
Estradiol (E2) Improves Glucose-Stimulated Insulin Secretion and Stabilizes GDM Progression in a Prediabetic Mouse Model


Abstract
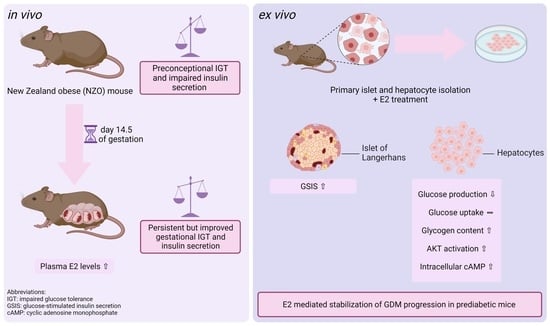
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Increased Plasma E2 Levels in NZO Mice during Gestation
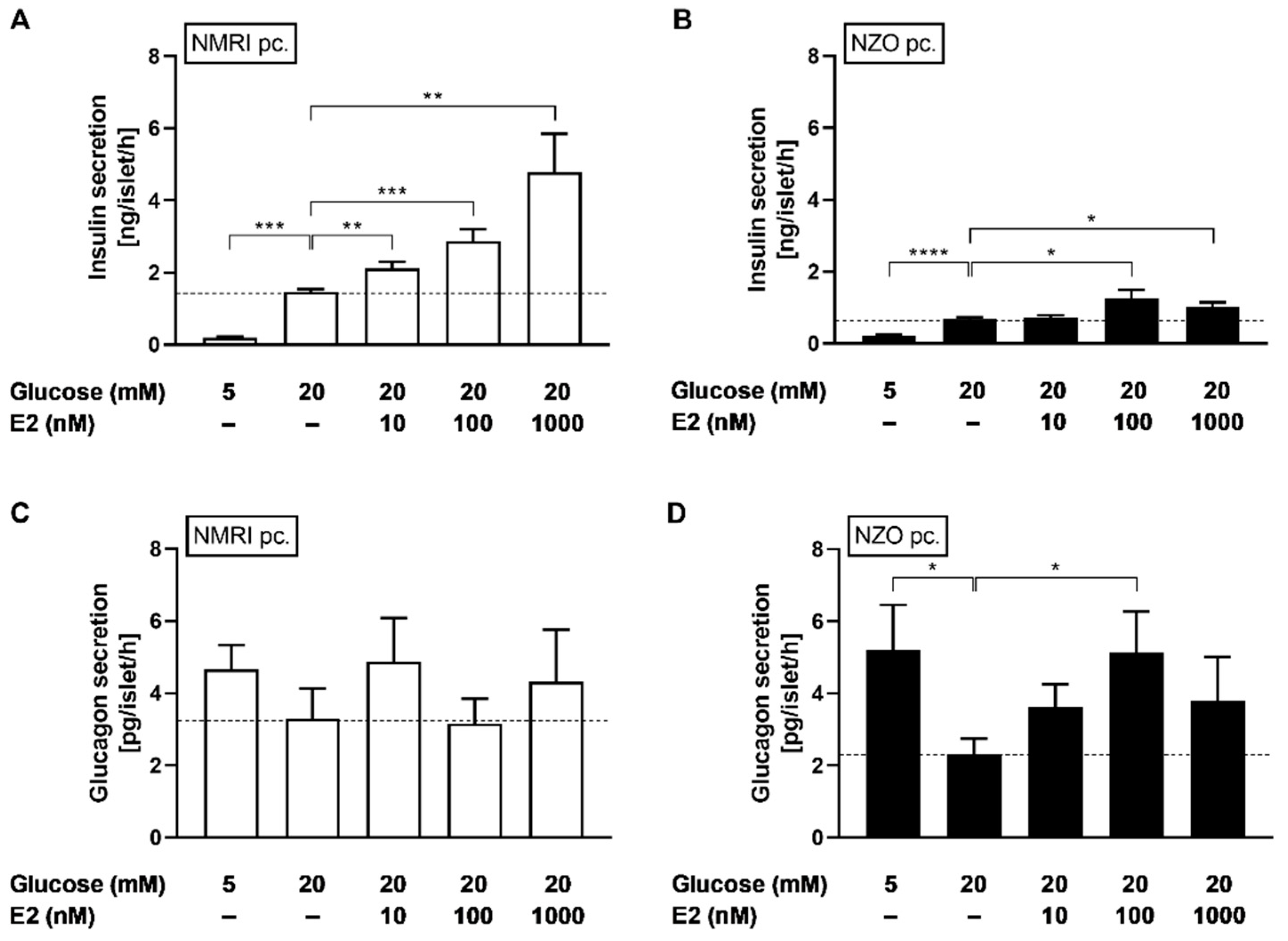
2.2. Effects of E2 on Insulin and Glucagon Secretion in Freshly Isolated Islets
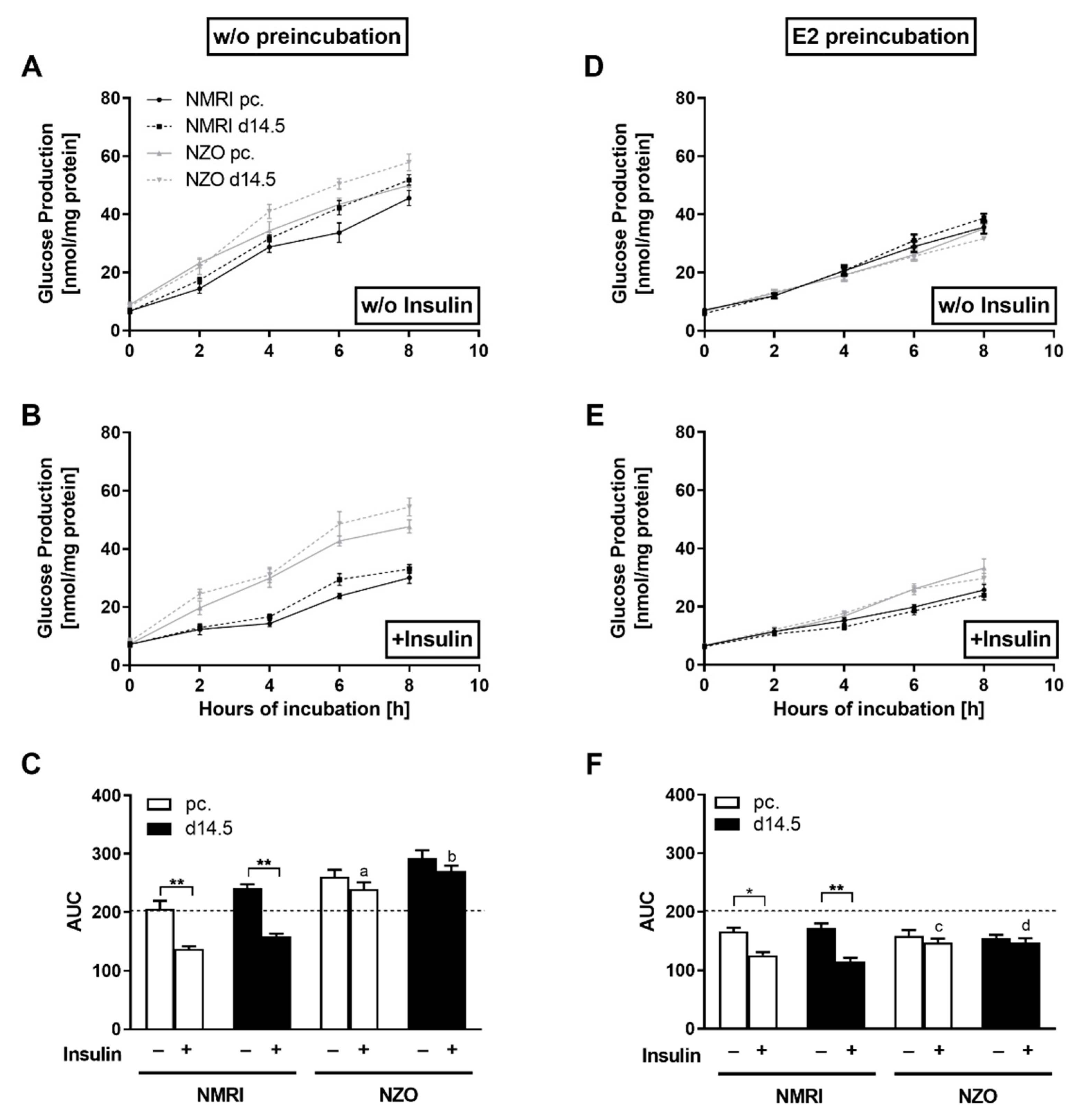
2.3. Effects of E2 on Hepatic Glucose Utilization in Primary Hepatocytes
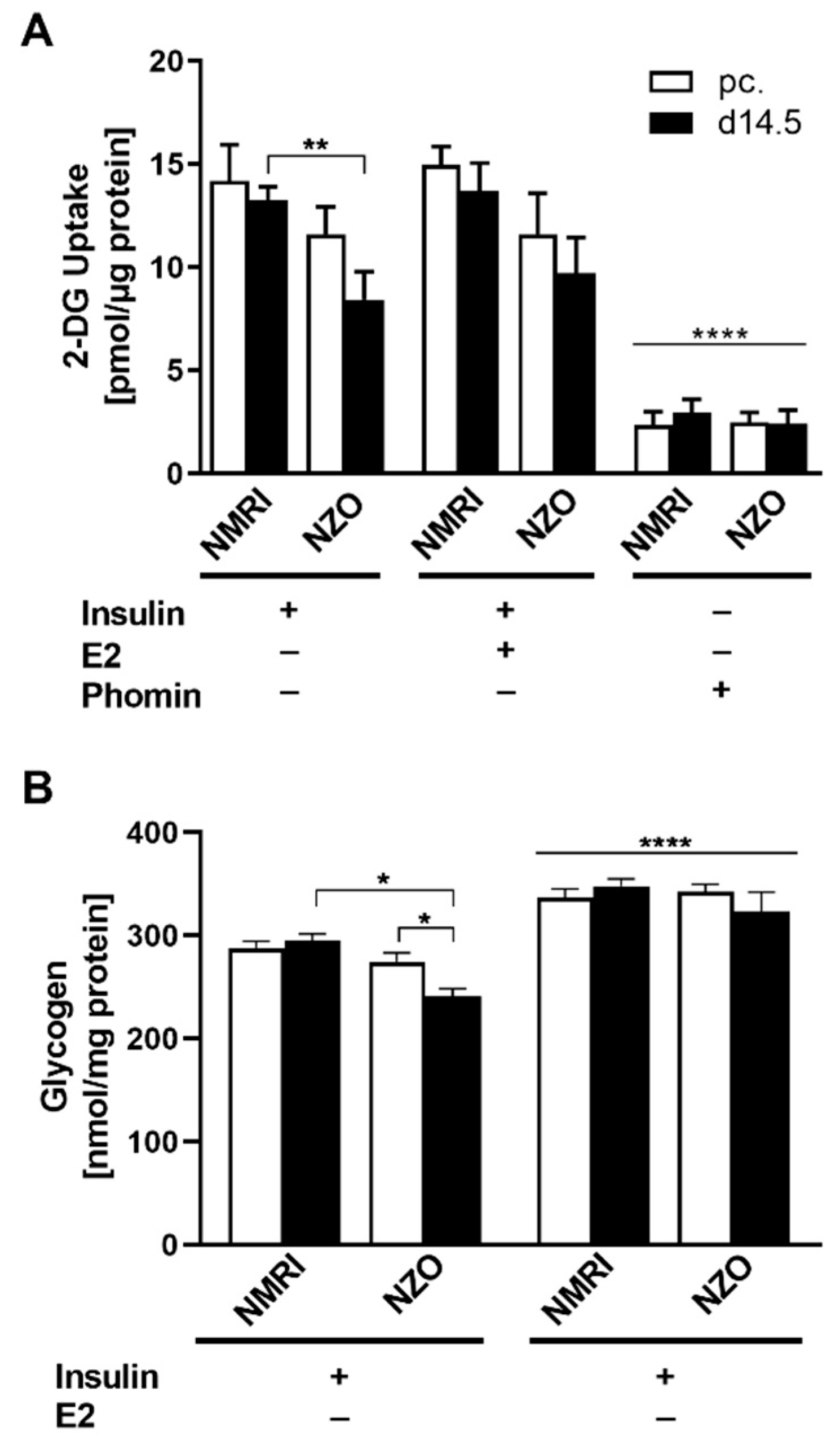
2.4. E2 Effect on Glucose Uptake and Glycogen Content in Primary Hepatocytes
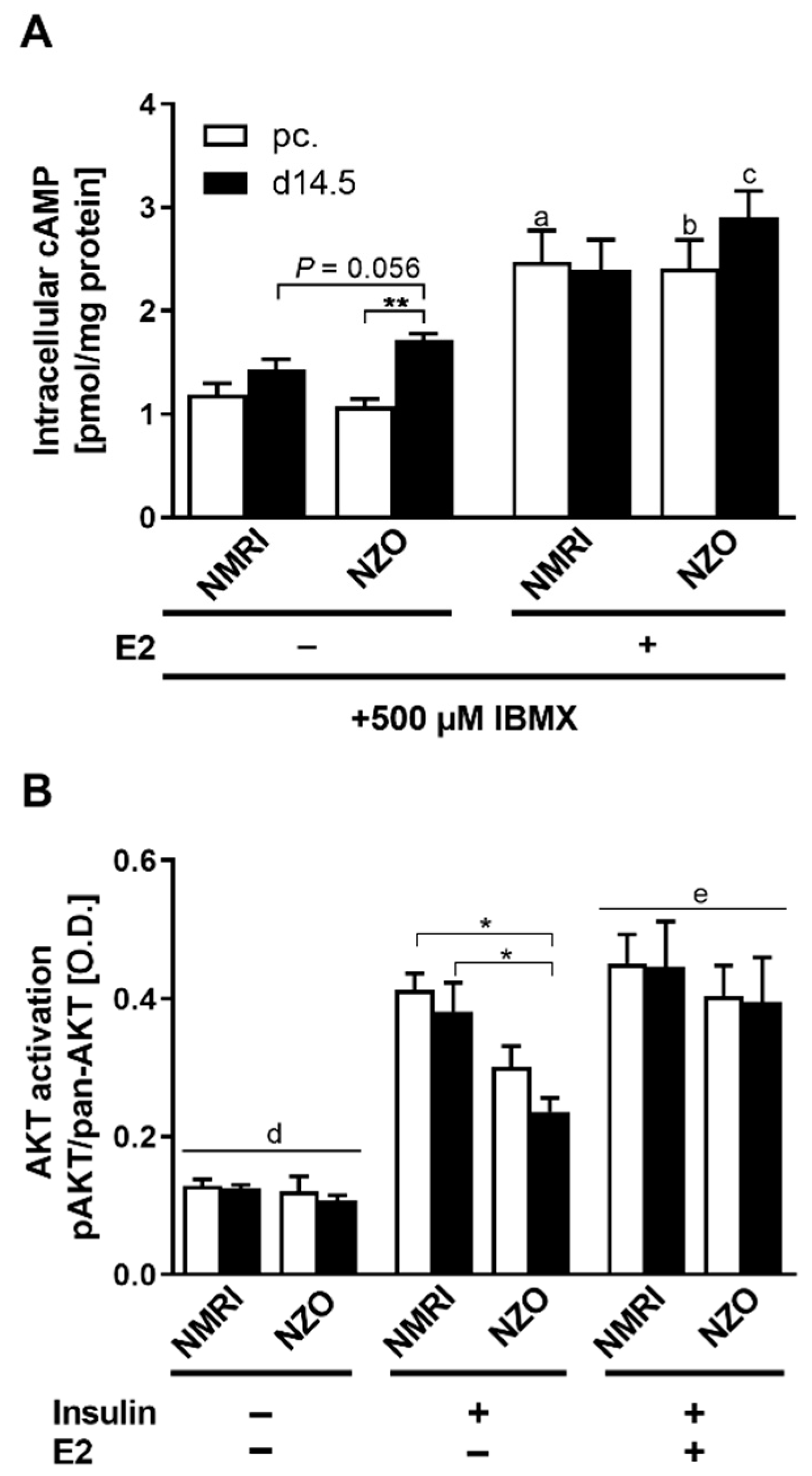
2.5. Modulatory Effects of E2 on cAMP and AKT Signaling
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Animals
5.2. Chemicals
5.3. Measurement of Plasma E2
5.4. Islet Isolation
5.5. Ex Vivo Stimulation and Hormone Secretion in Islets of Langerhans
5.6. Hepatocyte Isolation and Cell Culture
5.7. Ex Vivo Stimulation of Primary Hepatocytes
5.8. Glucose Production
5.9. Glucose Uptake
5.10. Glycogen Content
5.11. Measurement of Intracellular cAMP
5.12. Measurement of AKT Activation
5.13. Shared Control Groups
5.14. Data Handling and Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Arnetz, L.; Ekberg, N.R.; Alvarsson, M. Sex Differences in Type 2 Diabetes: Focus on Disease Course and Outcomes. Diabetes Metab. Syndr. Obes. 2014, 7, 409–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geer, E.B.; Shen, W. Gender Differences in Insulin Resistance, Body Composition, and Energy Balance. Gend. Med. 2009, 6 (Suppl. 1), 60–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bjune, J.-I.; Strømland, P.P.; Jersin, R.Å.; Mellgren, G.; Dankel, S.N. Metabolic and Epigenetic Regulation by Estrogen in Adipocytes. Front. Endocrinol. 2022, 13, 153. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.T.; Han, G.-Z.; Shim, J.-Y.; Wen, Y.; Jiang, X.-R. Quantitative Structure-Activity Relationship of Various Endogenous Estrogen Metabolites for Human Estrogen Receptor Alpha and Beta Subtypes: Insights into the Structural Determinants Favoring a Differential Subtype Binding. Endocrinology 2006, 147, 4132–4150. [Google Scholar] [CrossRef]
- Labhart, A. Clinical Endocrinology Theory and Practice; Springer: Berlin/Heidelberg, Germany, 1974; ISBN 978-3-642-96158-8. [Google Scholar]
- Coelingh Bennink, H.J.T. Are All Estrogens the Same? Maturitas 2004, 47, 269–275. [Google Scholar] [CrossRef]
- Park, S.K.; Harlow, S.D.; Zheng, H.; Karvonen-Gutierrez, C.; Thurston, R.C.; Ruppert, K.; Janssen, I.; Randolph, J.F. Association between Changes in Oestradiol and Follicle-Stimulating Hormone Levels during the Menopausal Transition and Risk of Diabetes. Diabet. Med. 2017, 34, 531–538. [Google Scholar] [CrossRef]
- Salpeter, S.R.; Walsh, J.M.E.; Ormiston, T.M.; Greyber, E.; Buckley, N.S.; Salpeter, E.E. Meta-Analysis: Effect of Hormone-Replacement Therapy on Components of the Metabolic Syndrome in Postmenopausal Women. Diabetes Obes. Metab. 2006, 8, 538–554. [Google Scholar] [CrossRef]
- Kim, C.; Kong, S.; Laughlin, G.A.; Golden, S.H.; Mather, K.J.; Nan, B.; Randolph, J.F.; Edelstein, S.L.; Labrie, F.; Buschur, E.; et al. Reductions in Glucose among Postmenopausal Women Who Use and Do Not Use Estrogen Therapy. Menopause 2013, 20, 393–400. [Google Scholar] [CrossRef] [Green Version]
- Mauvais-Jarvis, F.; Clegg, D.J.; Hevener, A.L. The Role of Estrogens in Control of Energy Balance and Glucose Homeostasis. Endocr. Rev. 2013, 34, 309–338. [Google Scholar] [CrossRef] [Green Version]
- Ciana, P.; Raviscioni, M.; Mussi, P.; Vegeto, E.; Que, I.; Parker, M.G.; Lowik, C.; Maggi, A. In Vivo Imaging of Transcriptionally Active Estrogen Receptors. Nat. Med. 2003, 9, 82–86. [Google Scholar] [CrossRef]
- Gao, H.; Fält, S.; Sandelin, A.; Gustafsson, J.-A.; Dahlman-Wright, K. Genome-Wide Identification of Estrogen Receptor Alpha-Binding Sites in Mouse Liver. Mol. Endocrinol. 2008, 22, 10–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoo, J.-J.; Lim, Y.S.; Kim, M.S.; Lee, B.; Kim, B.-Y.; Kim, Z.; Lee, J.E.; Lee, M.H.; Kim, S.G.; Kim, Y.S. Risk of Fatty Liver after Long-Term Use of Tamoxifen in Patients with Breast Cancer. PLoS ONE 2020, 15, e0236506. [Google Scholar] [CrossRef] [PubMed]
- Hart-Unger, S.; Arao, Y.; Hamilton, K.J.; Lierz, S.L.; Malarkey, D.E.; Hewitt, S.C.; Freemark, M.; Korach, K.S. Hormone Signaling and Fatty Liver in Females: Analysis of Estrogen Receptor α Mutant Mice. Int. J. Obes. 2017, 41, 945–954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Eon, T.M.; Souza, S.C.; Aronovitz, M.; Obin, M.S.; Fried, S.K.; Greenberg, A.S. Estrogen Regulation of Adiposity and Fuel Partitioning. Evidence of Genomic and Non-Genomic Regulation of Lipogenic and Oxidative Pathways. J. Biol. Chem. 2005, 280, 35983–35991. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Li, N.; Chivese, T.; Werfalli, M.; Sun, H.; Yuen, L.; Hoegfeldt, C.A.; Elise Powe, C.; Immanuel, J.; Karuranga, S.; et al. IDF Diabetes Atlas: Estimation of Global and Regional Gestational Diabetes Mellitus Prevalence for 2021 by International Association of Diabetes in Pregnancy Study Group’s Criteria. Diabetes Res. Clin. Pract. 2022, 183, 109050. [Google Scholar] [CrossRef]
- Haschka, S.J.; Gar, C.; Sacco, V.; Banning, F.; Ferrari, U.; Freibothe, I.; Kern-Matschilles, S.; Potzel, A.L.; Rauch, B.; Fueessl, L.U.; et al. Pre-Diabetes, Diabetes and Fluctuations of Glucose Tolerance after Gestational Diabetes Mellitus: 5-Year Follow-up of a Contemporary, Prospective Study in Germany. BMJ Open Diabetes Res. Care 2022, 10, e002621. [Google Scholar] [CrossRef]
- Tulchinsky, D.; Hobel, C.J.; Yeager, E.; Marshall, J.R. Plasma Estrone, Estradiol, Estriol, Progesterone, and 17-Hydroxyprogesterone in Human Pregnancy: I. Normal Pregnancy. Am. J. Obstet. Gynecol. 1972, 112, 1095–1100. [Google Scholar] [CrossRef]
- Garland, H.O.; Atherton, J.C.; Baylis, C.; Morgan, M.R.; Milne, C.M. Hormone Profiles for Progesterone, Oestradiol, Prolactin, Plasma Renin Activity, Aldosterone and Corticosterone during Pregnancy and Pseudopregnancy in Two Strains of Rat: Correlation with Renal Studies. J. Endocrinol. 1987, 113, 435–444. [Google Scholar] [CrossRef]
- Albrecht, E.D.; Aberdeen, G.W.; Pepe, G.J. The Role of Estrogen in the Maintenance of Primate Pregnancy. Am. J. Obstet. Gynecol. 2000, 182, 432–438. [Google Scholar] [CrossRef]
- Nadal, A.; Rovira, J.M.; Laribi, O.; Leon-quinto, T.; Andreu, E.; Ripoll, C.; Soria, B. Rapid Insulinotropic Effect of 17beta-Estradiol via a Plasma Membrane Receptor. FASEB J. 1998, 12, 1341–1348. [Google Scholar] [CrossRef] [Green Version]
- Alonso-Magdalena, P.; Morimoto, S.; Ripoll, C.; Fuentes, E.; Nadal, A. The Estrogenic Effect of Bisphenol A Disrupts Pancreatic Beta-Cell Function in Vivo and Induces Insulin Resistance. Env. Health Perspect 2006, 114, 106–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montelongo, A.; Lasunción, M.A.; Pallardo, L.F.; Herrera, E. Longitudinal Study of Plasma Lipoproteins and Hormones during Pregnancy in Normal and Diabetic Women. Diabetes 1992, 41, 1651–1659. [Google Scholar] [CrossRef]
- Villarroel, C.; Salinas, A.; López, P.; Kohen, P.; Rencoret, G.; Devoto, L.; Codner, E. Pregestational Type 2 Diabetes and Gestational Diabetes Exhibit Different Sexual Steroid Profiles during Pregnancy. Gynecol. Endocrinol. 2017, 33, 212–217. [Google Scholar] [CrossRef] [PubMed]
- Couch, S.C.; Philipson, E.H.; Bendel, R.B.; Pujda, L.M.; Milvae, R.A.; Lammi-Keefe, C.J. Elevated Lipoprotein Lipids and Gestational Hormones in Women With Diet-Treated Gestational Diabetes Mellitus Compared to Healthy Pregnant Controls. J. Diabetes Its Complicat. 1998, 12, 1–9. [Google Scholar] [CrossRef]
- Shi, Z.; Zhao, C.; Guo, X.; Ding, H.; Cui, Y.; Shen, R.; Liu, J. Differential Expression of MicroRNAs in Omental Adipose Tissue From Gestational Diabetes Mellitus Subjects Reveals MiR-222 as a Regulator of ERα Expression in Estrogen-Induced Insulin Resistance. Endocrinology 2014, 155, 1982–1990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deischinger, C.; Leitner, K.; Baumgartner-Parzer, S.; Bancher-Todesca, D.; Kautzky-Willer, A.; Harreiter, J. CTRP-1 Levels Are Related to Insulin Resistance in Pregnancy and Gestational Diabetes Mellitus. Sci. Rep. 2020, 10, 17345. [Google Scholar] [CrossRef] [PubMed]
- Nadal, A.; Alonso-Magdalena, P.; Soriano, S.; Quesada, I.; Ropero, A.B. The Pancreatic β-Cell as a Target of Estrogens and Xenoestrogens: Implications for Blood Glucose Homeostasis and Diabetes. Mol. Cell. Endocrinol. 2009, 304, 63–68. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Zheng, T.; Xu, Y.-J.; Yang, M.-N.; Wang, W.-J.; Huang, R.; Zhang, G.-H.; Guo, Y.-N.; Zhang, J.; Ouyang, F.; et al. Sex Dimorphic Associations of Gestational Diabetes Mellitus With Cord Plasma Fatty Acid Binding Protein 4 and Estradiol. Front. Endocrinol. 2021, 12, 740902. [Google Scholar] [CrossRef]
- Joost, H.-G. The Genetic Basis of Obesity and Type 2 Diabetes: Lessons from the New Zealand Obese Mouse, a Polygenic Model of the Metabolic Syndrome. Sens. Metab. Control Energy Balance 2010, 52, 1–11. [Google Scholar] [CrossRef]
- Vogel, H.; Mirhashemi, F.; Liehl, B.; Taugner, F.; Kluth, O.; Kluge, R.; Joost, H.-G.; Schürmann, A. Estrogen Deficiency Aggravates Insulin Resistance and Induces β-Cell Loss and Diabetes in Female New Zealand Obese Mice. Horm. Metab. Res. 2013, 45, 430–435. [Google Scholar] [CrossRef] [Green Version]
- Grupe, K.; Asuaje Pfeifer, M.; Dannehl, F.; Liebmann, M.; Rustenbeck, I.; Schürmann, A.; Scherneck, S. Metabolic Changes during Pregnancy in Glucose-intolerant NZO Mice: A Polygenic Model with Prediabetic Metabolism. Physiol. Rep. 2020, 8, e14417. [Google Scholar] [CrossRef] [PubMed]
- Abbassi-Ghanavati, M.; Greer, L.G.; Cunningham, F.G. Pregnancy and Laboratory Studies: A Reference Table for Clinicians. Obstet Gynecol. 2009, 114, 1326–1331. [Google Scholar] [CrossRef] [PubMed]
- Lubura, M.; Hesse, D.; Kraemer, M.; Hallahan, N.; Schupp, M.; von Löffelholz, C.; Kriebel, J.; Rudovich, N.; Pfeiffer, A.; John, C.; et al. Diabetes Prevalence in NZO Females Depends on Estrogen Action on Liver Fat Content. Am. J. Physiol. Endocrinol. Metab. 2015, 309, E968–E980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, E.L.; Song, Y.; Malik, V.S.; Liu, S. Sex Differences of Endogenous Sex Hormones and Risk of Type 2 Diabetes: A Systematic Review and Meta-Analysis. JAMA 2006, 295, 1288. [Google Scholar] [CrossRef]
- Radavelli-Bagatini, S.; Blair, A.R.; Proietto, J.; Spritzer, P.M.; Andrikopoulos, S. The New Zealand Obese Mouse Model of Obesity Insulin Resistance and Poor Breeding Performance: Evaluation of Ovarian Structure and Function. J. Endocrinol. 2011, 209, 307–315. [Google Scholar] [CrossRef] [Green Version]
- Walters, K.A.; Allan, C.M.; Handelsman, D.J. Rodent Models for Human Polycystic Ovary Syndrome. Biol. Reprod. 2012, 86, 1–12. [Google Scholar] [CrossRef]
- Pagán, Y.L.; Srouji, S.S.; Jimenez, Y.; Emerson, A.; Gill, S.; Hall, J.E. Inverse Relationship between Luteinizing Hormone and Body Mass Index in Polycystic Ovarian Syndrome: Investigation of Hypothalamic and Pituitary Contributions. J. Clin. Endocrinol. Metab. 2006, 91, 1309–1316. [Google Scholar] [CrossRef] [Green Version]
- Santos, R.S.; Batista, T.M.; Camargo, R.L.; Morato, P.N.; Borck, P.C.; Leite, N.C.; Kurauti, M.A.; Wanschel, A.C.B.A.; Nadal, Á.; Clegg, D.J.; et al. Lacking of Estradiol Reduces Insulin Exocytosis from Pancreatic β-Cells and Increases Hepatic Insulin Degradation. Steroids 2016, 114, 16–24. [Google Scholar] [CrossRef]
- Balhuizen, A.; Kumar, R.; Amisten, S.; Lundquist, I.; Salehi, A. Activation of G Protein-Coupled Receptor 30 Modulates Hormone Secretion and Counteracts Cytokine-Induced Apoptosis in Pancreatic Islets of Female Mice. Mol. Cell. Endocrinol. 2010, 320, 16–24. [Google Scholar] [CrossRef] [Green Version]
- Alonso-Magdalena, P.; Ropero, A.B.; Carrera, M.P.; Cederroth, C.R.; Baquié, M.; Gauthier, B.R.; Nef, S.; Stefani, E.; Nadal, A. Pancreatic Insulin Content Regulation by the Estrogen Receptor ERα. PLoS ONE 2008, 3, e2069. [Google Scholar] [CrossRef] [Green Version]
- Kumar, R.; Balhuizen, A.; Amisten, S.; Lundquist, I.; Salehi, A. Insulinotropic and Antidiabetic Effects of 17β-Estradiol and the GPR30 Agonist G-1 on Human Pancreatic Islets. Endocrinology 2011, 152, 2568–2579. [Google Scholar] [CrossRef] [PubMed]
- Costrini, N.V.; Kalkhoff, R.K. Relative Effects of Pregnancy, Estradiol, and Progesterone on Plasma Insulin and Pancreatic Islet Insulin Secretion. J. Clin. Investig. 1971, 50, 992–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soriano, S.; Ropero, A.B.; Alonso-Magdalena, P.; Ripoll, C.; Quesada, I.; Gassner, B.; Kuhn, M.; Gustafsson, J.-Å.; Nadal, A. Rapid Regulation of KATP Channel Activity by 17β-Estradiol in Pancreatic β-Cells Involves the Estrogen Receptor β and the Atrial Natriuretic Peptide Receptor. Mol. Endocrinol. 2009, 23, 1973–1982. [Google Scholar] [CrossRef] [Green Version]
- Mårtensson, U.E.A.; Salehi, S.A.; Windahl, S.; Gomez, M.F.; Swärd, K.; Daszkiewicz-Nilsson, J.; Wendt, A.; Andersson, N.; Hellstrand, P.; Grände, P.-O.; et al. Deletion of the G Protein-Coupled Receptor 30 Impairs Glucose Tolerance, Reduces Bone Growth, Increases Blood Pressure, and Eliminates Estradiol-Stimulated Insulin Release in Female Mice. Endocrinology 2009, 150, 687–698. [Google Scholar] [CrossRef]
- Sharma, G.; Prossnitz, E.R. Mechanisms of Estradiol-Induced Insulin Secretion by the G Protein-Coupled Estrogen Receptor GPR30/GPER in Pancreatic β-Cells. Endocrinology 2011, 152, 3030–3039. [Google Scholar] [CrossRef] [PubMed]
- Faure, A.; Sutter-Dub, M.-T.; Sutter, B.C.J.; Assan, R. Ovarian-Adrenal Interactions in Regulation of Endocrine Pancreatic Function in the Rat. Diabetologia 1983, 24, 122–127. [Google Scholar] [CrossRef] [Green Version]
- Herrera, E.; Amusquivar, E.; López-Soldado, I.; Ortega, H. Maternal Lipid Metabolism and Placental Lipid Transfer. Horm. Res. Paediatr. 2006, 65, 59–64. [Google Scholar] [CrossRef]
- Bryzgalova, G.; Gao, H.; Ahren, B.; Zierath, J.R.; Galuska, D.; Steiler, T.L.; Dahlman-Wright, K.; Nilsson, S.; Gustafsson, J.-A.; Efendic, S.; et al. Evidence That Oestrogen Receptor-Alpha Plays an Important Role in the Regulation of Glucose Homeostasis in Mice: Insulin Sensitivity in the Liver. Diabetologia 2006, 49, 588–597. [Google Scholar] [CrossRef] [Green Version]
- Ferrannini, E.; Gastaldelli, A.; Iozzo, P. Pathophysiology of Prediabetes. Med. Clin. 2011, 95, 327–339. [Google Scholar] [CrossRef] [Green Version]
- Qiu, S.; Vazquez, J.T.; Boulger, E.; Liu, H.; Xue, P.; Hussain, M.A.; Wolfe, A. Hepatic Estrogen Receptor α Is Critical for Regulation of Gluconeogenesis and Lipid Metabolism in Males. Sci. Rep. 2017, 7, 1661. [Google Scholar] [CrossRef] [Green Version]
- Carter, S.; McKenzie, S.; Mourtzakis, M.; Mahoney, D.J.; Tarnopolsky, M.A. Short-Term 17beta-Estradiol Decreases Glucose R(a) but Not Whole Body Metabolism during Endurance Exercise. J. Appl. Physiol. 2001, 90, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Sladek, C.D. The Effects of Human Chorionic Somatomammotropin and Estradiol on Gluconeogenesis and Hepatic Glycogen Formation in the Rat. Horm. Metab. Res. 1975, 7, 50–54. [Google Scholar] [CrossRef]
- Yan, H.; Yang, W.; Zhou, F.; Li, X.; Pan, Q.; Shen, Z.; Han, G.; Newell-Fugate, A.; Tian, Y.; Majeti, R.; et al. Estrogen Improves Insulin Sensitivity and Suppresses Gluconeogenesis via the Transcription Factor Foxo1. Diabetes 2018, 68, 291–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, H.; Mu, J.; Kim, J.K.; Thorvaldsen, J.L.; Chu, Q.; Crenshaw, E.B.; Kaestner, K.H.; Bartolomei, M.S.; Shulman, G.I.; Birnbaum, M.J. Insulin Resistance and a Diabetes Mellitus-Like Syndrome in Mice Lacking the Protein Kinase Akt2 (PKBβ). Science 2001, 292, 1728–1731. [Google Scholar] [CrossRef]
- Dummler, B.; Tschopp, O.; Hynx, D.; Yang, Z.-Z.; Dirnhofer, S.; Hemmings, B.A. Life with a Single Isoform of Akt: Mice Lacking Akt2 and Akt3 Are Viable but Display Impaired Glucose Homeostasis and Growth Deficiencies. Mol. Cell Biol. 2006, 26, 8042–8051. [Google Scholar] [CrossRef] [Green Version]
- Toda, K.; Toda, A.; Ono, M.; Saibara, T. Lack of 17β-Estradiol Reduces Sensitivity to Insulin in the Liver and Muscle of Male Mice. Heliyon 2018, 4, e00772. [Google Scholar] [CrossRef]
- Zhu, L.; Martinez, M.N.; Emfinger, C.H.; Palmisano, B.T.; Stafford, J.M. Estrogen Signaling Prevents Diet-Induced Hepatic Insulin Resistance in Male Mice with Obesity. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E1188–E1197. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.-Y.; Hong, T.; Wen, G.-B.; Han, J.; Zuo, D.; Liu, Z.; Cao, W. Increased Basal Level of Akt-Dependent Insulin Signaling May Be Responsible for the Development of Insulin Resistance. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E898–E906. [Google Scholar] [CrossRef] [Green Version]
- Camporez, J.P.G.; Jornayvaz, F.R.; Lee, H.-Y.; Kanda, S.; Guigni, B.A.; Kahn, M.; Samuel, V.T.; Carvalho, C.R.O.; Petersen, K.F.; Jurczak, M.J.; et al. Cellular Mechanism by Which Estradiol Protects Female Ovariectomized Mice From High-Fat Diet-Induced Hepatic and Muscle Insulin Resistance. Endocrinology 2013, 154, 1021–1028. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, Q.; Kang, S.-G.; Huang, K.; Tong, T. Dietary Bioactive Ingredients Modulating the CAMP Signaling in Diabetes Treatment. Nutrients 2021, 13, 3038. [Google Scholar] [CrossRef]
- Romere, C.; Duerrschmid, C.; Bournat, J.; Constable, P.; Jain, M.; Xia, F.; Saha, P.K.; Del Solar, M.; Zhu, B.; York, B.; et al. Asprosin, a Fasting-Induced Glucogenic Protein Hormone. Cell 2016, 165, 566–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumoto, M.; Pocai, A.; Rossetti, L.; Depinho, R.A.; Accili, D. Impaired Regulation of Hepatic Glucose Production in Mice Lacking the Forkhead Transcription Factor Foxo1 in Liver. Cell Metab. 2007, 6, 208–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakae, J.; Kitamura, T.; Silver, D.L.; Accili, D. The Forkhead Transcription Factor Foxo1 (Fkhr) Confers Insulin Sensitivity onto Glucose-6-Phosphatase Expression. J. Clin. Investig. 2001, 108, 1359–1367. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yu, J.; Zhou, Q.; Wang, X.; Mukhanova, M.; Du, W.; Sun, L.; Pajvani, U.B.; Accili, D. TOX4, an Insulin Receptor-Independent Regulator of Hepatic Glucose Production, Is Activated in Diabetic Liver. Cell Metab. 2022, 34, 158–170. [Google Scholar] [CrossRef] [PubMed]
- Pedram, A.; Razandi, M.; O’Mahony, F.; Harvey, H.; Harvey, B.J.; Levin, E.R. Estrogen Reduces Lipid Content in the Liver Exclusively from Membrane Receptor Signaling. Sci. Signal. 2013, 6, ra36. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Li, F.; Klussmann, E.; Stallone, J.N.; Han, G. G Protein-Coupled Estrogen Receptor 1 Mediates Relaxation of Coronary Arteries via CAMP/PKA-Dependent Activation of MLCP. Am. J. Physiol. Endocrinol. Metab. 2014, 307, E398–E407. [Google Scholar] [CrossRef] [Green Version]
- Zucchetti, A.E.; Barosso, I.R.; Boaglio, A.C.; Basiglio, C.L.; Miszczuk, G.; Larocca, M.C.; Ruiz, M.L.; Davio, C.A.; Roma, M.G.; Crocenzi, F.A.; et al. G-Protein-Coupled Receptor 30/Adenylyl Cyclase/Protein Kinase A Pathway Is Involved in Estradiol 17ß-D-Glucuronide-Induced Cholestasis. Hepatology 2014, 59, 1016–1029. [Google Scholar] [CrossRef]
- Zucchetti, A.E.; Barosso, I.R.; Boaglio, A.; Pellegrino, J.M.; Ochoa, E.J.; Roma, M.G.; Crocenzi, F.A.; Sánchez Pozzi, E.J. Prevention of Estradiol 17beta-D-Glucuronide-Induced Canalicular Transporter Internalization by Hormonal Modulation of CAMP in Rat Hepatocytes. Mol. Biol. Cell 2011, 22, 3902–3915. [Google Scholar] [CrossRef]
- Sharma, G.; Prossnitz, E.R. GPER/GPR30 Knockout Mice: Effects of GPER on Metabolism. Methods Mol. Biol. 2016, 1366, 489–502. [Google Scholar] [CrossRef] [Green Version]
- Prossnitz, E.R.; Barton, M. Estrogen Biology: New Insights into GPER Function and Clinical Opportunities. Mol. Cell Endocrinol. 2014, 389, 71–83. [Google Scholar] [CrossRef] [Green Version]
- Filardo, E.J.; Thomas, P. Minireview: G Protein-Coupled Estrogen Receptor-1, GPER-1: Its Mechanism of Action and Role in Female Reproductive Cancer, Renal and Vascular Physiology. Endocrinology 2012, 153, 2953–2962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Achard, C.S.; Laybutt, D.R. Lipid-Induced Endoplasmic Reticulum Stress in Liver Cells Results in Two Distinct Outcomes: Adaptation with Enhanced Insulin Signaling or Insulin Resistance. Endocrinology 2012, 153, 2164–2177. [Google Scholar] [CrossRef] [PubMed]
- Irimia, J.M.; Meyer, C.M.; Segvich, D.M.; Surendran, S.; DePaoli-Roach, A.A.; Morral, N.; Roach, P.J. Lack of Liver Glycogen Causes Hepatic Insulin Resistance and Steatosis in Mice. J. Biol. Chem. 2017, 292, 10455–10464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demers, L.M.; Jacobs, R.D. Hormonal Regulation of Rat Uterine Glycogen Metabolism. Biol. Reprod. 1973, 9, 272–278. [Google Scholar] [CrossRef] [Green Version]
- Choi, S.; Shin, H.; Song, H.; Lim, H.J. Suppression of Autophagic Activation in the Mouse Uterus by Estrogen and Progesterone. J. Endocrinol. 2014, 221, 39–50. [Google Scholar] [CrossRef]
- MacAulay, K.; Doble, B.W.; Patel, S.; Hansotia, T.; Sinclair, E.M.; Drucker, D.J.; Nagy, A.; Woodgett, J.R. Glycogen Synthase Kinase 3alpha-Specific Regulation of Murine Hepatic Glycogen Metabolism. Cell Metab. 2007, 6, 329–337. [Google Scholar] [CrossRef] [Green Version]
- Nozaki, Y.; Petersen, M.C.; Zhang, D.; Vatner, D.F.; Perry, R.J.; Abulizi, A.; Haedersdal, S.; Zhang, X.-M.; Butrico, G.M.; Samuel, V.T.; et al. Metabolic Control Analysis of Hepatic Glycogen Synthesis in Vivo. Proc. Natl. Acad. Sci. USA 2020, 117, 8166–8176. [Google Scholar] [CrossRef] [Green Version]
- Hatlapatka, K.; Willenborg, M.; Rustenbeck, I. Plasma Membrane Depolarization as a Determinant of the First Phase of Insulin Secretion. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E315–E322. [Google Scholar] [CrossRef]
- Baltrusch, S.; Francini, F.; Lenzen, S.; Tiedge, M. Interaction of Glucokinase With the Liver Regulatory Protein Is Conferred by Leucine-Asparagine Motifs of the Enzyme. Diabetes 2005, 54, 2829–2837. [Google Scholar] [CrossRef] [Green Version]
- Johanns, M.; Lai, Y.-C.; Hsu, M.-F.; Jacobs, R.; Vertommen, D.; Van Sande, J.; Dumont, J.E.; Woods, A.; Carling, D.; Hue, L.; et al. AMPK Antagonizes Hepatic Glucagon-Stimulated Cyclic AMP Signalling via Phosphorylation-Induced Activation of Cyclic Nucleotide Phosphodiesterase 4B. Nat. Commun. 2016, 7, 10856. [Google Scholar] [CrossRef]
- Titchenell, P.M.; Chu, Q.; Monks, B.R.; Birnbaum, M.J. Hepatic Insulin Signalling Is Dispensable for Suppression of Glucose Output by Insulin in Vivo. Nat. Commun. 2015, 6, 7078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prescott, M.J.; Lidster, K. Improving Quality of Science through Better Animal Welfare: The NC3Rs Strategy. Lab. Anim. 2017, 46, 152–156. [Google Scholar] [CrossRef] [PubMed]
- Törnqvist, E.; Annas, A.; Granath, B.; Jalkesten, E.; Cotgreave, I.; Öberg, M. Strategic Focus on 3R Principles Reveals Major Reductions in the Use of Animals in Pharmaceutical Toxicity Testing. PLoS ONE 2014, 9, e101638. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liebmann, M.; Asuaje Pfeifer, M.; Grupe, K.; Scherneck, S. Estradiol (E2) Improves Glucose-Stimulated Insulin Secretion and Stabilizes GDM Progression in a Prediabetic Mouse Model. Int. J. Mol. Sci. 2022, 23, 6693. https://doi.org/10.3390/ijms23126693
Liebmann M, Asuaje Pfeifer M, Grupe K, Scherneck S. Estradiol (E2) Improves Glucose-Stimulated Insulin Secretion and Stabilizes GDM Progression in a Prediabetic Mouse Model. International Journal of Molecular Sciences. 2022; 23(12):6693. https://doi.org/10.3390/ijms23126693
Chicago/Turabian StyleLiebmann, Moritz, Melissa Asuaje Pfeifer, Katharina Grupe, and Stephan Scherneck. 2022. "Estradiol (E2) Improves Glucose-Stimulated Insulin Secretion and Stabilizes GDM Progression in a Prediabetic Mouse Model" International Journal of Molecular Sciences 23, no. 12: 6693. https://doi.org/10.3390/ijms23126693