
Melatonin-Induced Postconditioning Suppresses NMDA Receptor through Opening of the Mitochondrial Permeability Transition Pore via Melatonin Receptor in Mouse Neurons

 , , and
, , and 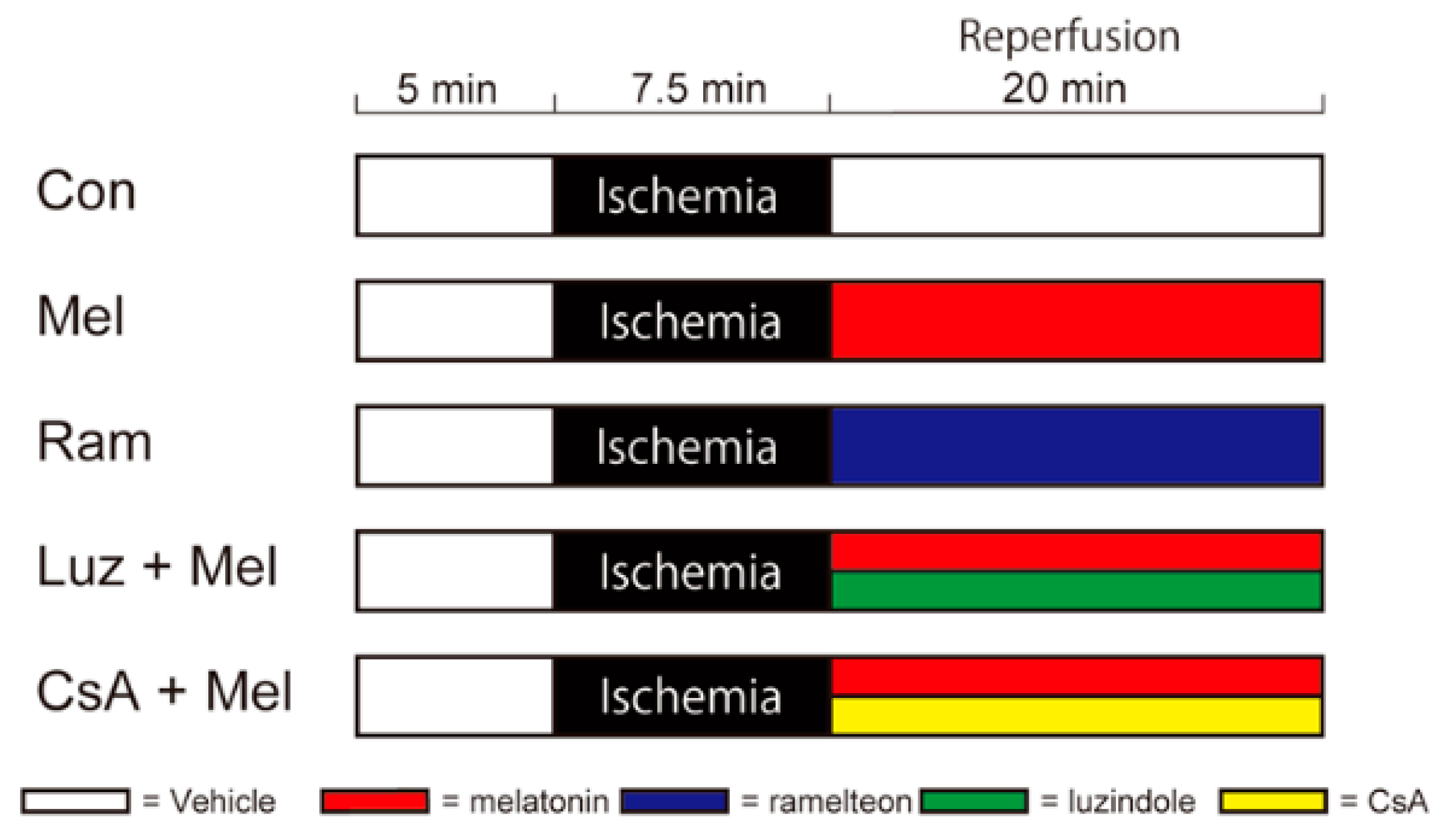{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
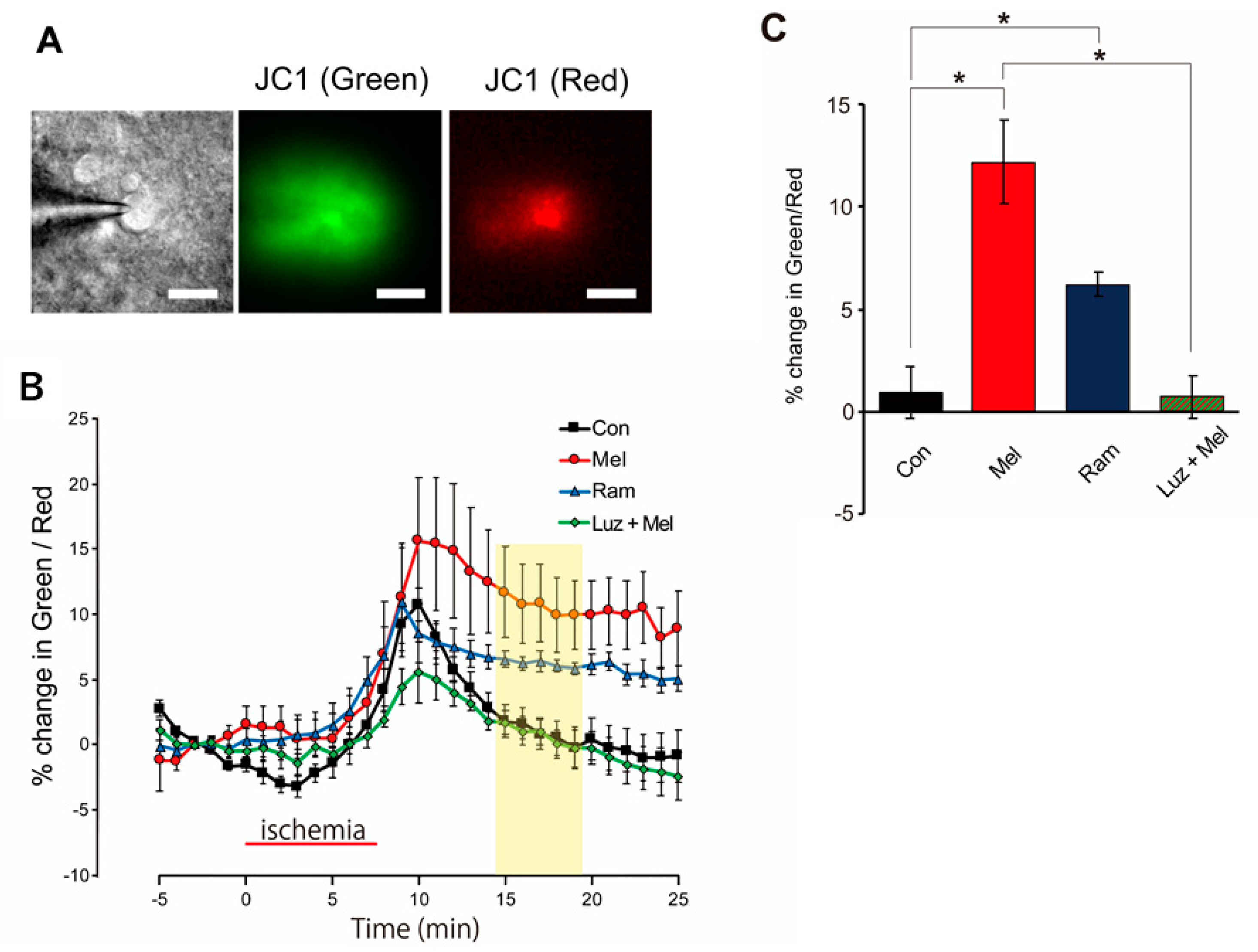{kind=link}
Abstract
:1. Introduction
2. Results
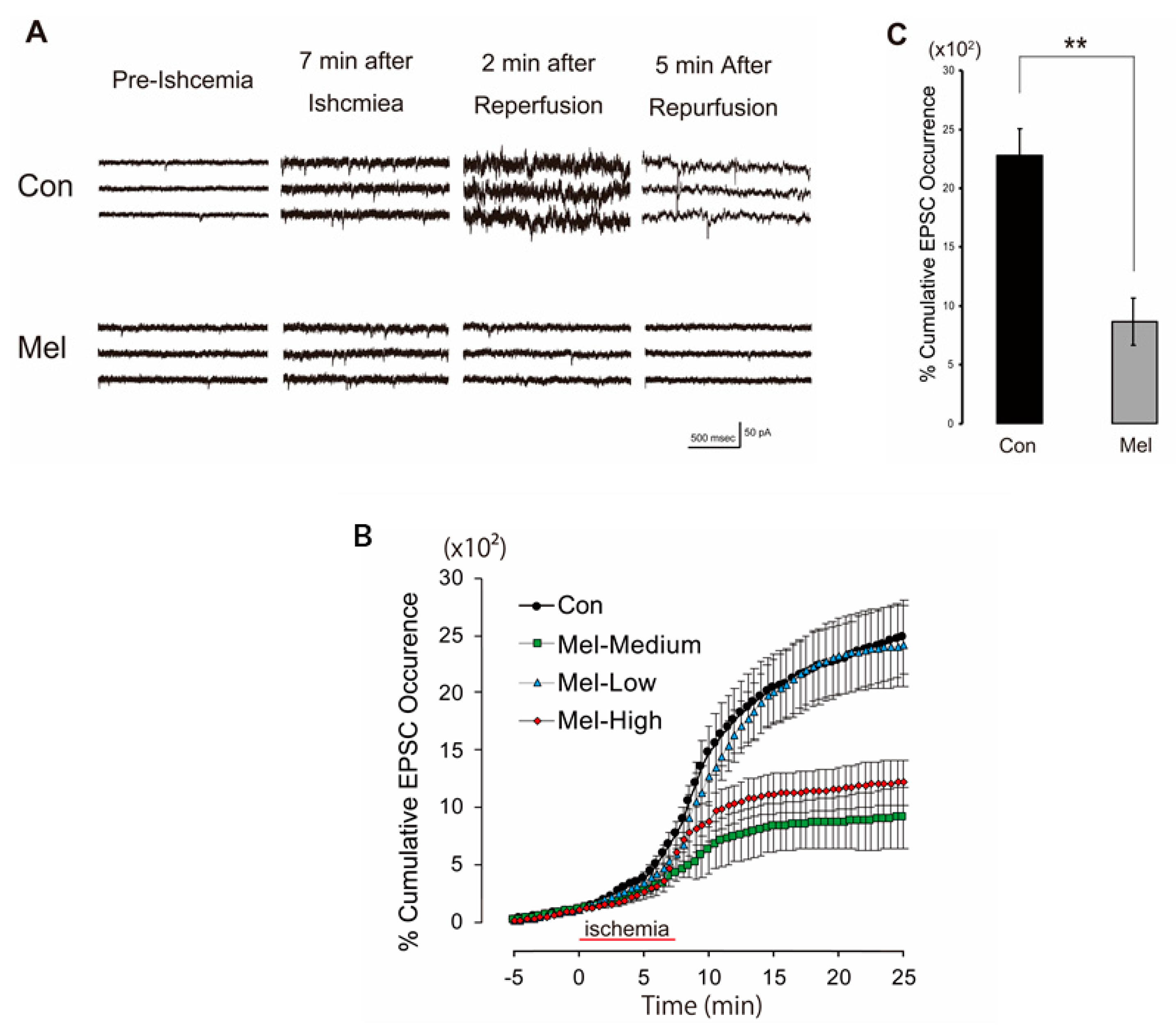
2.1. Melatonin-Induced PostC Suppresses the Surge of sEPSCs
2.2. Melatonin-Induced PostC Reduces the Number of Dead Hippocampal CA1 Neurons
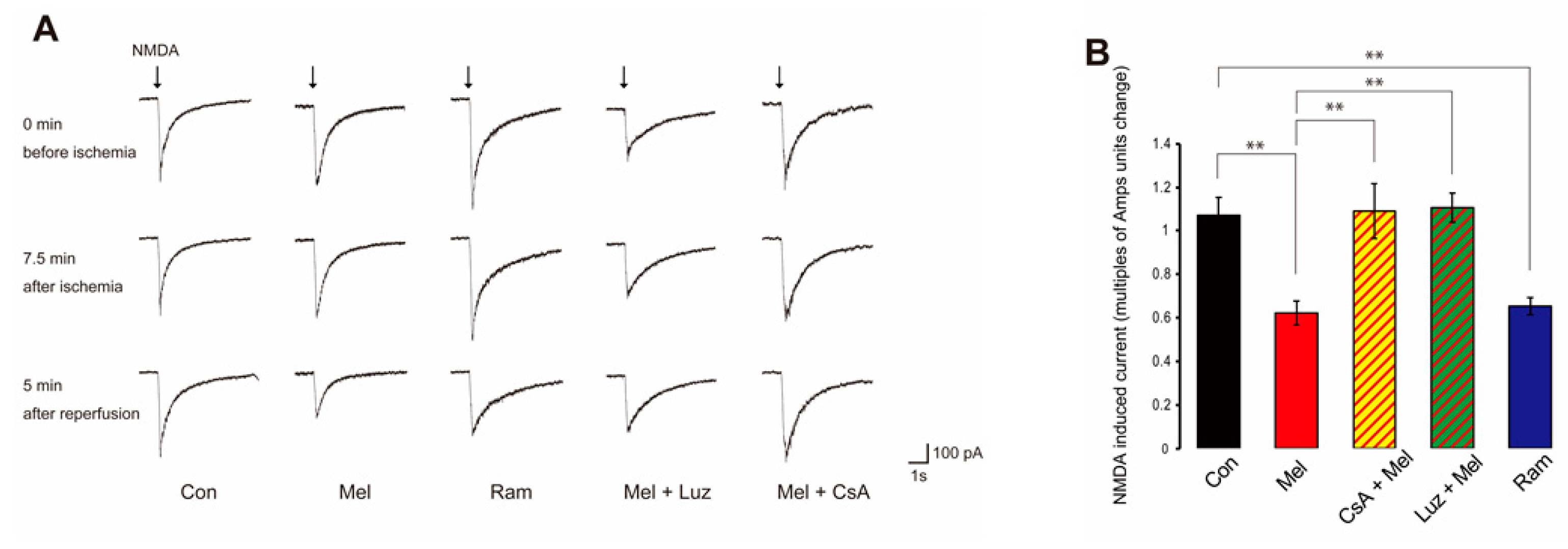
2.3. Melatonin-Induced PostC Silences NMDAR Currents after Reperfusion
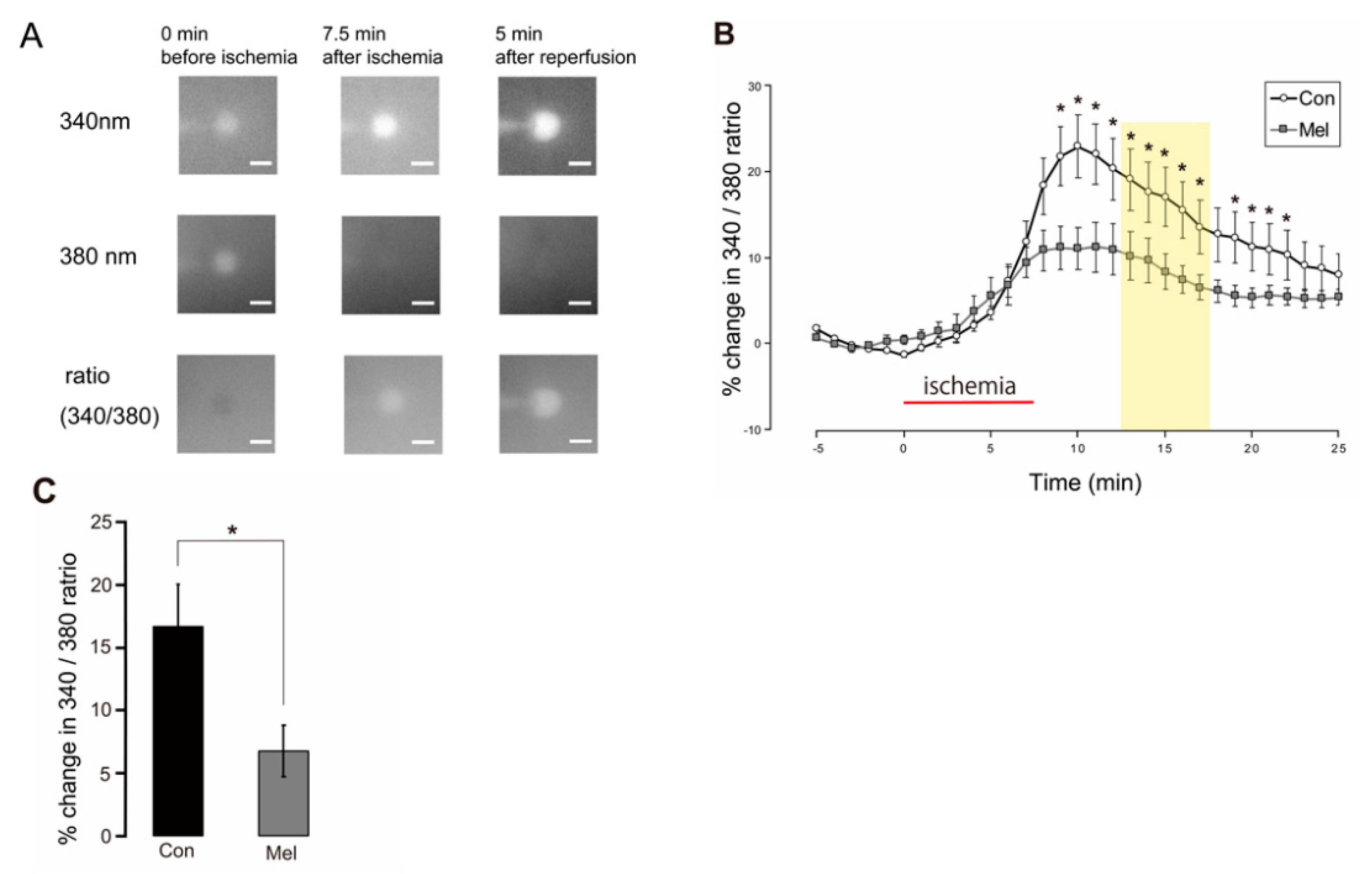
2.4. Postconditioning Suppresses Cytosolic Ca2+ Increase
2.5. Mitochondria Temporarily Depolarize during Melatonin-Induced PostC
3. Discussion
3.1. Melatonin-Induced PostC Reduces Excessive Accumulation of Extracellular Glutamate
3.2. Melatonin Suppresses Influx of Extracellular Ca2+ by Downregulating NMDARs after Ischemia
3.3. Melatonin Leads to Neuroprotection by Putting mPTP into Low-Conductance Mode
3.4. Role of Melatonin Receptors in Melatonin-Induced PostC
3.5. Conductance Control of mPTP and Melatonin-Induced PostC Mechanism
4. Materials and Methods
4.1. Preparation of Mouse Hippocampal Slices
4.2. Patch-Clamp Recording
4.3. Simulating Ischemia and Pharmacological Postconditioning in Hippocampal Slices
4.4. Perfusion Protocols
4.5. Recording of Whole-Cell Current Responses to NMDA Application
4.6. Fluorometric Evaluation of Cytosolic Ca2+ Changes
4.7. Fluorometric Evaluation of Mitochondrial Membrane Potential
4.8. Cell Staining
4.9. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kitagawa, K.; Matsumoto, M.; Tagaya, M.; Hata, R.; Ueda, H.; Niinobe, M.; Handa, N.; Fukunaga, R.; Kimura, K.; Mikoshiba, K.; et al. “Ischemic tolerance” phenomenon found in the brain. Brain Res. 1990, 528, 21–24. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, I.; Nakase, H.; Aketa, S.; Kamada, Y.; Yamashita, M.; Sakaki, T. ATP-dependent potassium channel mediates neuroprotection by chemical preconditioning with 3-nitropropionic acid in gerbil hippocampus. Neurosci. Lett. 2002, 320, 33–36. [Google Scholar] [CrossRef]
- Yin, X.H.; Zhang, Q.G.; Miao, B.; Zhang, G.Y. Neuroprotective effects of preconditioning ischaemia on ischaemic brain injury through inhibition of mixed-lineage kinase 3 via NMDA receptor-mediated Akt1 activation. J. Neurochem. 2005, 93, 1021–1029. [Google Scholar] [CrossRef] [PubMed]
- Xing, B.; Chen, H.; Zhang, M.; Zhao, D.; Jiang, R.; Liu, X.; Zhang, S. Ischemic post-conditioning protects brain and reduces inflammation in a rat model of focal cerebral ischemia/reperfusion. J. Neurochem. 2008, 105, 1737–1745. [Google Scholar] [CrossRef] [PubMed]
- Pignataro, G.; Scorziello, A.; Di Renzo, G.; Annunziato, L. Post-ischemic brain damage: Effect of ischemic preconditioning and postconditioning and identification of potential candidates for stroke therapy. FEBS J. 2009, 276, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Hawrysh, P.J.; Buck, L.T. Anoxia-Mediated calcium release through the mitochondrial permeability transition pore silences NMDA receptor currents in turtle neurons. J. Exp. Biol. 2013, 216, 4375–4387. [Google Scholar] [CrossRef] [Green Version]
- Brenner, C.; Moulin, M. Physiological roles of the permeability transition pore. Circ. Res. 2012, 111, 1237–1247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baines, C.P. The mitochondrial permeability transition pore and ischemia-reperfusion injury. Basic Res. Cardiol. 2009, 104, 181–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warne, J.; Pryce, G.; Hill, J.M.; Shi, X.; Lennerås, F.; Puentes, F.; Kip, M.; Hilditch, L.; Walker, P.; Simone, M.I.; et al. Selective inhibition of the mitochondrial permeability transition pore protects against neurodegeneration in experimental multiple sclerosis. J. Biol. Chem. 2016, 291, 4356–4373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokoyama, S.; Nakagawa, I.; Ogawa, Y.; Morisaki, Y.; Motoyama, Y.; Park, Y.S.; Saito, Y.; Nakase, H. Ischemic postconditioning prevents surge of presynaptic glutamate release by activating mitochondrial ATP-dependent potassium channels in the mouse hippocampus. PLoS ONE 2019, 14, e0215104. [Google Scholar] [CrossRef] [Green Version]
- Morisaki, Y.; Nakagawa, I.; Ogawa, Y.; Yokoyama, S.; Furuta, T.; Saito, Y.; Nakase, H. Ischemic Postconditioning Reduces NMDA Receptor Currents Through the Opening of the Mitochondrial Permeability Transition Pore and KATP Channel in Mouse Neurons. Cell. Mol. Neurobiol. 2020, 42, 1079–1089. [Google Scholar] [CrossRef] [PubMed]
- Shake, J.G.; Peck, E.A.; Marban, E.; Gott, V.L.; Johnston, M.V.; Troncoso, J.C.; Redmond, J.M.; Baumgartner, W.A. Pharmacologically induced preconditioning with diazoxide: A novel approach to brain protection. Ann. Thorac. Surg. 2001, 72, 1849–1854. [Google Scholar] [CrossRef]
- Farkas, E.; Timmer, N.M.; Domoki, F.; Mihály, A.; Luiten, P.G.M.; Bari, F. Post-ischemic administration of diazoxide attenuates long-term microglial activation in the rat brain after permanent carotid artery occlusion. Neurosci. Lett. 2005, 387, 168–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, L.; Shen, F.; Lin, L.; Zhang, X.; Bruce, I.C.; Xia, Q. The neuroprotection conferred by activating the mitochondrial ATP-sensitive K+ channel is mediated by inhibiting the mitochondrial permeability transition pore. Neurosci. Lett. 2006, 402, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Kumar, G.K.; Dastoor, F.C.; Robayo, J.R.; Razzaque, M.A. Side effects of diazoxide. JAMA 1976, 235, 275–276. [Google Scholar] [CrossRef]
- Claustrat, B.; Leston, J. Melatonin: Physiological effects in humans. Neurochirurgie 2015, 61, 77–84. [Google Scholar] [CrossRef]
- Patiño, P.; Parada, E.; Farré-Alins, V.; Molz, S.; Cacabelos, R.; Marco-Contelles, J.; López, M.G.; Tasca, C.I.; Ramos, E.; Romero, A.; et al. Melatonin protects against oxygen and glucose deprivation by decreasing extracellular glutamate and Nox-derived ROS in rat hippocampal slices. Neurotoxicology 2016, 57, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Hardeland, R.; Cardinali, D.P.; Brown, G.M.; Pandi-Perumal, S.R. Melatonin and brain inflammaging. Prog. Neurobiol. 2015, 127–128, 46–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pazar, A.; Kolgazi, M.; Memisoglu, A.; Bahadir, E.; Sirvanci, S.; Yaman, A.; Yeğen, B.; Ozek, E. The neuroprotective and anti-apoptotic effects of melatonin on hemolytic hyperbilirubinemia-induced oxidative brain damage. J. Pineal Res. 2016, 60, 74–83. [Google Scholar] [CrossRef]
- Roohbakhsh, A.; Shamsizadeh, A.; Hayes, A.W.; Reiter, R.J.; Karimi, G. Melatonin as an endogenous regulator of diseases: The role of autophagy. Pharmacol. Res. 2018, 133, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Andersen, L.P.H.; Gögenur, I.; Rosenberg, J.; Reiter, R.J. The Safety of Melatonin in Humans. Clin. Drug Investig. 2016, 36, 169–175. [Google Scholar] [CrossRef]
- Suofu, Y.; Li, W.; Jean-Alphonse, F.G.; Jia, J.; Khattar, N.K.; Li, J.; Baranov, S.V.; Leronni, D.; Mihalik, A.C.; He, Y.; et al. Dual role of mitochondria in producing melatonin and driving GPCR signaling to block cytochrome c release. Proc. Natl. Acad. Sci. USA 2017, 114, E7997–E8006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, H.Y.; Ng, K.Y.; Koh, R.Y.; Chye, S.M. Pharmacological Effects of Melatonin as Neuroprotectant in Rodent Model: A Review on the Current Biological Evidence. Cell. Mol. Neurobiol. 2020, 40, 25–51. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Clough, S.J.; Hutchinson, A.J.; Adamah-Biassi, E.B.; Popovska-Gorevski, M.; Dubocovich, M.L. MT1 and MT2 Melatonin Receptors: A Therapeutic Perspective. Annu. Rev. Pharmacol. Toxicol. 2016, 56, 361–383. [Google Scholar] [CrossRef] [Green Version]
- Ma, Z.; Xin, Z.; Di, W.; Yan, X.; Li, X.; Reiter, R.J.; Yang, Y. Melatonin and mitochondrial function during ischemia/reperfusion injury. Cell. Mol. Life Sci. 2017, 74, 3989–3998. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Lu, C.; Li, T.; Wang, W.; Ye, W.; Zeng, R.; Ni, L.; Lai, Z.; Wang, X.; Liu, C. The protective effect of melatonin on brain ischemia and reperfusion in rats and humans: In vivo assessment and a randomized controlled trial. J. Pineal Res. 2018, 65, e12521. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.L.; Lu, S.S.; Liu, M.R.; Tang, W.D.; Chen, J.Z.; Zheng, Y.R.; Ahsan, A.; Cao, M.; Jiang, L.; Hu, W.W.; et al. Melatonin receptor agonist ramelteon attenuates mouse acute and chronic ischemic brain injury. Acta Pharmacol. Sin. 2020, 41, 1016–1024. [Google Scholar] [CrossRef] [PubMed]
- Bonova, P.; Burda, J.; Danielisova, V.; Nemethova, M.; Gottlieb, M. Delayed post-conditioning reduces post-ischemic glutamate level and improves protein synthesis in brain. Neurochem. Int. 2013, 62, 854–860. [Google Scholar] [CrossRef] [PubMed]
- Dávalos, A.; Castillo, J.; Serena, J.; Noya, M. Duration of glutamate release after acute ischemic stroke. Stroke 1997, 28, 708–710. [Google Scholar] [CrossRef] [PubMed]
- Soria, F.N.; Pérez-Samartín, A.; Martin, A.; Gona, K.B.; Llop, J.; Szczupak, B.; Chara, J.C.; Matute, C.; Domercq, M. Extrasynaptic glutamate release through cystine/glutamate antiporter contributes to ischemic damage. J. Clin. Investig. 2014, 124, 3645–3655. [Google Scholar] [CrossRef] [Green Version]
- Kristián, T.; Siesjö, B.K. Calcium in ischemic cell death. Stroke 1998, 29, 705–718. [Google Scholar] [CrossRef] [PubMed]
- Lau, A.; Tymianski, M. Glutamate receptors, neurotoxicity and neurodegeneration. Pflug. Arch. Eur. J. Physiol. 2010, 460, 525–542. [Google Scholar] [CrossRef]
- Curcio, M.; Salazar, I.L.; Mele, M.; Canzoniero, L.M.T.; Duarte, C.B. Calpains and neuronal damage in the ischemic brain: The swiss knife in synaptic injury. Prog. Neurobiol. 2016, 143, 1–35. [Google Scholar] [CrossRef] [PubMed]
- Eulenburg, V.; Gomeza, J. Neurotransmitter transporters expressed in glial cells as regulators of synapse function. Brain Res. Rev. 2010, 63, 103–112. [Google Scholar] [CrossRef]
- Lehre, K.P.; Danbolt, N.C. The number of glutamate transport subtype molecules at glutamatergic synapses: Chemical and stereological quantification in young adult rat brain. J. Neurosci. 1998, 18, 8751–8757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torp, R.; Lekieffre, D.; Levy, L.M.; Haug, F.M.; Danbolt, N.C.; Meldrum, B.S.; Ottersen, O.P. Reduced postischemic expression of a glial glutamate transporter, GLT1, in the rat hippocampus. Exp. Brain Res. 1995, 103, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Harvey, B.K.; Airavaara, M.; Hinzman, J.; Wires, E.M.; Chiocco, M.J.; Howard, D.B.; Shen, H.; Gerhardt, G.; Hoffer, B.J.; Wang, Y. Targeted over-expression of glutamate transporter 1 (GLT-1) reduces ischemic brain injury in a rat model of stroke. PLoS ONE 2011, 6, e22135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.N.; Hao, L.; Guo, Y.S.; Wang, H.Y.; Li, L.L.; Liu, L.Z.; Li, W. Bin Are glutamate transporters neuroprotective or neurodegenerative during cerebral ischemia? J. Mol. Med. 2019, 97, 281–289. [Google Scholar] [CrossRef]
- Kirichok, Y.; Krapivinsky, G.; Clapham, D.E. The mitochondrial calcium uniporter is a highly selective ion channel. Nature 2004, 427, 360–364. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Yao, Y.; Guo, M.; Li, J.; Yang, P.; Xu, H.; Lin, D. Sevoflurane increases intracellular calcium to induce mitochondrial injury and neuroapoptosis. Toxicol. Lett. 2021, 336, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Pan, M.; Ma, J.; Song, X.; Cao, W.; Zhang, P. Recent progress in the use of mitochondrial membrane permeability transition pore in mitochondrial dysfunction-related disease therapies. Mol. Cell. Biochem. 2021, 476, 493–506. [Google Scholar] [CrossRef] [PubMed]
- Starkov, A.A.; Chinopoulos, C.; Fiskum, G. Mitochondrial calcium and oxidative stress as mediators of ischemic brain injury. Cell Calcium 2004, 36, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Gouriou, Y.; Demaurex, N.; Bijlenga, P.; De Marchi, U. Mitochondrial calcium handling during ischemia-induced cell death in neurons. Biochimie 2011, 93, 2060–2067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, D.W. Ionic dependence of glutamate neurotoxicity. J. Neurosci. 1987, 7, 369–379. [Google Scholar] [CrossRef] [Green Version]
- Escames, G.; León, J.; López, L.C.; Acuña-Castroviejo, D. Mechanisms of N-methyl-D-aspartate receptor inhibition by melatonin in the rat striatum. J. Neuroendocrinol. 2004, 16, 929–935. [Google Scholar] [CrossRef] [PubMed]
- Szabo, I.; Bernardi, P.; Zoratti, M. Modulation of the mitochondrial megachannel by divalent cations and protons. J. Biol. Chem. 1992, 267, 2940–2946. [Google Scholar] [CrossRef]
- Rasola, A.; Bernardi, P. The mitochondrial permeability transition pore and its involvement in cell death and in disease pathogenesis. Apoptosis 2007, 12, 815–833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giorgio, V.; Von Stockum, S.; Antoniel, M.; Fabbro, A.; Fogolari, F.; Forte, M.; Glick, G.D.; Petronilli, V.; Zoratti, M.; Szabó, I.; et al. Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc. Natl. Acad. Sci. USA 2013, 110, 5887–5892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez, M.J.; Quintanilla, R.A. Development or disease: Duality of the mitochondrial permeability transition pore. Dev. Biol. 2017, 426, 1–7. [Google Scholar] [CrossRef]
- Li, S.; Wang, T.; Zhai, L.; Ge, K.; Zhao, J.; Cong, W.; Guo, Y. Picroside II Exerts a Neuroprotective Effect by Inhibiting mPTP Permeability and EndoG Release after Cerebral Ischemia/Reperfusion Injury in Rats. J. Mol. Neurosci. 2018, 64, 144–155. [Google Scholar] [CrossRef]
- Ow, Y.L.P.; Green, D.R.; Hao, Z.; Mak, T.W. Cytochrome c: Functions beyond respiration. Nat. Rev. Mol. Cell Biol. 2008, 9, 532–542. [Google Scholar] [CrossRef]
- Neginskaya, M.A.; Solesio, M.E.; Berezhnaya, E.V.; Amodeo, G.F.; Mnatsakanyan, N.; Jonas, E.A.; Pavlov, E. V ATP Synthase C-Subunit-Deficient Mitochondria Have a Small Cyclosporine A-Sensitive Channel, but Lack the Permeability Transition Pore. Cell Rep. 2019, 26, 11–17.e2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giorgio, V.; Bisetto, E.; Soriano, M.E.; Dabbeni-Sala, F.; Basso, E.; Petronilli, V.; Forte, M.A.; Bernardi, P.; Lippe, G. Cyclophilin D modulates mitochondrial F0F1-ATP synthase by interacting with the lateral stalk of the complex. J. Biol. Chem. 2009, 284, 33982–33988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chinopoulos, C.; Konràd, C.; Kiss, G.; Metelkin, E.; Töröcsik, B.; Zhang, S.F.; Starkov, A.A. Modulation of F0F1-ATP synthase activity by cyclophilin D regulates matrix adenine nucleotide levels. FEBS J. 2011, 278, 1112–1125. [Google Scholar] [CrossRef] [Green Version]
- Bernardi, P. The mitochondrial permeability transition pore: A mystery solved? Front. Physiol. 2013, 4, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giorgio, V.; Soriano, M.E.; Basso, E.; Bisetto, E.; Lippe, G.; Forte, M.A.; Bernardi, P. Cyclophilin D in mitochondrial pathophysiology. Biochim. Biophys. Acta-Bioenerg. 2010, 1797, 1113–1118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nighoghossian, N.; Ovize, M.; Mewton, N.; Ong, E.; Cho, T.H. Cyclosporine A, a Potential Therapy of Ischemic Reperfusion Injury. A Common History for Heart and Brain. Cerebrovasc. Dis. 2016, 42, 309–318. [Google Scholar] [CrossRef]
- Sun, J.; Luan, Q.; Dong, H.; Song, W.; Xie, K.; Hou, L.; Xiong, L. Inhibition of mitochondrial permeability transition pore opening contributes to the neuroprotective effects of ischemic postconditioning in rats. Brain Res. 2012, 1436, 101–110. [Google Scholar] [CrossRef]
- Wang, H.; Chen, S.; Zhang, Y.; Xu, H.; Sun, H. Electroacupuncture ameliorates neuronal injury by Pink1/Parkin-mediated mitophagy clearance in cerebral ischemia-reperfusion. Nitric Oxide-Biol. Chem. 2019, 91, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Okahara, A.; Koga, J.; Matoba, T.; Fujiwara, M.; Tokutome, M.; Ikeda, G.; Nakano, K.; Tachibana, M.; Ago, T.; Kitazono, T.; et al. Simultaneous targeting of mitochondria and monocytes enhances neuroprotection against ischemia–reperfusion injury. Sci. Rep. 2020, 10, 14435. [Google Scholar] [CrossRef] [PubMed]
- Wacquier, B.; Combettes, L.; Dupont, G. Dual dynamics of mitochondrial permeability transition pore opening. Sci. Rep. 2020, 10, 3924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cervantes, M.; Moralí, G.; Letechipía-Vallejo, G. Melatonin and ischemia-reperfusion injury of the brain. J. Pineal Res. 2008, 45, 1–7. [Google Scholar] [CrossRef]
- Paterniti, I.; Cordaro, M.; Esposito, E.; Cuzzocrea, S. The antioxidative property of melatonin against brain ischemia. Expert Rev. Neurother. 2016, 16, 841–848. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.; Joh, T.H.; Baik, H.H.; Dibinis, C.; Volpe, B.T. Melatonin administration protects CA1 hippocampal neurons after transient forebrain ischemia in rats. Brain Res. 1997, 755, 335–338. [Google Scholar] [CrossRef]
- Tan, D.X.; Manchester, L.C.; Reiter, R.J.; Qi, W.B.; Karbownik, M.; Calvoa, J.R. Significance of melatonin in antioxidative defense system: Reactions and products. Neurosignals 2000, 9, 137–159. [Google Scholar] [CrossRef] [PubMed]
- Kilic, U.; Yilmaz, B.; Ugur, M.; Yüksel, A.; Reiter, R.J.; Hermann, D.M.; Kilic, E. Evidence that membrane-bound G protein-coupled melatonin receptors MT1 and MT2 are not involved in the neuroprotective effects of melatonin in focal cerebral ischemia. J. Pineal Res. 2012, 52, 228–235. [Google Scholar] [CrossRef]
- Andrabi, S.A.; Sayeed, I.; Siemen, D.; Wolf, G.; Horn, T.F.W. Direct inhibition of the mitochondrial permeability transition pore: A possible mechanism responsible for anti-apoptotic effects of melatonin. FASEB J. 2004, 18, 869–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stroethoff, M.; Behmenburg, F.; Spittler, K.; Raupach, A.; Heinen, A.; Hollmann, M.W.; Huhn, R.; Mathes, A. Activation of melatonin receptors by ramelteon induces cardioprotection by postconditioning in the rat heart. Anesth. Analg. 2018, 126, 2112–2115. [Google Scholar] [CrossRef] [PubMed]
- Bazil, J.N.; Buzzard, G.T.; Rundell, A.E. A bioenergetic model of the mitochondrial population undergoing permeability transition. J. Theor. Biol. 2010, 265, 672–690. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Furuta, T.; Nakagawa, I.; Yokoyama, S.; Morisaki, Y.; Saito, Y.; Nakase, H. Melatonin-Induced Postconditioning Suppresses NMDA Receptor through Opening of the Mitochondrial Permeability Transition Pore via Melatonin Receptor in Mouse Neurons. Int. J. Mol. Sci. 2022, 23, 3822. https://doi.org/10.3390/ijms23073822
Furuta T, Nakagawa I, Yokoyama S, Morisaki Y, Saito Y, Nakase H. Melatonin-Induced Postconditioning Suppresses NMDA Receptor through Opening of the Mitochondrial Permeability Transition Pore via Melatonin Receptor in Mouse Neurons. International Journal of Molecular Sciences. 2022; 23(7):3822. https://doi.org/10.3390/ijms23073822
Chicago/Turabian StyleFuruta, Takanori, Ichiro Nakagawa, Shohei Yokoyama, Yudai Morisaki, Yasuhiko Saito, and Hiroyuki Nakase. 2022. "Melatonin-Induced Postconditioning Suppresses NMDA Receptor through Opening of the Mitochondrial Permeability Transition Pore via Melatonin Receptor in Mouse Neurons" International Journal of Molecular Sciences 23, no. 7: 3822. https://doi.org/10.3390/ijms23073822