
Integrative Analysis of Expression Profiles of mRNA and MicroRNA Provides Insights of Cotton Response to Verticillium dahliae

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Differential Expression of mRNAs between J11 and Z2 Response to V. dahliae Stress
2.2. Functional Classifications of DEGs in Response to V. dahliae Stress
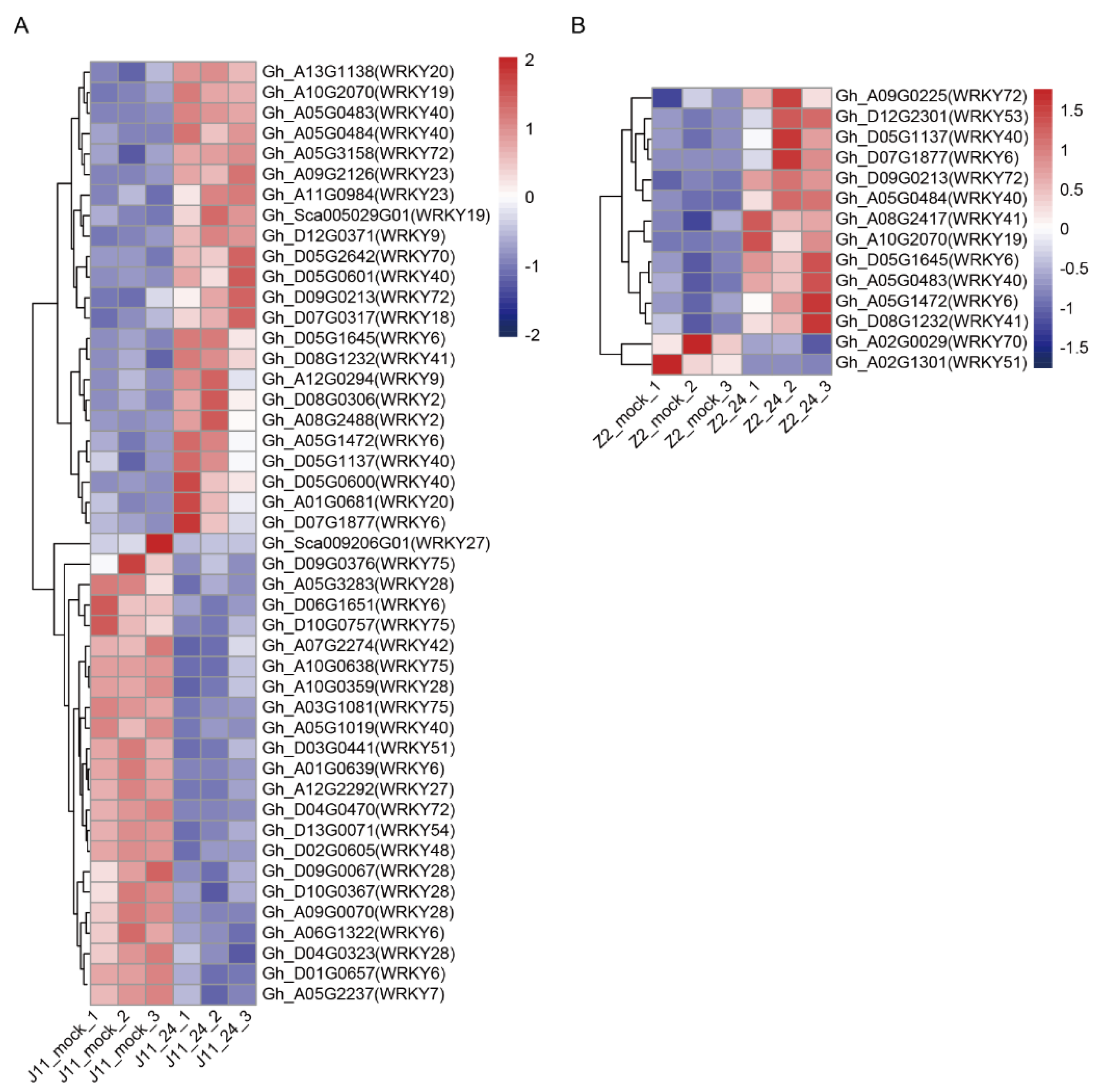
2.3. DEGs of WRKY Transcription Factors in Response to V. dahliae Stress
2.4. Identification of Cotton miRNAs in Response to V. dahliae Stress
2.5. Target Prediction and Construction of a Regulatory and Interaction Network
2.6. Validation of Gene Expression by qRT-PCR
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Pathogen Treatment
4.2. RNA Preparation and Sequencing
4.3. Identification of DEGs and DEMs
4.4. Integrated Analysis of mRNA and miRNA Sequencing
4.5. Expression Analyses of mRNA and miRNA
4.6. 5’-RNA Ligase-Mediated Rapid Amplification of cDNA Ends (5’ RLM-RACE)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yang, Z.; Qanmber, G.; Wang, Z.; Yang, Z.; Li, F. Gossypium genomics: Trends, scope, and utilization for cotton improvement. Trends Plant Sci. 2020, 25, 488–500. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Sun, Y.; Zhu, Q.H. Breeding next-generation naturally colored cotton. Trends Plant Sci. 2021, 26, 539–542. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Ma, X.; Li, N.; Zhou, L.; Liu, Z.; Han, H.; Gui, Y.; Bao, Y.; Chen, J.; Dai, X. Genome-wide association study discovered candidate genes of Verticillium wilt resistance in upland cotton (Gossypium hirsutum L.). Plant Biotechnol. J. 2017, 15, 1520–1532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, R.; Li, J.; Xie, C.; Jian, W.; Yang, X. An overview of the molecular genetics of plant resistance to the Verticillium wilt pathogen Verticillium dahliae. Int. J. Mol. Sci. 2020, 21, 1120. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Sun, L.; Wassan, G.M.; He, X.; Shaban, M.; Zhang, L.; Zhu, L.; Zhang, X. GbSOBIR1 confers Verticillium wilt resistance by phosphorylating the transcriptional factor GbbHLH171 in Gossypium barbadense. Plant Biotechnol. J. 2019, 17, 152–163. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Hu, X.; Wang, P.; Gao, L.; Pei, Y.; Ge, Z.; Ge, X.; Li, F.; Hou, Y. GhPLP2 positively regulates cotton resistance to Verticillium wilt by modulating fatty acid accumulation and jasmonic acid signaling pathway. Front. Plant Sci. 2021, 12, 749630. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wu, L.; Wang, X.; Chen, B.; Zhao, J.; Cui, J.; Li, Z.; Yang, J.; Wu, L.; Wu, J.; et al. The cotton laccase gene GhLAC15 enhances Verticillium wilt resistance via an increase in defence-induced lignification and lignin components in the cell walls of plants. Mol. Plant Pathol. 2019, 20, 309–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, M.; Zhang, Z.; Ren, Z.; Wang, X.; Sun, W.; Feng, H.; Zhao, J.; Zhang, F.; Li, W.; Ma, X.; et al. The GhSWEET42 glucose transporter participates in Verticillium dahliae infection in cotton. Front. Plant Sci. 2021, 12, 690754. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Zhou, L.; Jamieson, P.; Zhang, L.; Zhao, Z.; Babilonia, K.; Shao, W.; Wu, L.; Mustafa, R.; Amin, I.; et al. The cotton wall-associated kinase GhWAK7A mediates responses to fungal wilt pathogens by complexing with the chitin sensory receptors. Plant Cell 2020, 32, 3978–4001. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Zhu, L.; Zhang, X.; Guan, Q.; Xiao, S.; Min, L.; Zhang, X. GhCPK33 negatively regulates defense against Verticillium dahliae by phosphorylating GhOPR3. Plant Physiol. 2018, 178, 876–889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Y.; Zhai, Y.; Li, L.; Yang, Z.; Ge, X.; Yang, Z.; Zhang, C.; Li, F.; Ren, M. BIN2 negatively regulates plant defence against Verticillium dahliae in Arabidopsis and cotton. Plant Biotechnol. J. 2021, 19, 2097–2112. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Wang, G.; Huang, Z.; Hu, L.; Fu, T.; Wang, X. Silencing GhIAA43, a member of cotton AUX/IAA genes, enhances wilt resistance via activation of salicylic acid-mediated defenses. Plant Sci. 2022, 314, 111126. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Zhao, Y.L.; Zhao, J.H.; Wang, S.; Jin, Y.; Chen, Z.Q.; Fang, Y.Y.; Hua, C.L.; Ding, S.W.; Guo, H.S. Cotton plants export microRNAs to inhibit virulence gene expression in a fungal pathogen. Nat. Plants 2016, 2, 16153. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Li, Y.; Cao, X.; Qi, Y. MicroRNAs and their regulatory roles in plant-environment interactions. Annu. Rev. Plant Biol. 2019, 70, 489–525. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Sun, Q.; Jiang, H.; Zhu, X.; Mo, J.; Long, L.; Xiang, L.; Xie, Y.; Shi, Y.; Yuan, Y.; et al. Identification of novel microRNAs in the Verticillium wilt-resistant upland cotton variety KV-1 by high-throughput sequencing. SpringerPlus 2014, 3, 564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Wang, W.; Chen, J.; Liu, J.; Xia, M.; Shen, F. Identification of miRNAs and their targets in cotton inoculated with Verticillium dahliae by high-throughput sequencing and degradome analysis. Int. J. Mol. Sci. 2015, 16, 14749–14768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, G.; Hao, M.; Wang, L.; Liu, J.; Zhang, Z.; Tang, Y.; Peng, Q.; Yang, Z.; Wu, J. The cotton miR477-CBP60A module participates in plant defense against Verticillium dahlia. Mol. Plant Microbe. Interact. 2020, 33, 624–636. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Lei, Y.; Liu, J.; Hao, M.; Zhang, Z.; Tang, Y.; Chen, A.; Wu, J. The ghr-miR164 and GhNAC100 modulate cotton plant resistance against Verticillium dahlia. Plant Sci. 2020, 293, 110438. [Google Scholar] [CrossRef]
- Wei, T.; Tang, Y.; Jia, P.; Zeng, Y.; Wang, B.; Wu, P.; Quan, Y.; Chen, A.; Li, Y.; Wu, J. A cotton lignin biosynthesis gene, GhLAC4, fine-tuned by ghr-miR397 modulates plant resistance against Verticillium dahliae. Front. Plant Sci. 2021, 12, 743795. [Google Scholar] [CrossRef]
- Wani, S.H.; Anand, S.; Singh, B.; Bohra, A.; Joshi, R. WRKY transcription factors and plant defense responses: Latest discoveries and future prospects. Plant Cell Rep. 2021, 40, 1071–1085. [Google Scholar] [CrossRef]
- Abeysinghe, J.K.; Lam, K.M.; Ng, D.W. Differential regulation and interaction of homoeologous WRKY18 and WRKY40 in Arabidopsis allotetraploids and biotic stress responses. Plant J. 2019, 97, 352–367. [Google Scholar] [CrossRef] [PubMed]
- Segal, L.M.; Wilson, R.A. Reactive oxygen species metabolism and plant-fungal interactions. Fungal Genet. Biol. 2018, 110, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Zechmann, B. Subcellular roles of glutathione in mediating plant defense during biotic stress. Plants 2020, 9, 1067. [Google Scholar] [CrossRef]
- Steinberg, G. Tracks for traffic: Microtubules in the plant pathogen Ustilago maydis. New Phytol. 2007, 174, 721–733. [Google Scholar] [CrossRef] [PubMed]
- Erickson, J.L.; Adlung, N.; Lampe, C.; Bonas, U.; Schattat, M.H. The Xanthomonas effector XopL uncovers the role of microtubules in stromule extension and dynamics in Nicotiana benthamiana. Plant J. 2018, 93, 856–870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verbon, E.H.; Trapet, P.L.; Stringlis, I.A.; Kruijs, S.; Bakker, P.A.H.M.; Pieterse, C.M.J. Iron and Immunity. Annu Rev. Phytopathol. 2017, 55, 355–375. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.; Wei, Z.; Shao, Z.; Friman, V.P.; Cao, K.; Yang, T.; Kramer, J.; Wang, X.; Li, M.; Mei, X.; et al. Competition for iron drives phytopathogen control by natural rhizosphere microbiomes. Nat. Microbiol. 2020, 5, 1002–1010. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Fernando, W.G.D. Hormonal responses to susceptible, intermediate, and resistant interactions in the Brassica napus-Leptosphaeria maculans pathosystem. Int. J. Mol. Sci. 2021, 22, 4714. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; He, X.; Li, Y.; Wang, L.; Guo, X.; Guo, X. The cotton MAPK kinase GhMPK20 negatively regulates resistance to Fusarium oxysporum by mediating the MKK4-MPK20-WRKY40 cascade. Mol. Plant Pathol. 2018, 19, 1624–1638. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Li, L.; Zhou, K.; Zhang, Y.; Han, X.; Din, Y.; Ge, X.; Qin, W.; Wang, P.; Li, F.; et al. GhWRKY6 acts as a negative regulator in both transgenic Arabidopsis and cotton during drought and salt stress. Front. Genet. 2019, 10, 392. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Zhao, P.; Fang, Y.Y.; Gao, F.; Guo, H.S.; Zhao, J.H. Genome-wide profiling of sRNAs in the Verticillium dahliae-infected Arabidopsis roots. Mycology 2018, 9, 155–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Zhang, L.; Yang, Y.; Schmid, M.; Wang, Y. MiRNA mediated regulation and interaction between plants and pathogens. Int. J. Mol. Sci. 2021, 22, 2913. [Google Scholar] [CrossRef]
- Künstler, A.; Gullner, G.; Ádám, A.L.; Kolozsváriné Nagy, J.; Király, L. The versatile roles of sulfur-containing biomolecules in plant defense-a road to disease resistance. Plants 2020, 9, 1705. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Hui, S.; Lv, Y.; Zhang, M.; Chen, D.; Tian, J.; Zhang, H.; Liu, H.; Cao, J.; Xie, W.; et al. MiR395-regulated sulfate metabolism exploits pathogen sensitivity to sulfate to boost immunity in rice. Mol. Plant 2022, 15, 671–688. [Google Scholar] [CrossRef] [PubMed]
- Du, Q.; Wang, H. The role of HD-ZIP III transcription factors and miR165/166 in vascular development and secondary cell wall formation. Plant Signal. Behav. 2015, 10, e1078955. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Wu, Y.; Zhang, X.; Zhao, L.; Feng, Z.; Wei, F.; Zhang, Y.; Feng, H.; Zhou, Y.; Zhu, H. MPK homolog GhNTF6 was involved in cotton against Verticillium wilt by interacted with VdEPG1. Int. J. Biol. Macromol. 2022, 195, 456–465. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Ke, L.; Zhang, D.; Wu, Y.; Sun, Y.; Mei, J.; Sun, J.; Sun, Y. Multi-omics assisted identification of the key and species-specific regulatory components of drought-tolerant mechanisms in Gossypium stocksii. Plant Biotechnol. J. 2021, 19, 1690–1692. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zhang, D.; Zheng, H.; Wu, Y.; Mei, J.; Ke, L.; Yu, D.; Sun, Y. Biochemical and expression analyses revealed the involvement of proanthocyanidins and/or their derivatives in fiber pigmentation of Gossypium stocksii. Int. J. Mol. Sci. 2022, 23, 1008. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mei, J.; Wu, Y.; Niu, Q.; Miao, M.; Zhang, D.; Zhao, Y.; Cai, F.; Yu, D.; Ke, L.; Feng, H.; et al. Integrative Analysis of Expression Profiles of mRNA and MicroRNA Provides Insights of Cotton Response to Verticillium dahliae. Int. J. Mol. Sci. 2022, 23, 4702. https://doi.org/10.3390/ijms23094702
Mei J, Wu Y, Niu Q, Miao M, Zhang D, Zhao Y, Cai F, Yu D, Ke L, Feng H, et al. Integrative Analysis of Expression Profiles of mRNA and MicroRNA Provides Insights of Cotton Response to Verticillium dahliae. International Journal of Molecular Sciences. 2022; 23(9):4702. https://doi.org/10.3390/ijms23094702
Chicago/Turabian StyleMei, Jun, Yuqing Wu, Qingqing Niu, Meng Miao, Diandian Zhang, Yanyan Zhao, Fangfang Cai, Dongliang Yu, Liping Ke, Hongjie Feng, and et al. 2022. "Integrative Analysis of Expression Profiles of mRNA and MicroRNA Provides Insights of Cotton Response to Verticillium dahliae" International Journal of Molecular Sciences 23, no. 9: 4702. https://doi.org/10.3390/ijms23094702