
Novel OPN1LW/OPN1MW Exon 3 Haplotype-Associated Splicing Defect in Patients with X-Linked Cone Dysfunction

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Results
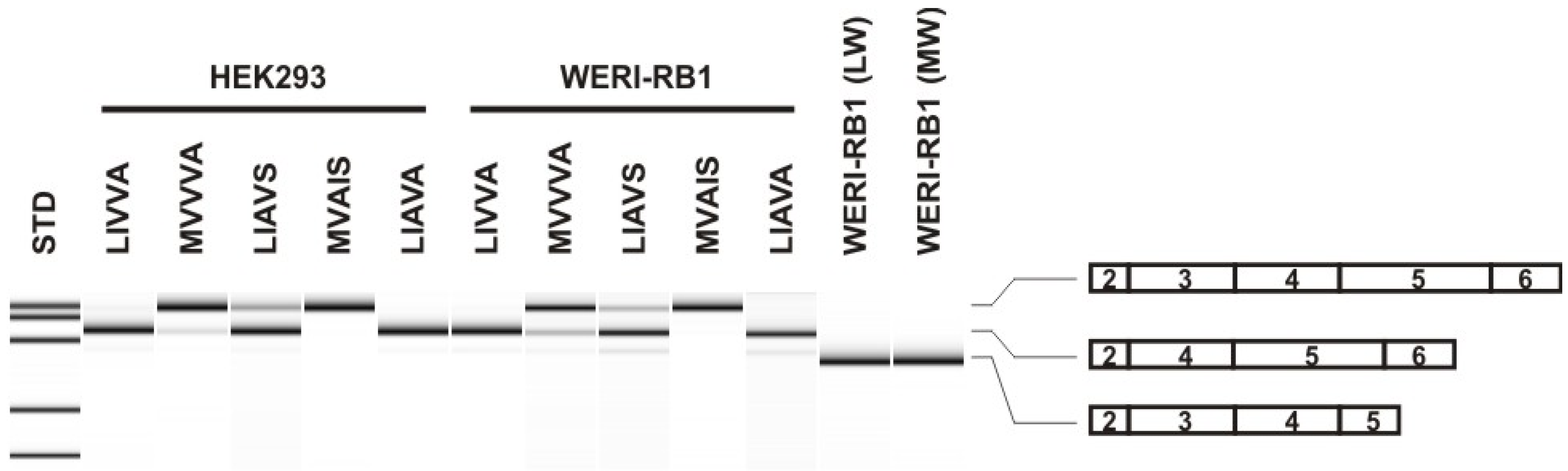
2.1. Identification of Patients with the Novel LIVVA Exon 3 Haplotype in the OPN1LW and/or the OPN1MW Gene and Functional Minigene Splicing Assays
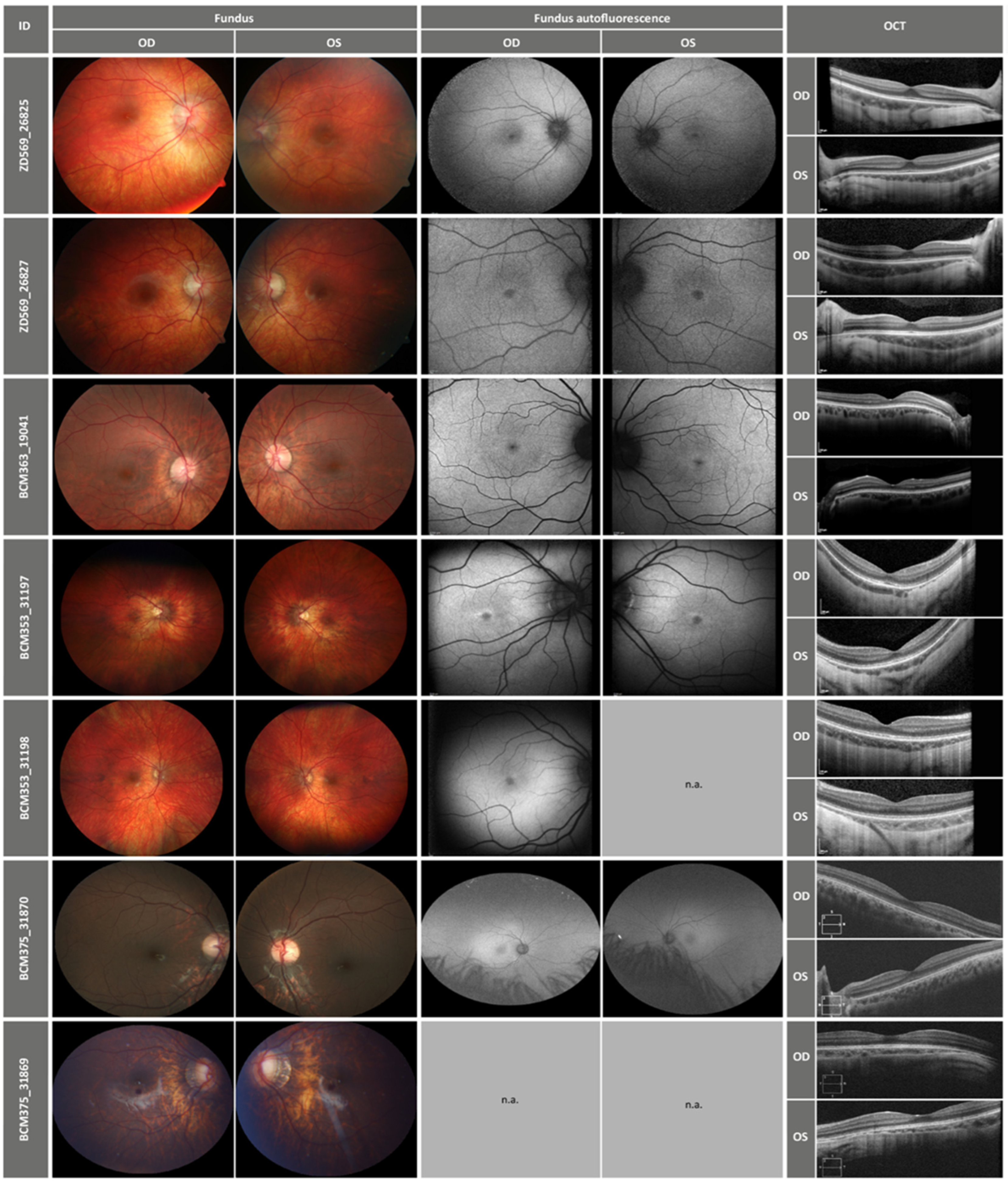
2.2. Clinical Findings
3. Discussion
4. Materials and Methods
4.1. Patient Recruitment and Clinical Evaluation
4.2. Genotyping of the OPN1LW/OPN1MW Gene Cluster
4.3. Genotyping of the OPN1LW/OPN1MW Gene Cluster
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nathans, J.; Thomas, D.; Hogness, D.S. Molecular genetics of human color vision: The genes encoding blue, green, and red pigments. Science 1986, 232, 193–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smallwood, P.M.; Wang, Y.; Nathans, J. Role of a locus control region in the mutually exclusive expression of human red and green cone pigment genes. Proc. Natl. Acad. Sci. USA 2002, 99, 1008–1011. [Google Scholar] [CrossRef] [Green Version]
- Nathans, J.; Piantanida, T.P.; Eddy, R.L.; Shows, T.B.; Hogness, D.S. Molecular genetics of inherited variation in human color vision. Science 1986, 232, 203–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nathans, J.; Davenport, C.M.; Maumenee, I.H.; Lewis, R.A.; Hejtmancik, J.F.; Litt, M.; Lovrien, E.; Weleber, R.; Bachynski, B.; Zwas, F.; et al. Molecular genetics of human blue cone monochromacy. Science 1989, 245, 831–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizrahi-Meissonnier, L.; Merin, S.; Banin, E.; Sharon, D. Variable retinal phenotypes caused by mutations in the X-linked photopigment gene array. Investig. Ophthalmol. Vis. Sci. 2010, 51, 3884–3892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michaelides, M.; Johnson, S.; Bradshaw, K.; Holder, G.E.; Simunovic, M.P.; Mollon, J.D.; Moore, A.T.; Hunt, D.M. X-linked cone dysfunction syndrome with myopia and protanopia. Ophthalmology 2005, 112, 1448–1454. [Google Scholar] [CrossRef]
- Ueyama, H.; Muraki-Oda, S.; Yamade, S.; Tanabe, S.; Yamashita, T.; Shichida, Y.; Ogita, H. Unique haplotype in exon 3 of cone opsin mRNA affects splicing of its precursor, leading to congenital color vision defect. Biochem. Biophys. Res. Commun. 2012, 424, 152–157. [Google Scholar] [CrossRef]
- Gardner, J.C.; Liew, G.; Quan, Y.-H.; Ermetal, B.; Ueyama, H.; Davidson, A.E.; Schwarz, N.; Kanuga, N.; Chana, R.; Maher, E.R.; et al. Three Different Cone Opsin Gene Array Mutational Mechanisms with Genotype–Phenotype Correlation and Functional Investigation of Cone Opsin Variants. Hum. Mutat. 2014, 35, 1354–1362. [Google Scholar] [CrossRef] [Green Version]
- Buena-Atienza, E.; Rüther, K.; Baumann, B.; Bergholz, R.; Birch, D.; Baere, E.D.; Dollfus, H.; Greally, M.T.; Gustavsson, P.; Hamel, C.P.; et al. De novo intrachromosomal gene conversion from OPN1MW to OPN1LW in the male germline results in Blue Cone Monochromacy. Sci. Rep. 2016, 6, 28253. [Google Scholar] [CrossRef] [Green Version]
- Greenwald, S.H.; Kuchenbecker, J.A.; Rowlan, J.S.; Neitz, J.; Neitz, M. Role of a Dual Splicing and Amino Acid Code in Myopia, Cone Dysfunction and Cone Dystrophy Associated with L/M Opsin Interchange Mutations. Transl. Vis. Sci. Technol. 2017, 6, 2. [Google Scholar] [CrossRef] [Green Version]
- Holmquist, D.; Epstein, D.; Olsson, M.; Wissinger, B.; Kohl, S.; Hengstler, J.; Tear-Fahnehjelm, K. Visual and ocular findings in a family with X-linked cone dysfunction and protanopia. Ophthalmic Genet. 2021, 42, 570–576. [Google Scholar] [CrossRef] [PubMed]
- Khateb, S.; Shemesh, A.; Offenheim, A.; Sheffer, R.; Ben-Yosef, T.; Chowers, I.; Leibu, R.; Baumann, B.; Wissinger, B.; Kohl, S.; et al. Relatively mild blue cone monochromacy phenotype caused by various haplotypes in the L- and M-cone opsin genes. Mol. Vis. 2022, 28, 21–28. [Google Scholar] [PubMed]
- McFall, R.C.; Sery, T.W.; Makadon, M. Characterization of a new continuous cell line derived from a human retinoblastoma. Cancer Res. 1977, 37, 1003–1010. [Google Scholar] [PubMed]
- Shaaban, S.A.; Deeb, S.S. Functional analysis of the promoters of the human red and green visual pigment genes. Investig. Ophthalmol. Vis. Sci. 1998, 39, 885–896. [Google Scholar]
- Patterson, E.J.; Wilk, M.; Langlo, C.S.; Kasilian, M.; Ring, M.; Hufnagel, R.B.; Dubis, A.M.; Tee, J.J.; Kalitzeos, A.; Gardner, J.C.; et al. Cone Photoreceptor Structure in Patients With X-Linked Cone Dysfunction and Red-Green Color Vision Deficiency. Investig. Ophthalmol. Vis. Sci. 2016, 57, 3853–3863. [Google Scholar] [CrossRef] [Green Version]
- McClements, M.; Davies, W.I.; Michaelides, M.; Young, T.; Neitz, M.; MacLaren, R.E.; Moore, A.T.; Hunt, D.M. Variations in opsin coding sequences cause x-linked cone dysfunction syndrome with myopia and dichromacy. Investig. Ophthalmol. Vis. Sci. 2013, 54, 1361–1369. [Google Scholar] [CrossRef] [Green Version]
- Haim, M.; Fledelius, H.C.; Skarsholm, D. X-linked myopia in Danish family. Acta Ophthalmol. 1988, 66, 450–456. [Google Scholar] [CrossRef]
- Schwartz, M.; Haim, M.; Skarsholm, D. X-linked myopia: Bornholm eye disease. Linkage to DNA markers on the distal part of Xq. Clin. Genet. 1990, 38, 281–286. [Google Scholar] [CrossRef]
- Blackwell, H.R.; Blackwell, O.M. Blue mono-cone monochromacy: A new color vision defect. J. Opt. Soc. Am. 1957, 47, 338. [Google Scholar]
- Alpern, A.; Lee, G.B.; Spivey, B.E. π1 cone monochromatism. Arch. Ophthalmol. 1965, 74, 334–337. [Google Scholar] [CrossRef]
- Orosz, O.; Rajta, I.; Vajas, A.; Takács, L.; Csutak, A.; Fodor, M.; Kolozsvári, B.; Resch, M.; Sényi, K.; Lesch, B.; et al. Myopia and Late-Onset Progressive Cone Dystrophy Associate to LVAVA/MVAVA Exon 3 Interchange Haplotypes of Opsin Genes on Chromosome X. Investig. Ophthalmol. Vis. Sci. 2017, 58, 1834–1842. [Google Scholar] [CrossRef] [Green Version]
- Young, T.L.; Deeb, S.S.; Ronan, S.M.; Dewan, A.T.; Alvear, A.B.; Scavello, G.S.; Paluru, P.C.; Brott, M.S.; Hayashi, T.; Holleschau, A.M.; et al. X-linked high myopia associated with cone dysfunction. Arch. Ophthalmol. 2004, 122, 897–908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patterson, E.J.; Kalitzeos, A.; Kasilian, M.; Gardner, J.C.; Neitz, J.; Hardcastle, A.J.; Neitz, M.; Carroll, J.; Michaelides, M. Residual Cone Structure in Patients With X-Linked Cone Opsin Mutations. Investig. Ophthalmol. Vis. Sci. 2018, 59, 4238–4248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cideciyan, A.V.; Hufnagel, R.B.; Carroll, J.; Sumaroka, A.; Luo, X.; Schwartz, S.B.; Dubra, A.; Land, M.; Michaelides, M.; Gardner, J.C.; et al. Human cone visual pigment deletions spare sufficient photoreceptors to warrant gene therapy. Hum. Gene Ther. 2013, 24, 993–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Subject/Proband | OPN1LW Exon 3 Haplotype (nt/aa) 1 | OPN1MW Exon 3 Haplotype (nt/aa) 1 | ∑ Gene Copies 2 | ||
|---|---|---|---|---|---|
| BCM363_19041 | G-C-G-A-T-T-G-G | L-I-V-V-A | G-C-G-A-T-T-G-G | L-I-V-V-A | 2 |
| BCM353_31197 | G-C-G-A-T-C-G-T | L-I-A-V-S | G-C-G-A-T-T-G-G | L-I-V-V-A | 2 |
| BCM353_31198 | G-C-G-A-T-C-G-T | L-I-A-V-S | G-C-G-A-T-T-G-G | L-I-V-V-A | 2 |
| ZD569_26825 | G-C-G-A-T-T-G-G | L-I-V-V-A | A-A-C-G-G-T-G-G | M-V-V-V-A | 2 |
| ZD569_26827 | G-C-G-A-T-T-G-G | L-I-V-V-A | A-A-C-G-G-T-G-G | M-V-V-V-A | 2 |
| BCM375_31869 | G-C-G-A-T-T-G-G | L-I-V-V-A | A-A-C-G-G-T-G-G | M-V-V-V-A | 2 |
| BCM375_31870 | G-C-G-A-T-T-G-G | L-I-V-V-A | A-A-C-G-G-T-G-G | M-V-V-V-A | 2 |
| Exon 3 Haplotype (nt/aa) | % Correctly Spliced | ||
|---|---|---|---|
| HEK293 1 | WERI-Rb1 1 | ||
| G-C-G-A-T-T-G-G | L-I-V-V-A | 5.28 ± 0.088 | 3.89 ± 0.18 |
| G-C-G-A-T-C-G-T | L-I-A-V-S | 28.36 ± 0.5 | 18.21 ± 0.24 |
| A-A-C-G-G-T-G-G | M-V-V-V-A | 74.41 ± 0.5 | 69.44 ± 0.17 |
| G-C-G-A-T-C-G-G | L-I-A-V-A | 0.00/ndt. | 0.00/ndt. |
| A-A-C-G-G-C-A-T | M-V-A-I-S | 98.12 ± 0.07 | 98.22 ± 0.16 |
| Patient-ID | Age at Onset 1 | First Symptom | Age (Years) 1 | BCVA 2 (Decimal) | Refraction 2 | Night Blindness | Photo- Phobia | Full-Field ERG | Color Test (Farnsworth 15 Hues) | Visual Field | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| OD | OS | OD | OS | |||||||||
| ZD569 _26825 | not known | no symptoms | 10 | 1.0 | 0.63 | −1.25/−2.5/151 | −0.5/−2.0/180 | no | yes | sctotopic normal, photopic very reduced | scotopic, red and green color confusions | III4e borders normal |
| 13 | 1.0 | 0.8 | −7.75/−3.25/21 | −7.0/−33.5/98 | no | yes | photopic very reduced | n.a. | III4e borders normal | |||
| ZD569 _26827 | 6 | decreased visual acuity | 8 | 0.4 | 0.5 | −7.5/−4.25/29 | −7.0/−3.75/174 | no | yes | sctotopic normal, photopic very reduced | red and green color confusions | III4e borders normal |
| 11 | 0.8 | 0.6 | −9.5/−4.25/23 | −9.25/−3.25/160 | no | yes | sctotopic normal, photopic very reduced | n.a. | III4e borders normal | |||
| BCM363 _19041 | 10 | dyschromatspia in brightness | 14 | 0.4 | 0.4 | emmetropia | no | yes | sctotopic normal, photopic very reduced | green color confusions | III4e borders normal | |
| BCM353 _31197 | 3 | decreased visual acuity | 6 | 0.3 | 0.4 | −9/−2.5/15 | −9.5/−1/20 | no | no | photopic and flicker reduced, prolonged flicker implicit times | red and green color confusions (desatured test only) | Grossly normal (confrontation perimetry) |
| 11 | 0.4 | 0.5 | −7/−2/140 | −9/−1/145 | no | no | n.a. | red and green color confusions (saturated and desatured test) | relative central scotoma (automated perimetry) | |||
| BCM353 _31198 | not known | no symptoms | 4 | 0.4 | 0.4 | +5.5/−1/15 | +4.25/−2/175 | no | no | normal except flicker: reduced and prolonged implicit time | n.a. | n.a. |
| 10 | 0.8 | 0.6 | emmetropia | no | no | n.a. | green-color deficits (Ishihara) | n.a. | ||||
| BCM375 _31870 | 4 | decreased visual acuity | 9 | 0.63 | 0.8 | −9.75 | −8.00/+1.00/90 | no | mild hemeralopia | scotopic normal, photopic reduced | strong red and green color deficits | well preserved |
| 15 | 0.5 | 0.8 | −11.50/+1.25/25 | −9.75/+1.00/95 | no | mild photophobia | n.a. | strong red green color deficits | n.a. | |||
| BCM375 _31869 | 3 | decreased visual acuity | 7 | 0.25 | 0.16 | −20.00/+1.75/125 | −20.00/+2.00/90 | no | yes | scotopic normal, photopic severyl reduced | moderate red and green color deficits | well preserved |
| 14 | 0.5 | 0.2 | −23.00/+3.00/100 | −24.00/+1.00/90 | no | yes | n.a. | moderate red and green color deficits | n.a. | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stingl, K.; Baumann, B.; De Angeli, P.; Vincent, A.; Héon, E.; Cordonnier, M.; De Baere, E.; Raskin, S.; Sato, M.T.; Shiokawa, N.; et al. Novel OPN1LW/OPN1MW Exon 3 Haplotype-Associated Splicing Defect in Patients with X-Linked Cone Dysfunction. Int. J. Mol. Sci. 2022, 23, 6868. https://doi.org/10.3390/ijms23126868
Stingl K, Baumann B, De Angeli P, Vincent A, Héon E, Cordonnier M, De Baere E, Raskin S, Sato MT, Shiokawa N, et al. Novel OPN1LW/OPN1MW Exon 3 Haplotype-Associated Splicing Defect in Patients with X-Linked Cone Dysfunction. International Journal of Molecular Sciences. 2022; 23(12):6868. https://doi.org/10.3390/ijms23126868
Chicago/Turabian StyleStingl, Katarina, Britta Baumann, Pietro De Angeli, Ajoy Vincent, Elise Héon, Monique Cordonnier, Elfriede De Baere, Salmo Raskin, Mario Teruo Sato, Naoye Shiokawa, and et al. 2022. "Novel OPN1LW/OPN1MW Exon 3 Haplotype-Associated Splicing Defect in Patients with X-Linked Cone Dysfunction" International Journal of Molecular Sciences 23, no. 12: 6868. https://doi.org/10.3390/ijms23126868