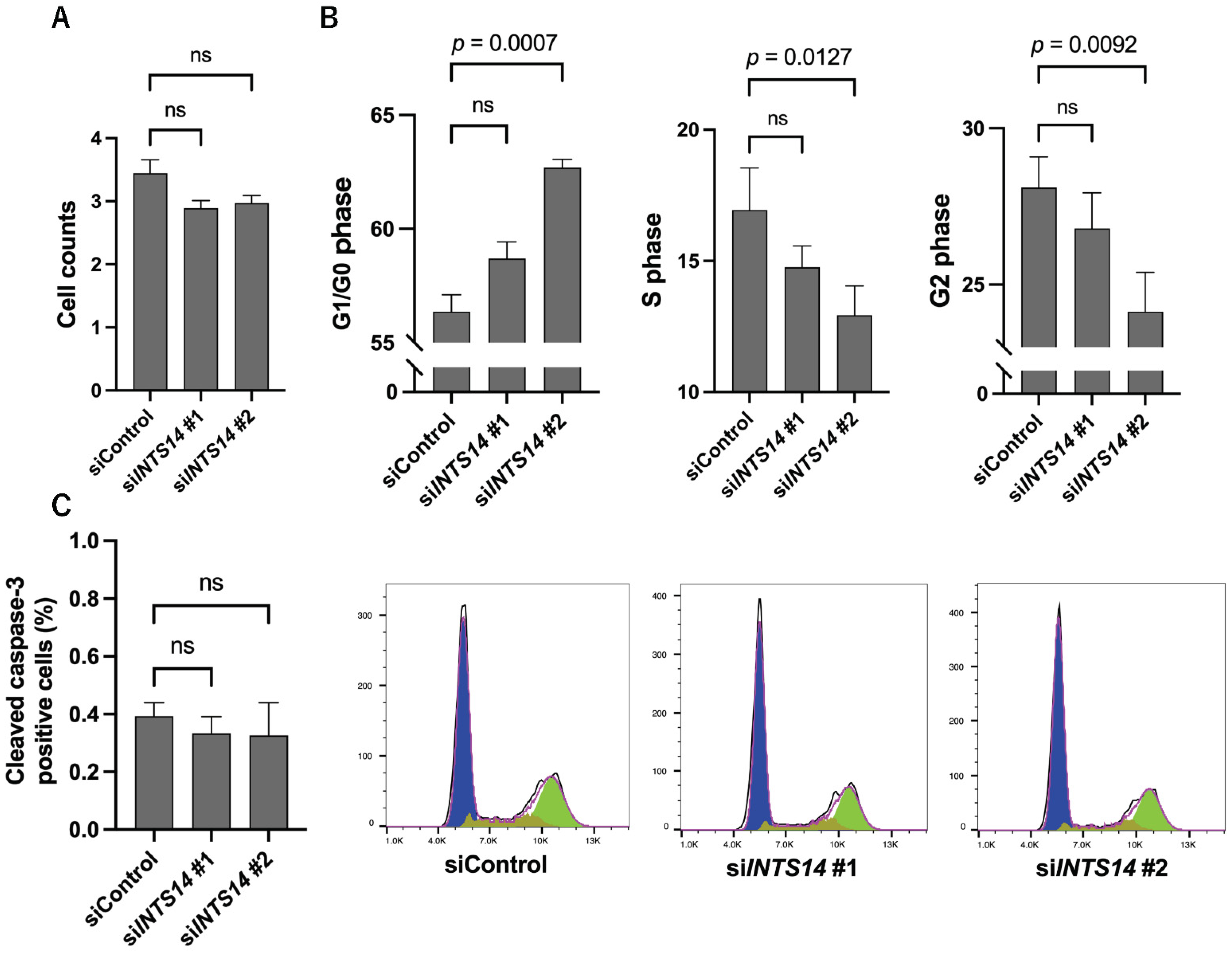
2.5. Knockdown of INTS14 Induces G0/1 Arrest in Prostate Cancer Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. SAM Library Screening
4.3. Plasmids
4.4. Reporter Assay
4.5. Database Analysis
4.6. Short Interference RNAs
4.7. Western Blots
4.8. Cell Cycle and Apoptosis Analysis
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Duffy, M.J.; O’Grady, S.; Tang, M.H.; Crown, J. MYC as a target for cancer treatment. Cancer Treat. Rev. 2021, 94, 102154. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V. A Time for MYC: Metabolism and Therapy. Cold Spring Harb. Symp. Quant. Biol. 2016, 81, 79–83. [Google Scholar] [CrossRef]
- Beaulieu, M.E.; Castillo, F.; Soucek, L. Structural and Biophysical Insights into the Function of the Intrinsically Disordered Myc Oncoprotein. Cells 2020, 9, 1038. [Google Scholar] [CrossRef] [PubMed]
- Carroll, P.A.; Freie, B.W.; Mathsyaraja, H.; Eisenman, R.N. The MYC transcription factor network: Balancing metabolism, proliferation and oncogenesis. Front Med. 2018, 12, 412–425. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.Y.; Loven, J.; Rahl, P.B.; Paranal, R.M.; Burge, C.B.; Bradner, J.E.; Lee, T.I.; Young, R.A. Transcriptional Amplification in Tumor Cells with Elevated c-Myc. Cell 2012, 151, 56–67. [Google Scholar] [CrossRef] [Green Version]
- Heidemann, M.; Hintermair, C.; Voss, K.; Eick, D. Dynamic phosphorylation patterns of RNA polymerase II CTD during transcription. Biochim. Et Biophys. Acta-Gene Regul. Mech. 2013, 1829, 55–62. [Google Scholar] [CrossRef]
- Shiekhattar, R.; Mermelstein, F.; Fisher, R.P.; Drapkin, R.; Dynlacht, B.; Wessling, H.C.; Morgan, D.O.; Reinberg, D. CDK-activating kinase complex is a component of human transcription factor tfiih. Nature 1995, 374, 283–287. [Google Scholar] [CrossRef] [PubMed]
- Guidi, B.W.; Bjornsdottir, G.; Hopkins, D.C.; Lacomis, L.; Erdjument-Bromage, H.; Tempst, P.; Myers, L.C. Mutual targeting of mediator and the TFIIH kinase Kin28. J. Biol. Chem. 2004, 279, 29114–29120. [Google Scholar] [CrossRef] [Green Version]
- Boeing, S.; Rigault, C.; Heidemann, M.; Eick, D.; Meisterernst, M. RNA Polymerase II C-terminal Heptarepeat Domain Ser-7 Phosphorylation Is Established in a Mediator-dependent Fashion. J. Biol. Chem. 2010, 285, 188–196. [Google Scholar] [CrossRef] [Green Version]
- Wullschleger, S.; Loewith, R.; Hall, M.N. TOR signaling in growth and metabolism. Cell 2006, 124, 471–484. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.H.; Sarbassov, D.D.; Ali, S.M.; King, J.E.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. MTOR interacts with Raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 2002, 110, 163–175. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Miranda, G.; Mendez, R. The CPEB-family of proteins, translational control in senescence and cancer. Ageing Res. Rev. 2012, 11, 460–472. [Google Scholar] [CrossRef] [PubMed]
- Groisman, I.; Ivshina, M.; Marin, V.; Kennedy, N.J.; Davis, R.J.; Richter, J.D. Control of cellular senescence by CPEB. Genes Dev. 2006, 20, 2701–2712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Liu, H.D.; Qing, G.L. Targeting oncogenic Myc as a strategy for cancer treatment. Signal Transduct. Target. Ther. 2018, 3, 5. [Google Scholar] [CrossRef] [Green Version]
- Taira, N.; Mimoto, R.; Kurata, M.; Yamaguchi, T.; Kitagawa, M.; Miki, Y.; Yoshida, K. DYRK2 priming phosphorylation of c-Jun and c-Myc modulates cell cycle progression in human cancer cells. J. Clin. Investig. 2012, 122, 859–872. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, A.; Kurata, M.; Onishi, I.; Sugita, K.; Matsumura, M.; Ishibashi, S.; Ikeda, M.; Yamamoto, K.; Kitagawa, M. CRISPR screening identifies M1AP as a new MYC regulator with a promoter-reporter system. PeerJ 2020, 8, e9046. [Google Scholar] [CrossRef]
- Kulkarni, V.; Naqvi, A.R.; Uttamani, J.R.; Nares, S. MiRNA-Target Interaction Reveals Cell-Specific Post-Transcriptional Regulation in Mammalian Cell Lines. Int. J. Mol. Sci. 2016, 17, 72. [Google Scholar] [CrossRef]
- Takwi, A.A.; Li, Y.; Becker Buscaglia, L.E.; Zhang, J.; Choudhury, S.; Park, A.K.; Liu, M.; Young, K.H.; Park, W.Y.; Martin, R.C.; et al. A statin-regulated microRNA represses human c-Myc expression and function. EMBO Mol. Med. 2012, 4, 896–909. [Google Scholar] [CrossRef]
- Baillat, D.; Wagner, E.J. Integrator: Surprisingly diverse functions in gene expression. Trends Biochem. Sci. 2015, 40, 257–264. [Google Scholar] [CrossRef] [Green Version]
- Guiro, J.; Murphy, S. Regulation of expression of human RNA polymerase II-transcribed snRNA genes. Open Biol. 2017, 7, 170073. [Google Scholar] [CrossRef] [Green Version]
- Baillat, D.; Hakimi, M.A.; Naar, A.M.; Shilatifard, A.; Cooch, N.; Shiekhattar, R. Integrator, a multiprotein mediator of small nuclear RNA processing, associates with the C-terminal repeat of RNA polymerase II. Cell 2005, 123, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Jurica, M.S.; Moore, M.J. Pre-mRNA splicing: Awash in a sea of proteins. Mol. Cell 2003, 12, 5–14. [Google Scholar] [CrossRef]
- Tatomer, D.C.; Elrod, N.D.; Liang, D.; Xiao, M.S.; Jiang, J.Z.; Jonathan, M.; Huang, K.L.; Wagner, E.J.; Cherry, S.; Wilusz, J.E. The integrator complex cleaves nascent mRNAs to attenuate transcription. Genes Dev 2019, 33, 1525–1538. [Google Scholar] [CrossRef]
- Sabath, K.; Staubli, M.L.; Marti, S.; Leitner, A.; Moes, M.; Jonas, S. INTS10-INTS13-INTS14 form a functional module of Integrator that binds nucleic acids and the cleavage module. Nat. Commun. 2020, 11, 3422. [Google Scholar] [CrossRef] [PubMed]
- Pfleiderer, M.M.; Galej, W.P. Structure of the catalytic core of the Integrator complex. Mol. Cell 2021, 81, 1246–1259.e8. [Google Scholar] [CrossRef] [PubMed]
- Ruan, G.X.; Li, Y.X.; Chen, W.J.; Huang, H.J.; Zhang, R.; Chen, C.X.; Lam, K.P.; Xu, S.L.; Ou, X.J. The spliceosome component Usp39 controls B cell development by regulating immunoglobulin gene rearrangement. Cell Rep. 2022, 38, 110338. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Kurata, M.; Wolf, N.K.; Lahr, W.S.; Weg, M.T.; Kluesner, M.G.; Lee, S.; Hui, K.; Shiraiwa, M.; Webber, B.R.; Moriarity, B.S. Highly multiplexed genome engineering using CRISPR/Cas9 gRNA arrays. PLoS ONE 2018, 13, e0198714. [Google Scholar] [CrossRef] [Green Version]
- Cerami, E.; Gao, J.J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tanaka, Y.; Kambayashi, H.; Yamamoto, A.; Onishi, I.; Sugita, K.; Matsumura, M.; Ishibashi, S.; Ikeda, M.; Yamamoto, K.; Kitagawa, M.; et al. Efficient Identification of the MYC Regulator with the Use of the CRISPR Library and Context-Matched Database Screenings. Int. J. Mol. Sci. 2022, 23, 7723. https://doi.org/10.3390/ijms23147723
Tanaka Y, Kambayashi H, Yamamoto A, Onishi I, Sugita K, Matsumura M, Ishibashi S, Ikeda M, Yamamoto K, Kitagawa M, et al. Efficient Identification of the MYC Regulator with the Use of the CRISPR Library and Context-Matched Database Screenings. International Journal of Molecular Sciences. 2022; 23(14):7723. https://doi.org/10.3390/ijms23147723
Chicago/Turabian StyleTanaka, Yosuke, Hidetaka Kambayashi, Akiko Yamamoto, Iichiroh Onishi, Keisuke Sugita, Miwa Matsumura, Sachiko Ishibashi, Masumi Ikeda, Kouhei Yamamoto, Masanobu Kitagawa, and et al. 2022. "Efficient Identification of the MYC Regulator with the Use of the CRISPR Library and Context-Matched Database Screenings" International Journal of Molecular Sciences 23, no. 14: 7723. https://doi.org/10.3390/ijms23147723