
Pangenomes as a Resource to Accelerate Breeding of Under-Utilised Crop Species

 , , , , , , , and
, , , , , , , and
Abstract
:1. Introduction
2. Under-Utilised Species
2.1. Cereal Grains
2.2. Vegetable/Pulse Crops
2.3. Tuberous Crops
2.4. Industrial Crops
2.5. Fruits
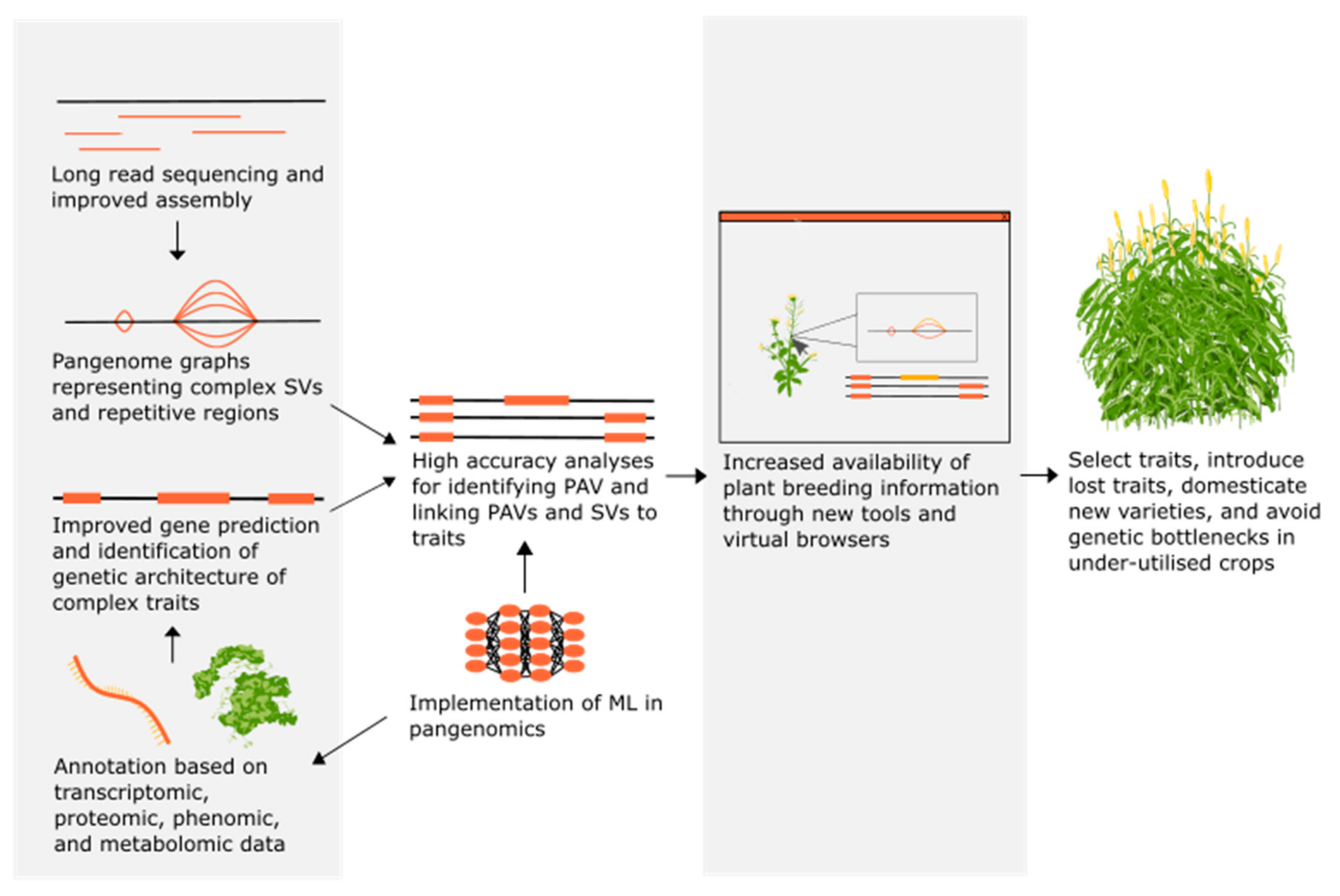
3. Developments in Pangenome Resources to Aid in the Breeding of Under-Utilised Crops
4. The Breeding Potential of Under-Utilised Crop Species
5. The Future of Pangenomics in Breeding Under-Utilised Crops

{kind=link}
{kind=link}
| Species Name | # of Individual Genomes | Assembly Method | References |
|---|---|---|---|
| Amborella trichopoda | 10 | Iterative mapping and assembly | [76,77] |
| Arabidopsis thaliana | 7 | De novo assembly | [157] |
| Brachypodium distachyon | 54 | De novo assembly | [33,158] |
| Brachypodium hybridum | 4 | De novo assembly | [158] |
| Brassica napus | 53 | Iterative mapping and assembly | [34] |
| Brassica napus | 8 | De novo assembly | [11] |
| Brassica oleracea | 10 | Iterative mapping and assembly | [32] |
| Cajanus cajan | 89 | Iterative mapping and assembly | [37] |
| Capsicum | 5 | Iterative mapping and assembly | [156] |
| Glycine max | 29 | Graph-based de novo assembly | [9] |
| Glycine max | 1110 | Iterative mapping and assembly | [10] |
| Gossypium | 1961 | De novo assembly | [38] |
| Hordeum vulgare | 20 | De novo assembly | [8] |
| Helianthus annuus | 287 | De novo assembly | [159] |
| Malus domestica | 91 | De novo assembly | [160] |
| Manihot esculenta | 57 | Practical haplotype graphs | [161] |
| Medicago truncatula | 15 | De novo assembly | [162] |
| Oryza sativa | 3 | De novo assembly | [31] |
| Oryza | 31 | De novo assembly | [6] |
| Poplar | 10 | De novo assembly | [163] |
| Sesamum indicum | 5 | De novo assembly | [35] |
| Solanum lycopersicum | 725 | De novo assembly | [36] |
| Sorghum bicolor | 398 | Practical haplotype graphs | [92] |
| Sorghum bicolor | 176 | Iterative mapping and assembly | [12] |
| Triticum aestivum | 18 | Iterative mapping and assembly | [20] |
| Zea mays | 4705 | Practical haplotype graphs | [96] |
| Scientific Names | Common Names | Type of Resource | References |
|---|---|---|---|
| Basella alba | Malabar spinach | Reports of viruses infecting Malbar spinach | [164,165] |
| Chromosome counts/Nuclear DNA quantification | [166] | ||
| Calathea allouia | Guinea arrowroot | Future prospects for underutilised medicinally valuable plants | [167] |
| Couma utilis | Milk tree | Identifying pollinators in edible Amazon fruit plants | [168] |
| Crambe cordifolia | Greater sea-kale | Ancestral chromosomal blocks in Brassiceae species | [169] |
| Leopoldia comosa | Tassel grape hyacinth | Identifying physiological responses | [170] |
| Mineral content and chemical analysis | [171] | ||
| Schinziophyton rautanenii | Mongongo tree | Sustainability review | [172] |
| Chemical composition of oil | [173] | ||
| Ullucus tuberosus | Ulluco | Viruses detected in ulluco | [174] |
| High throughput sequencing to detect novel viruses in ulluco | [175] |
| Scientific Names | Common Names | Type of Genomic Resources | References |
|---|---|---|---|
| Cereal grains | |||
| Canna edulis | African arrowroot | Chloroplast genome sequence | [68] |
| Digitaria exilis | White fonio | Genome assembly and annotation | [17,47] |
| Genotype-by-sequencing and SNP data | [48] | ||
| Panicum sumatrense | Little Millet | Chloroplast genome sequences | [43] |
| De novo transcriptome assembly | [44] | ||
| Vegetable/Pulse crops | |||
| Lablab purpureus | Hyacinth bean/Lablab bean | Chloroplast genome assembly | [61] |
| Draft genome assembly | [60] | ||
| Upregulation of drought tolerant genes | [58] | ||
| RFLP markers | [176] | ||
| Solanum nigrum | Black nightshade plant | Transcriptome sequence | [177,178] |
| Chloroplast genome sequence | [179,180] | ||
| Vigna aconitifolia | Moth bean | Genetic linkage map | [54] |
| Novel Vigna genetic resources | [53] | ||
| Tuberous crops | |||
| Pachyrhizus erosus | Yam bean | Draft genome assembly | [15] |
| Vigna vexillata | Zombi pea or Wild cowpea | Anti-inflammatory bioactivity | [181] |
| QTL analysis | [182] | ||
| Molecular linkage analysis | [183] | ||
| Hybridisation accession analysis | [184] | ||
| Industrial Crops | |||
| Carthamus tinctorius | Safflowers | Transcriptome sequencing | [185,186] |
| Chromosome-scale reference genome | [73] | ||
| Chloroplast genome sequence | [187] | ||
| Genetic mapping of SNPs | [71] | ||
| Hibiscus cannabinus | Kenaf | Mitochondrial genome assembly | [70] |
| Genome assembly and annotation | [16] | ||
| De novo transcriptome assembly | [69] | ||
| Fruit/Nuts | |||
| Bactris gasipaes | Peach palm | Chloroplast DNA for phylogenetic study | [188] |
| Macaúba palm transcriptome sequencing | [189] | ||
| RNA-seq of tropical palms | [190] | ||
| Plastome sequence | [191] | ||
| Citrullus colocynthis | Desert Watermelon or Wild watermelon | Gene markers | [192] |
| Transcriptome assembly | [193] | ||
| Genome Resequencing | [194] | ||
| Elaeagnus angustifolia | Russian olive or wild olive | Geographic study using machine learning | [195] |
| Hi-C assembly | [196] | ||
| Transciptome profiling | [197] | ||
| Plant signalling regarding salt | [198] | ||
| Ensete ventricosum | Ethiopian Banana | Genome assembly | [199,200] |
| Pangenome assembly | [80] | ||
| Markers/Microsatellites | [201] | ||
| Metabolite data | [202] | ||
| Euterpe oleracea | Açaí | Chemical genomic profiling | [203] |
| Karyotype and genome size | [204] | ||
| Psidium guajava | Guava | Genome assembly | [76,77] |
| Genome Markers | [76] | ||
| RNA-seq/transcriptome assembly | [78] | ||
| Vaccinium meridionale | Agraz or Colombian Berry | Phylogenetic relationships within the Vaccinieae tribe | [205] |
| Chemical, antimicrobial and molecular characterisation | [206] | ||
| Characterisation of phenotypic plasticity | [207] | ||
| Antiproliferative potential of Agraz juice | [208] | ||
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Anderson, R.; Bayer, P.E.; Edwards, D. Climate change and the need for agricultural adaptation. Curr. Opin. Plant Biol. 2020, 56, 197–202. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. The State of Food Security and Nutrition in the World 2019: Safeguarding Against Economic Slowdowns and Downturns; Food and Agriculture Organization of the United Nations: Rome, Italy, 2019. [Google Scholar]
- Ray, D.K.; Mueller, N.D.; West, P.C.; Foley, J.A. Yield trends are insufficient to double global crop production by 2050. PLoS ONE 2013, 8, e66428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Hu, S.; Wang, J.; Wong, G.K.S.; Li, S.; Liu, B.; Deng, Y.; Dai, L.; Zhou, Y.; Zhang, X.; et al. A draft sequence of the rice genome (Oryza sativa L. ssp. japonica). Science 2002, 296, 92–100. [Google Scholar] [CrossRef] [PubMed]
- International Wheat Genome Sequencing Consortium (IWGSC); Appels, R.; Eversole, K.; Stein, N.; Feuillet, C.; Keller, B.; Rogers, J.; Pozniak, C.J.; Choulet, F.; Distelfeld, A.; et al. Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science 2018, 361, eaar7191. [Google Scholar]
- Qin, P.; Lu, H.; Du, H.; Wang, H.; Chen, W.; Chen, Z.; He, Q.; Ou, S.; Zhang, H.; Li, X.; et al. Pan-genome analysis of 33 genetically diverse rice accessions reveals hidden genomic variations. Cell 2021, 184, 3542–3558.e3516. [Google Scholar] [CrossRef]
- Lu, F.; Romay, M.C.; Glaubitz, J.C.; Bradbury, P.J.; Elshire, R.J.; Wang, T.; Li, Y.; Li, Y.; Semagn, K.; Zhang, X.; et al. High-resolution genetic mapping of maize pan-genome sequence anchors. Nat. Commun. 2015, 6, 6914. [Google Scholar] [CrossRef] [Green Version]
- Jayakodi, M.; Padmarasu, S.; Haberer, G.; Bonthala, V.S.; Gundlach, H.; Monat, C.; Lux, T.; Kamal, N.; Lang, D.; Himmelbach, A.; et al. The barley pan-genome reveals the hidden legacy of mutation breeding. Nature 2020, 588, 284–289. [Google Scholar] [CrossRef]
- Liu, Y.; Du, H.; Li, P.; Shen, Y.; Peng, H.; Liu, S.; Zhou, G.A.; Zhang, H.; Liu, Z.; Shi, M.; et al. Pan-genome of wild and cultivated soybeans. Cell 2020, 182, 162–176.e113. [Google Scholar] [CrossRef]
- Bayer, P.E.; Valliyodan, B.; Hu, H.; Marsh, J.I.; Yuan, Y.; Vuong, T.D.; Patil, G.; Song, Q.; Batley, J.; Varshney, R.K.; et al. Sequencing the USDA core soybean collection reveals gene loss during domestication and breeding. Plant Genome 2021, e20109. [Google Scholar] [CrossRef]
- Song, J.-M.; Guan, Z.; Hu, J.; Guo, C.; Yang, Z.; Wang, S.; Liu, D.; Wang, B.; Lu, S.; Zhou, R.; et al. Eight high-quality genomes reveal pan-genome architecture and ecotype differentiation of Brassica napus. Nat. Plants 2020, 6, 34–45. [Google Scholar] [CrossRef]
- Ruperao, P.; Thirunavukkarasu, N.; Gandham, P.; Selvanayagam, S.; Govindaraj, M.; Nebie, B.; Manyasa, E.; Gupta, R.; Das, R.R.; Odeny, D.A.; et al. Sorghum pan-genome explores the functional utility for genomic-assisted breeding to accelerate the genetic gain. Front. Plant Sci. 2021, 12, 666342. [Google Scholar] [CrossRef]
- Pratap, A.; Prajapati, U.; Singh, C.M.; Gupta, S.; Rathore, M.; Malviya, N.; Tomar, R.; Gupta, A.K.; Tripathi, S.; Singh, N.P. Potential, constraints and applications of in vitro methods in improving grain legumes. Plant Breed. 2018, 137, 235–249. [Google Scholar] [CrossRef]
- Abewoy, D. Review on genetics and breeding of tomato (Lycopersicon esculentum Mill). Adv. Crop Sci. Technol. 2017, 5, 306. [Google Scholar]
- Tay Fernandez, C.G.; Pati, K.; Severn-Ellis, A.A.; Batley, J.; Edwards, D. Studying the genetic diversity of yam bean using a new draft genome assembly. Agronomy 2021, 11, 953. [Google Scholar] [CrossRef]
- Zhang, L.; Xu, Y.; Zhang, X.; Ma, X.; Zhang, L.; Liao, Z.; Zhang, Q.; Wan, X.; Cheng, Y.; Zhang, J.; et al. The genome of kenaf (Hibiscus cannabinus L.) provides insights into bast fibre and leaf shape biogenesis. Plant Biotechnol. J. 2020, 18, 1796–1809. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Chen, S.; Ma, X.; Yssel, A.E.J.; Chaluvadi, S.R.; Johnson, M.S.; Gangashetty, P.; Hamidou, F.; Sanogo, M.D.; Zwaenepoel, A.; et al. Genome sequence and genetic diversity analysis of an under-domesticated orphan crop, white fonio (Digitaria exilis). GigaScience 2021, 10, giab013. [Google Scholar] [CrossRef]
- Bayer, P.E.; Golicz, A.A.; Scheben, A.; Batley, J.; Edwards, D. Plant pan-genomes are the new reference. Nat. Plants 2020, 6, 914–920. [Google Scholar] [CrossRef]
- Gage, J.L.; Vaillancourt, B.; Hamilton, J.P.; Manrique-Carpintero, N.C.; Gustafson, T.J.; Barry, K.; Lipzen, A.; Tracy, W.F.; Mikel, M.A.; Kaeppler, S.M.; et al. Multiple maize reference genomes impact the identification of variants by genome-wide association study in a diverse inbred panel. Plant Genome 2019, 12, 180069. [Google Scholar] [CrossRef] [Green Version]
- Montenegro, J.D.; Golicz, A.A.; Bayer, P.E.; Hurgobin, B.; Lee, H.; Chan, C.-K.K.; Visendi, P.; Lai, K.; Doležel, J.; Batley, J.; et al. The pangenome of hexaploid bread wheat. Plant J. 2017, 90, 1007–1013. [Google Scholar] [CrossRef] [Green Version]
- The Rice Chromosomes 11 and 12 Sequencing Consortia. The sequence of rice chromosomes 11 and 12, rich in disease resistance genes and recent gene duplications. BMC Biol. 2005, 3, 20. [Google Scholar]
- Woodhouse, M.R.; Schnable, J.C.; Pedersen, B.S.; Lyons, E.; Lisch, D.; Subramaniam, S.; Freeling, M. Following tetraploidy in maize, a short deletion mechanism removed genes preferentially from one of the two homeologs. PLoS Biol. 2010, 8, e1000409. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Tehrim, S.; Zhang, F.; Tong, C.; Huang, J.; Cheng, X.; Dong, C.; Zhou, Y.; Qin, R.; Hua, W.; et al. Genome-wide comparative analysis of NBS-encoding genes between Brassica species and Arabidopsis thaliana. BMC Genom. 2014, 15, 3. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.-Y.; Huang, J.-Q.; Li, N.-Y.; Ma, X.-F.; Wang, J.-L.; Liu, C.; Liu, Y.-F.; Liang, Y.; Bao, Y.-M.; Dai, X.-F. Genome-wide analysis of the gene families of resistance gene analogues in cotton and their response to Verticillium wilt. BMC Plant Biol. 2015, 15, 148. [Google Scholar] [CrossRef] [Green Version]
- Bennetzen, J.L. Transposable element contributions to plant gene and genome evolution. Plant Mol. Biol. 2000, 42, 251–269. [Google Scholar] [CrossRef]
- Zhang, L.; Ren, Y.; Yang, T.; Li, G.; Chen, J.; Gschwend, A.R.; Yu, Y.; Hou, G.; Zi, J.; Zhou, R.; et al. Rapid evolution of protein diversity by de novo origination in Oryza. Nat. Ecol. Evol. 2019, 3, 679–690. [Google Scholar] [CrossRef]
- Dunning, L.T.; Olofsson, J.K.; Parisod, C.; Choudhury, R.R.; Moreno-Villena, J.J.; Yang, Y.; Dionora, J.; Quick, W.P.; Park, M.; Bennetzen, J.L.; et al. Lateral transfers of large DNA fragments spread functional genes among grasses. Proc. Natl. Acad. Sci. USA 2019, 116, 4416. [Google Scholar] [CrossRef] [Green Version]
- Tettelin, H.; Masignani, V.; Cieslewicz, M.J.; Donati, C.; Medini, D.; Ward, N.L.; Angiuoli, S.V.; Crabtree, J.; Jones, A.L.; Durkin, A.S.; et al. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: Implications for the microbial “pan-genome”. Proc. Natl. Acad. Sci. USA 2005, 102, 13950. [Google Scholar] [CrossRef] [Green Version]
- Morgante, M.; De Paoli, E.; Radovic, S. Transposable elements and the plant pan-genomes. Curr. Opin. Plant Biol. 2007, 10, 149–155. [Google Scholar] [CrossRef]
- Li, Y.H.; Zhou, G.; Ma, J.; Jiang, W.; Jin, L.G.; Zhang, Z.; Guo, Y.; Zhang, J.; Sui, Y.; Zheng, L.; et al. De novo assembly of soybean wild relatives for pan-genome analysis of diversity and agronomic traits. Nat. Biotechnol. 2014, 32, 1045–1052. [Google Scholar] [CrossRef] [Green Version]
- Schatz, M.C.; Maron, L.G.; Stein, J.C.; Wences, A.H.; Gurtowski, J.; Biggers, E.; Lee, H.; Kramer, M.; Antoniou, E.; Ghiban, E.; et al. Whole genome de novo assemblies of three divergent strains of rice, Oryza sativa, document novel gene space of aus and indica. Genome Biol. 2014, 15, 506. [Google Scholar]
- Golicz, A.A.; Bayer, P.E.; Barker, G.C.; Edger, P.P.; Kim, H.; Martinez, P.A.; Chan, C.K.K.; Severn-Ellis, A.; McCombie, W.R.; Parkin, I.A.P.; et al. The pangenome of an agronomically important crop plant Brassica oleracea. Nat. Commun. 2016, 7, 13390. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S.P.; Contreras-Moreira, B.; Woods, D.P.; Des Marais, D.L.; Burgess, D.; Shu, S.; Stritt, C.; Roulin, A.C.; Schackwitz, W.; Tyler, L.; et al. Extensive gene content variation in the Brachypodium distachyon pan-genome correlates with population structure. Nat. Commun. 2017, 8, 2184. [Google Scholar] [CrossRef] [PubMed]
- Hurgobin, B.; Golicz, A.A.; Bayer, P.E.; Chan, C.-K.K.; Tirnaz, S.; Dolatabadian, A.; Schiessl, S.V.; Samans, B.; Montenegro, J.D.; Parkin, I.A.P.; et al. Homoeologous exchange is a major cause of gene presence/absence variation in the amphidiploid Brassica napus. Plant Biotechnol. J. 2017, 16, 1265–1274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Golicz, A.A.; Lu, K.; Dossa, K.; Zhang, Y.; Chen, J.; Wang, L.; You, J.; Fan, D.; Edwards, D.; et al. Insight into the evolution and functional characteristics of the pan-genome assembly from sesame landraces and modern cultivars. Plant Biotechnol. J. 2019, 17, 881–892. [Google Scholar] [CrossRef] [Green Version]
- Gao, L.; Gonda, I.; Sun, H.; Ma, Q.; Bao, K.; Tieman, D.M.; Burzynski-Chang, E.A.; Fish, T.L.; Stromberg, K.A.; Sacks, G.L.; et al. The tomato pan-genome uncovers new genes and a rare allele regulating fruit flavor. Nat. Genet. 2019, 51, 1044–1051. [Google Scholar] [CrossRef]
- Zhao, J.; Bayer, P.E.; Ruperao, P.; Saxena, R.K.; Khan, A.W.; Golicz, A.A.; Nguyen, H.T.; Batley, J.; Edwards, D.; Varshney, R.K. Trait associations in the pangenome of pigeon pea (Cajanus cajan). Plant Biotechnol. J. 2020, 18, 1946–1954. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Yuan, D.; Wang, P.; Wang, Q.; Sun, M.; Liu, Z.; Si, H.; Xu, Z.; Ma, Y.; Zhang, B.; et al. Cotton pan-genome retrieves the lost sequences and genes during domestication and selection. Genome Biol. 2021, 22, 119. [Google Scholar] [CrossRef]
- Golicz, A.A.; Bayer, P.E.; Bhalla, P.L.; Batley, J.; Edwards, D. Pangenomics comes of age: From bacteria to plant and animal applications. Trends Genet. 2020, 36, 132–145. [Google Scholar] [CrossRef]
- Kamal, N.M.; Gorafi, Y.S.A.; Abdelrahman, M.; Abdellatef, E.; Tsujimoto, H. Stay-green trait: A prospective approach for yield potential, and drought and heat stress adaptation in globally important cereals. Int. J. Mol. Sci. 2019, 20, 5837. [Google Scholar] [CrossRef] [Green Version]
- Muthamilarasan, M.; Prasad, M. Small millets for enduring food security amidst pandemics. Trends Plant Sci. 2021, 26, 33–40. [Google Scholar] [CrossRef]
- Chauhan, M.; Sonawane, S.K.; Arya, S. Nutritional and nutraceutical properties of millets: A review. Clin. J. Nutr. Diet. 2018, 1, 1–10. [Google Scholar]
- Sebastin, R.; Lee, G.A.; Lee, K.J.; Shin, M.J.; Cho, G.T.; Lee, J.R.; Ma, K.H.; Chung, J.W. The complete chloroplast genome sequences of little millet (Panicum sumatrense Roth ex Roem. and Schult.) (Poaceae). Mitochondrial DNA Part B 2018, 3, 719–720. [Google Scholar] [CrossRef] [Green Version]
- Das, R.R.; Pradhan, S.; Parida, A. De-novo transcriptome analysis unveils differentially expressed genes regulating drought and salt stress response in Panicum sumatrense. Sci. Rep. 2020, 10, 21251. [Google Scholar] [CrossRef]
- Ballogou, V.; Soumanou, M.; Toukourou, F.; Hounhouigan, J. Structure and nutritional composition of fonio (Digitaria exilis) grains: A review. Int. Res. J. Biol. Sci. 2013, 2, 73–79. [Google Scholar]
- National Research Council. Lost Crops of Africa: Volume 1: Grains; National Academies Press: Washington, DC, USA, 1996. [Google Scholar]
- Abrouk, M.; Ahmed, H.I.; Cubry, P.; Šimoníková, D.; Cauet, S.; Pailles, Y.; Bettgenhaeuser, J.; Gapa, L.; Scarcelli, N.; Couderc, M.; et al. Fonio millet genome unlocks African orphan crop diversity for agriculture in a changing climate. Nat. Commun. 2020, 11, 4488. [Google Scholar] [CrossRef]
- Ibrahim Bio Yerima, A.R.; Issoufou, K.A.; Adje, C.A.; Mamadou, A.; Oselebe, H.; Gueye, M.C.; Billot, C.; Achigan-Dako, E.G. Genome-wide scanning enabled SNP discovery, linkage disequilibrium patterns and population structure in a panel of fonio (Digitaria exilis [Kippist] Stapf) germplasm. Front. Sustain. Food Syst. 2021, 5, 699549. [Google Scholar] [CrossRef]
- Bennetzen, J.L.; Schmutz, J.; Wang, H.; Percifield, R.; Hawkins, J.; Pontaroli, A.C.; Estep, M.; Feng, L.; Vaughn, J.N.; Grimwood, J. Reference genome sequence of the model plant Setaria. Nat. Biotechnol. 2012, 30, 555–561. [Google Scholar] [CrossRef] [Green Version]
- Varshney, R.K.; Shi, C.; Thudi, M.; Mariac, C.; Wallace, J.; Qi, P.; Zhang, H.; Zhao, Y.; Wang, X.; Rathore, A.; et al. Pearl millet genome sequence provides a resource to improve agronomic traits in arid environments. Nat. Biotechnol. 2017, 35, 969–976. [Google Scholar] [CrossRef] [Green Version]
- Zou, C.; Li, L.; Miki, D.; Li, D.; Tang, Q.; Xiao, L.; Rajput, S.; Deng, P.; Peng, L.; Jia, W. The genome of broomcorn millet. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Bennetzen, J.L.; Freeling, M. The unified grass genome: Synergy in synteny. Genome Res. 1997, 7, 301–306. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, Y.; Somta, P.; Muto, C.; Iseki, K.; Naito, K.; Pandiyan, M.; Natesan, S.; Tomooka, N. Novel genetic resources in the genus vigna unveiled from gene bank accessions. PLoS ONE 2016, 11, e0147568. [Google Scholar] [CrossRef]
- Yundaeng, C.; Somta, P.; Amkul, K.; Kongjaimun, A.; Kaga, A.; Tomooka, N. Construction of genetic linkage map and genome dissection of domestication-related traits of moth bean (Vigna aconitifolia), a legume crop of arid areas. Mol. Genet. Genom. 2019, 294, 621–635. [Google Scholar] [CrossRef]
- Minde, J.J.; Venkataramana, P.B.; Matemu, A.O. Dolichos Lablab-an underutilized crop with future potentials for food and nutrition security: A review. Crit. Rev. Food Sci. Nutr. 2021, 61, 2249–2261. [Google Scholar] [CrossRef]
- Chakoma, I.; Manyawu, G.J.; Gwiriri, L.; Moyo, S.; Dube, S. The Agronomy and Use of Lablab Purpureus in Smallholder Farming Systems of Southern Africa; International Livestock Research Institute: Nairobi, Kenya, 2016. [Google Scholar]
- Missanga, J.S.; Venkataramana, P.B.; Ndakidemi, P.A. Recent developments in Lablab purpureus genomics: A focus on drought stress tolerance and use of genomic resources to develop stress-resilient varieties. Legume Sci. 2021, 3, e99. [Google Scholar] [CrossRef]
- Wang, B.; Zhao, M.; Yao, L.; Joao, V.M.d.S.; Babu, V.; Wu, T.; Nguyen, H.T. Identification of drought-inducible regulatory factors in Lablab purpureus by a comparative genomic approach. Crop Pasture Sci. 2018, 69, 632–641. [Google Scholar] [CrossRef]
- Rai, K.K.; Rai, N.; Rai, S.P. Recent advancement in modern genomic tools for adaptation of Lablab purpureus L to biotic and abiotic stresses: Present mechanisms and future adaptations. Acta Physiol. Plant. 2018, 40, 164. [Google Scholar] [CrossRef]
- Chang, Y.; Liu, H.; Liu, M.; Liao, X.; Sahu, S.K.; Fu, Y.; Song, B.; Cheng, S.; Kariba, R.; Muthemba, S.; et al. The draft genomes of five agriculturally important African orphan crops. GigaScience 2019, 8, giy152. [Google Scholar] [CrossRef]
- Li, N.; Bai, J.-Q.; Gao, S.; Yang, L.; Li, J.; Du, S.-B.; Wang, X.-P. The complete molecular sequence of chloroplast genome of Lablab purpureus (L.) Sweet. Mitochondrial DNA Part B 2021, 6, 758–759. [Google Scholar] [CrossRef]
- Sørensen, M. Observations on distribution, ecology and cultivation of the tuber-bearing legume genus Pachyrhizus Rich. ex DC. Agric. Univ. Wagening. Pap. 1990, 3, 38. [Google Scholar]
- Ade-Omowaye, B.; Tucker, G.; Smetanska, I. Nutritional potential of nine underexploited legumes in Southwest Nigeria. Int. Food Res. J. 2015, 22, 798. [Google Scholar]
- Pati, K.; Zhang, F.; Batley, J. First report of genome size and ploidy of the underutilized leguminous tuber crop Yam Bean (Pachyrhizus erosus and P. tuberosus) by flow cytometry. Plant Genet. Resour. Charact. Util. 2019, 17, 456–459. [Google Scholar] [CrossRef]
- Sørensen, M. Yam Bean: Pachyrhizus DC.-Promoting the Conservation and Use of Underutilized and Neglected Crops. 2; Bioversity International: Roma, Italy, 1996; Volume 2. [Google Scholar]
- Zhang, W.E.; Wang, F.; Pan, X.J.; Tian, Z.G.; Zhao, X.M. Antioxidant enzymes and photosynthetic responses to drought stress of three Canna edulis Cultivars. Hortic. Sci. Technol. 2013, 31, 677–686. [Google Scholar] [CrossRef] [Green Version]
- Sandoval, L.; Zamora-Castro, S.A.; Vidal-Álvarez, M.; Marín-Muñiz, J.L. Role of wetland plants and use of ornamental flowering plants in constructed wetlands for wastewater treatment: A review. Appl. Sci. 2019, 9, 685. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Q.; Cai, L.; Li, H.; Zhang, Y.; Su, W.; Zhou, Q. The complete chloroplast genome sequence of the Canna edulis Ker Gawl. (Cannaceae). Mitochondrial DNA Part B Resour. 2020, 5, 2427–2428. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wan, X.; Xu, J.; Lin, L.; Qi, J. De novo assembly of kenaf (Hibiscus cannabinus) transcriptome using Illumina sequencing for gene discovery and marker identification. Mol. Breed. 2015, 35, 192. [Google Scholar] [CrossRef]
- Liao, X.; Zhao, Y.; Kong, X.; Khan, A.; Zhou, B.; Liu, D.; Kashif, M.H.; Chen, P.; Wang, H.; Zhou, R. Complete sequence of kenaf (Hibiscus cannabinus) mitochondrial genome and comparative analysis with the mitochondrial genomes of other plants. Sci. Rep. 2018, 8, 12714. [Google Scholar] [CrossRef]
- Bowers, J.E.; Pearl, S.A.; Burke, J.M. Genetic mapping of millions of SNPs in safflower (Carthamus tinctorius L.) via whole-genome resequencing. G3 Genes|Genomes|Genet. 2016, 6, 2203–2211. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Wang, Z.; Ma, C.; Tian, J.; Fu, F.; Li, C.; Guo, D.; Roeder, E.; Liu, K. Neuroprotective effects of hydroxysafflor yellow A: In vivo and in vitro studies. Planta Med. 2003, 69, 429–433. [Google Scholar]
- Wu, Z.; Liu, H.; Zhan, W.; Yu, Z.; Qin, E.; Liu, S.; Yang, T.; Xiang, N.; Kudrna, D.; Chen, Y.; et al. The chromosome-scale reference genome of safflower (Carthamus tinctorius) provides insights into linoleic acid and flavonoid biosynthesis. Plant Biotechnol. J. 2021, 19, 1725–1742. [Google Scholar] [CrossRef]
- Ray, P.K. Breeding Tropical and Subtropical Fruits; Springer Science & Business Media: Berlin, Germany, 2002. [Google Scholar]
- Upadhyay, R.; Dass, J.F.P.; Chauhan, A.K.; Yadav, P.; Singh, M.; Singh, R.B. Chapter 21—Guava enriched functional foods: Therapeutic potentials and technological challenges. In The Role of Functional Food Security in Global Health; Singh, R.B., Watson, R.R., Takahashi, T., Eds.; Academic Press: Cambridge, MA, USA, 2019; pp. 365–378. [Google Scholar]
- Thakur, S.; Yadav, I.S.; Jindal, M.; Sharma, P.K.; Dhillon, G.S.; Boora, R.S.; Arora, N.K.; Gill, M.I.S.; Chhuneja, P.; Mittal, A. Development of genome-wide functional markers using draft genome assembly of guava (Psidium guajava L.) cv. Allahabad safeda to expedite molecular breeding. Front. Plant Sci. 2021, 12, 708332. [Google Scholar] [CrossRef]
- Feng, C.; Feng, C.; Lin, X.; Liu, S.; Li, Y.; Kang, M. A chromosome-level genome assembly provides insights into ascorbic acid accumulation and fruit softening in guava (Psidium guajava). Plant Biotechnol. J. 2021, 19, 717–730. [Google Scholar] [CrossRef]
- Mittal, A.; Yadav, I.S.; Arora, N.K.; Boora, R.S.; Mittal, M.; Kaur, P.; Erskine, W.; Chhuneja, P.; Gill, M.I.S.; Singh, K. RNA-sequencing based gene expression landscape of guava cv. Allahabad Safeda and comparative analysis to colored cultivars. BMC Genom. 2020, 21, 484. [Google Scholar] [CrossRef]
- Borrell, J.S.; Biswas, M.K.; Goodwin, M.; Blomme, G.; Schwarzacher, T.; Heslop-Harrison, J.S.; Wendawek, A.M.; Berhanu, A.; Kallow, S.; Janssens, S.; et al. Enset in Ethiopia: A poorly characterized but resilient starch staple. Ann. Bot. 2019, 123, 747–766. [Google Scholar] [CrossRef] [Green Version]
- Rijzaani, H.; Bayer, P.E.; Rouard, M.; Doležel, J.; Batley, J.; Edwards, D. The pangenome of banana highlights differences between genera and genomes. Plant Genome 2021, e20100. [Google Scholar] [CrossRef]
- Golicz, A.A.; Batley, J.; Edwards, D. Towards plant pangenomics. Plant Biotechnol. J. 2016, 14, 1099–1105. [Google Scholar] [CrossRef]
- Danilevicz, M.F.; Tay Fernandez, C.G.; Marsh, J.I.; Bayer, P.E.; Edwards, D. Plant pangenomics: Approaches, applications and advancements. Curr. Opin. Plant Biol. 2020, 54, 18–25. [Google Scholar] [CrossRef]
- Garrison, E.; Sirén, J.; Novak, A.M.; Hickey, G.; Eizenga, J.M.; Dawson, E.T.; Jones, W.; Garg, S.; Markello, C.; Lin, M.F.; et al. Variation graph toolkit improves read mapping by representing genetic variation in the reference. Nat. Biotechnol. 2018, 36, 875–879. [Google Scholar] [CrossRef]
- Marcus, S.; Lee, H.; Schatz, M.C. SplitMEM: A graphical algorithm for pan-genome analysis with suffix skips. Bioinformatics 2014, 30, 3476–3483. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Feng, X.; Chu, C. The design and construction of reference pangenome graphs with minigraph. Genome Biol. 2020, 21, 265. [Google Scholar] [CrossRef]
- Eizenga, J.M.; Novak, A.M.; Sibbesen, J.A.; Heumos, S.; Ghaffaari, A.; Hickey, G.; Chang, X.; Seaman, J.D.; Rounthwaite, R.; Ebler, J.; et al. Pangenome graphs. Annu. Rev. Genom. Hum. Genet. 2020, 21, 139–162. [Google Scholar] [CrossRef]
- Paten, B.; Novak, A.M.; Eizenga, J.M.; Garrison, E. Genome graphs and the evolution of genome inference. Genome Res. 2017, 27, 665–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hickey, G.; Heller, D.; Monlong, J.; Sibbesen, J.A.; Sirén, J.; Eizenga, J.; Dawson, E.T.; Garrison, E.; Novak, A.M.; Paten, B. Genotyping structural variants in pangenome graphs using the vg toolkit. Genome Biol. 2020, 21, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabbani, L.; Müller, J.; Weigel, D. An algorithm to build a multi-genome reference. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- The Computational Pan-Genomics Consortium. Computational pan-genomics: Status, promises and challenges. Brief. Bioinform. 2018, 19, 118–135. [Google Scholar]
- Rakocevic, G.; Semenyuk, V.; Lee, W.-P.; Spencer, J.; Browning, J.; Johnson, I.J.; Arsenijevic, V.; Nadj, J.; Ghose, K.; Suciu, M.C.; et al. Fast and accurate genomic analyses using genome graphs. Nat. Genet. 2019, 51, 354–362. [Google Scholar] [CrossRef]
- Jensen, S.E.; Charles, J.R.; Muleta, K.; Bradbury, P.J.; Casstevens, T.; Deshpande, S.P.; Gore, M.A.; Gupta, R.; Ilut, D.C.; Johnson, L.; et al. A sorghum Practical Haplotype Graph facilitates genome-wide imputation and cost-effective genomic prediction. Plant Genome 2020, 13, e20009. [Google Scholar] [CrossRef] [Green Version]
- Zanini, S.F.; Bayer, P.E.; Wells, R.; Snowdon, R.J.; Batley, J.; Varshney, R.K.; Nguyen, H.T.; Edwards, D.; Golicz, A.A. Pangenomics in crop improvement—From coding structural variations to finding regulatory variants with pangenome graphs. Plant Genome 2021, 13, e20177. [Google Scholar] [CrossRef]
- Bradbury, P.J.; Casstevens, T.; Jensen, S.E.; Johnson, L.C.; Miller, Z.R.; Monier, B.; Romay, M.C.; Song, B.; Buckler, E.S. The practical haplotype graph, a platform for storing and using pangenomes for imputation. bioRxiv 2021. [Google Scholar] [CrossRef]
- Long, E.M.; Bradbury, P.J.; Romay, M.C.; Buckler, E.S.; Robbins, K.R. Genome-wide imputation using the practical haplotype graph in the heterozygous crop cassava. G3 Genes|Genomes|Genet. 2021, 12, jkab383. [Google Scholar] [CrossRef]
- Franco, J.A.V.; Gage, J.L.; Bradbury, P.J.; Johnson, L.C.; Miller, Z.R.; Buckler, E.S.; Romay, M.C. A maize practical haplotype graph leverages diverse NAM assemblies. bioRxiv 2020. [Google Scholar] [CrossRef]
- Maistrenko, O.M.; Mende, D.R.; Luetge, M.; Hildebrand, F.; Schmidt, T.S.B.; Li, S.S.; Rodrigues, J.F.M.; von Mering, C.; Pedro Coelho, L.; Huerta-Cepas, J.; et al. Disentangling the impact of environmental and phylogenetic constraints on prokaryotic within-species diversity. ISME J. 2020, 14, 1247–1259. [Google Scholar] [CrossRef] [Green Version]
- Khan, A.W.; Garg, V.; Roorkiwal, M.; Golicz, A.A.; Edwards, D.; Varshney, R.K. Super-pangenome by integrating the wild side of a species for accelerated crop improvement. Trends Plant Sci. 2020, 25, 148–158. [Google Scholar] [CrossRef] [Green Version]
- He, Z.; Ji, R.; Havlickova, L.; Wang, L.; Li, Y.; Lee, H.T.; Song, J.; Koh, C.; Yang, J.; Zhang, M.; et al. Genome structural evolution in Brassica crops. Nat. Plants 2021, 7, 757–765. [Google Scholar] [CrossRef]
- Mohd Saad, N.S.; Severn-Ellis, A.A.; Pradhan, A.; Edwards, D.; Batley, J. Genomics armed with diversity leads the way in Brassica improvement in a changing global environment. Front Genet 2021, 12, 110. [Google Scholar] [CrossRef]
- Yuan, Y.; Bayer, P.E.; Batley, J.; Edwards, D. Current status of structural variation studies in plants. Plant Biotechnol. J. 2021, 19, 2153–2163. [Google Scholar] [CrossRef]
- Sekhwal, M.K.; Li, P.; Lam, I.; Wang, X.; Cloutier, S.; You, F.M. Disease resistance gene analogs (RGAs) in plants. Int. J. Mol. Sci. 2015, 16, 19248–19290. [Google Scholar] [CrossRef] [Green Version]
- Bayer, P.E.; Golicz, A.A.; Tirnaz, S.; Chan, C.-K.K.; Edwards, D.; Batley, J. Variation in abundance of predicted resistance genes in the Brassica oleracea pangenome. Plant Biotechnol. J. 2019, 17, 789–800. [Google Scholar] [CrossRef] [Green Version]
- Dolatabadian, A.; Bayer, P.E.; Tirnaz, S.; Hurgobin, B.; Edwards, D.; Batley, J. Characterization of disease resistance genes in the Brassica napus pangenome reveals significant structural variation. Plant Biotechnol. J. 2020, 18, 969–982. [Google Scholar] [CrossRef] [Green Version]
- Cantila, A.Y.; Saad, N.S.M.; Amas, J.C.; Edwards, D.; Batley, J. Recent findings unravel genes and genetic factors underlying leptosphaeria maculans resistance in Brassica napus and its relatives. Int. J. Mol. Sci. 2020, 22, 313. [Google Scholar] [CrossRef]
- Zhang, Y.; Thomas, W.; Bayer, P.E.; Edwards, D.; Batley, J. Frontiers in dissecting and managing Brassica diseases: From reference-based RGA candidate identification to building Pan-RGAomes. Int. J. Mol. Sci. 2020, 21, 8964. [Google Scholar] [CrossRef]
- Bakker, E.G.; Toomajian, C.; Kreitman, M.; Bergelson, J. A genome-wide survey of R gene polymorphisms in Arabidopsis. Plant Cell 2006, 18, 1803–1818. [Google Scholar] [CrossRef] [Green Version]
- Stam, R.; Silva-Arias, G.A.; Tellier, A. Subsets of NLR genes show differential signatures of adaptation during colonization of new habitats. New Phytol. 2019, 224, 367–379. [Google Scholar] [CrossRef]
- Garg, B.K.; Kathju, S.; Burman, U. Influence of Water Stress on Water Relations, Photosynthetic Parameters and Nitrogen Metabolism of Moth Bean Genotypes. Biol. Plant. 2001, 44, 289–292. [Google Scholar] [CrossRef]
- Garg, B.K.; Burman, U.; Kathju, S. The influence of phosphorus nutrition on the physiological response of moth bean genotypes to drought. J. Plant Nutr. Soil Sci. 2004, 167, 503–508. [Google Scholar] [CrossRef]
- Yao, L.-M.; Wang, B.; Cheng, L.-J.; Wu, T.-L. Identification of key drought stress-related genes in the hyacinth bean. PLoS ONE 2013, 8, e58108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naeem, M.; Shabbir, A.; Ansari, A.A.; Aftab, T.; Khan, M.M.A.; Uddin, M. Hyacinth bean (Lablab purpureus L.)—An underutilised crop with future potential. Sci. Hortic. 2020, 272, 109551. [Google Scholar] [CrossRef]
- Hu, H.; Scheben, A.; Verpaalen, B.; Tirnaz, S.; Bayer, P.E.; Hodel, R.G.J.; Batley, J.; Soltis, D.E.; Soltis, P.S.; Edwards, D. Amborella gene presence/absence variation is associated with abiotic stress responses that may contribute to environmental adaptation. New Phytol. 2021, 233, 1548–1555. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, T.; Wang, J.; Wang, P.; Qiu, Y.; Zhao, W.; Pang, S.; Li, X.; Wang, H.; Song, J.; et al. Pan-genome of Raphanus highlights genetic variation and introgression among domesticated, wild, and weedy radishes. Mol. Plant 2021, 14, 2032–2055. [Google Scholar] [CrossRef]
- Alonge, M.; Wang, X.; Benoit, M.; Soyk, S.; Pereira, L.; Zhang, L.; Suresh, H.; Ramakrishnan, S.; Maumus, F.; Ciren, D.; et al. Major impacts of widespread structural variation on gene expression and crop improvement in tomato. Cell 2020, 182, 145–161.e123. [Google Scholar] [CrossRef]
- Varshney, R.K.; Bohra, A.; Roorkiwal, M.; Barmukh, R.; Cowling, W.A.; Chitikineni, A.; Lam, H.-M.; Hickey, L.T.; Croser, J.S.; Bayer, P.E.; et al. Fast-forward breeding for a food-secure world. Trends Genet. 2021, 37, 1124–1136. [Google Scholar] [CrossRef]
- Tanaka, N.; Shenton, M.; Kawahara, Y.; Kumagai, M.; Sakai, H.; Kanamori, H.; Yonemaru, J.; Fukuoka, S.; Sugimoto, K.; Ishimoto, M.; et al. Whole-genome sequencing of the NARO world rice core collection (WRC) as the Basis for diversity and association studies. Plant Cell Physiol. 2020, 61, 922–932. [Google Scholar] [CrossRef] [Green Version]
- Marsh, J.I.; Hu, H.; Gill, M.; Batley, J.; Edwards, D. Crop breeding for a changing climate: Integrating phenomics and genomics with bioinformatics. Theor. Appl. Genet. 2021, 134, 1677–1690. [Google Scholar] [CrossRef]
- Shook, J.M.; Zhang, J.; Jones, S.E.; Singh, A.; Diers, B.W.; Singh, A.K. Meta-GWAS for quantitative trait loci identification in soybean. G3 Genes|Genomes|Genet. 2021, 11, kab117. [Google Scholar] [CrossRef] [PubMed]
- Zheng, B.; Zhao, Q.; Wu, H.; Wang, S.; Zou, M. A Comparative metabolomics analysis of guava (Psidium guajava L.) fruit with different colors. ACS Food Sci. Technol. 2021, 1, 96–106. [Google Scholar] [CrossRef]
- Lee, S.; Choi, H.-K.; Cho, S.K.; Kim, Y.-S. Metabolic analysis of guava (Psidium guajava L.) fruits at different ripening stages using different data-processing approaches. J. Chromatogr. B 2010, 878, 2983–2988. [Google Scholar] [CrossRef]
- Moon, P.; Fu, Y.; Bai, J.; Plotto, A.; Crane, J.; Chambers, A. Assessment of fruit aroma for twenty-seven guava (Psidium guajava) accessions through three fruit developmental stages. Sci. Hortic. 2018, 238, 375–383. [Google Scholar] [CrossRef]
- Zanklan, A.S.; Becker, H.C.; Sørensen, M.; Pawelzik, E.; Grüneberg, W.J. Genetic diversity in cultivated yam bean (Pachyrhizus spp.) evaluated through multivariate analysis of morphological and agronomic traits. Genet. Resour. Crop Evol. 2018, 65, 811–843. [Google Scholar] [CrossRef] [Green Version]
- Tapia, C.; Sørensen, M. Morphological characterization of the genetic variation existing in a Neotropical collection of yam bean, Pachyrhizus tuberosus (Lam.) Spreng. Genet. Resour. Crop Evol. 2003, 50, 681–692. [Google Scholar] [CrossRef]
- Silva, E.; Filho, D.; Ticona-Benavente, C. Diversity of yam bean (Pachyrhizus spp. Fabaceae) based on morphoagronomic traits in the Brazilian Amazon. Acta Amaz. 2016, 233, 233–240. [Google Scholar] [CrossRef] [Green Version]
- Martina, M.; Tikunov, Y.; Portis, E.; Bovy, A.G. The genetic basis of tomato aroma. Genes 2021, 12, 226. [Google Scholar] [CrossRef]
- Pereira, L.; Sapkota, M.; Alonge, M.; Zheng, Y.; Zhang, Y.; Razifard, H.; Taitano, N.K.; Schatz, M.C.; Fernie, A.R.; Wang, Y.; et al. Natural genetic diversity in tomato flavor genes. Front. Plant Sci. 2021, 12, 914. [Google Scholar] [CrossRef] [PubMed]
- Sharma, D.; Tiwari, A.; Sood, S.; Jamra, G.; Singh, N.K.; Meher, P.K.; Kumar, A. Genome wide association mapping of agro-morphological traits among a diverse collection of finger millet (Eleusine coracana L.) genotypes using SNP markers. PLoS ONE 2018, 13, e0199444. [Google Scholar] [CrossRef] [PubMed]
- Puranik, S.; Sahu, P.P.; Beynon, S.; Srivastava, R.K.; Sehgal, D.; Ojulong, H.; Yadav, R. Genome-wide association mapping and comparative genomics identifies genomic regions governing grain nutritional traits in finger millet (Eleusine coracana L. Gaertn.). Plants People Planet 2020, 2, 649–662. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, D.; He, F.; Wang, J.; Joshi, T.; Xu, D. Phenotype prediction and genome-wide association study using deep convolutional neural network of soybean. Front. Genet. 2019, 10, 1091. [Google Scholar] [CrossRef]
- Bayer, P.E.; Petereit, J.; Danilevicz, M.F.; Anderson, R.; Batley, J.; Edwards, D. The application of pangenomics and machine learning in genomic selection in plants. Plant Genome 2021, 14, e20112. [Google Scholar] [CrossRef]
- Min, S.; Lee, B.; Yoon, S. Deep learning in bioinformatics. Brief. Bioinform. 2017, 18, 851–869. [Google Scholar] [CrossRef] [Green Version]
- Gao, S.; Wu, J.; Stiller, J.; Zheng, Z.; Zhou, M.; Wang, Y.-G.; Liu, C. Identifying barley pan-genome sequence anchors using genetic mapping and machine learning. Theor. Appl. Genet. 2020, 133, 2535–2544. [Google Scholar] [CrossRef]
- Bayer, P.E.; Scheben, A.; Golicz, A.A.; Yuan, Y.; Faure, S.; Lee, H.; Chawla, H.S.; Anderson, R.; Bancroft, I.; Raman, H.; et al. Modelling of gene loss propensity in the pangenomes of three Brassica species suggests different mechanisms between polyploids and diploids. Plant Biotechnol. J. 2021, 19, 2488–2500. [Google Scholar] [CrossRef]
- Gabur, I.; Chawla, H.S.; Lopisso, D.T.; von Tiedemann, A.; Snowdon, R.J.; Obermeier, C. Gene presence-absence variation associates with quantitative Verticillium longisporum disease resistance in Brassica napus. Sci. Rep. 2020, 10, 4131. [Google Scholar] [CrossRef] [Green Version]
- Merker, J.D.; Wenger, A.M.; Sneddon, T.; Grove, M.; Zappala, Z.; Fresard, L.; Waggott, D.; Utiramerur, S.; Hou, Y.; Smith, K.S.; et al. Long-read genome sequencing identifies causal structural variation in a Mendelian disease. Genet. Med. 2018, 20, 159–163. [Google Scholar] [CrossRef] [Green Version]
- Jain, M.; Olsen, H.E.; Paten, B.; Akeson, M. The oxford nanopore MinION: Delivery of nanopore sequencing to the genomics community. Genome Biol. 2016, 17, 239. [Google Scholar] [CrossRef] [Green Version]
- Jain, M.; Koren, S.; Miga, K.H.; Quick, J.; Rand, A.C.; Sasani, T.A.; Tyson, J.R.; Beggs, A.D.; Dilthey, A.T.; Fiddes, I.T.; et al. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat. Biotechnol. 2018, 36, 338–345. [Google Scholar] [CrossRef] [Green Version]
- Wenger, A.M.; Peluso, P.; Rowell, W.J.; Chang, P.-C.; Hall, R.J.; Concepcion, G.T.; Ebler, J.; Fungtammasan, A.; Kolesnikov, A.; Olson, N.D.; et al. Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nat. Biotechnol. 2019, 37, 1155–1162. [Google Scholar] [CrossRef]
- Lan, T.; Renner, T.; Ibarra-Laclette, E.; Farr, K.M.; Chang, T.-H.; Cervantes-Pérez, S.A.; Zheng, C.; Sankoff, D.; Tang, H.; Purbojati, R.W.; et al. Long-read sequencing uncovers the adaptive topography of a carnivorous plant genome. Proc. Natl. Acad. Sci. USA 2017, 114, E4435. [Google Scholar] [CrossRef] [Green Version]
- Mahmoud, M.; Gobet, N.; Cruz-Dávalos, D.I.; Mounier, N.; Dessimoz, C.; Sedlazeck, F.J. Structural variant calling: The long and the short of it. Genome Biol. 2019, 20, 246. [Google Scholar] [CrossRef]
- Bhat, J.A.; Ali, S.; Salgotra, R.K.; Mir, Z.A.; Dutta, S.; Jadon, V.; Tyagi, A.; Mushtaq, M.; Jain, N.; Singh, P.K.; et al. Genomic selection in the Era of next generation sequencing for complex traits in plant breeding. Front Genet 2016, 7, 221. [Google Scholar] [CrossRef] [Green Version]
- Midha, M.K.; Wu, M.; Chiu, K.P. Long-read sequencing in deciphering human genetics to a greater depth. Hum. Genet. 2019, 138, 1201–1215. [Google Scholar] [CrossRef]
- Laing, C.; Buchanan, C.; Taboada, E.N.; Zhang, Y.; Kropinski, A.; Villegas, A.; Thomas, J.E.; Gannon, V.P.J. Pan-genome sequence analysis using Panseq: An online tool for the rapid analysis of core and accessory genomic regions. BMC Bioinform. 2010, 11, 461. [Google Scholar] [CrossRef] [Green Version]
- Xiao, T.; Zhou, W. The third generation sequencing: The advanced approach to genetic diseases. Transl. Pediatr. 2020, 9, 163–173. [Google Scholar] [CrossRef]
- Amarasinghe, S.L.; Su, S.; Dong, X.; Zappia, L.; Ritchie, M.E.; Gouil, Q. Opportunities and challenges in long-read sequencing data analysis. Genome Biol. 2020, 21, 30. [Google Scholar] [CrossRef] [Green Version]
- Brůna, T.; Hoff, K.J.; Lomsadze, A.; Stanke, M.; Borodovsky, M. BRAKER2: Automatic eukaryotic genome annotation with GeneMark-EP+ and AUGUSTUS supported by a protein database. NAR Genom. Bioinform. 2021, 3, lqaa108. [Google Scholar] [CrossRef] [PubMed]
- Campbell, M.S.; Holt, C.; Moore, B.; Yandell, M. Genome annotation and curation using MAKER and MAKER-P. Curr. Protoc. Bioinform. 2014, 48, 4.11.11–14.11.39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salzberg, S.L. Next-generation genome annotation: We still struggle to get it right. Genome Biol. 2019, 20, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golicz, A.A.; Bhalla, P.L.; Singh, M.B. MCRiceRepGP: A framework for the identification of genes associated with sexual reproduction in rice. Plant J. 2018, 96, 188–202. [Google Scholar] [CrossRef] [Green Version]
- Scheben, A.; Edwards, D. Bottlenecks for genome-edited crops on the road from lab to farm. Genome Biol. 2018, 19, 178. [Google Scholar] [CrossRef] [Green Version]
- The UniProt Consortium. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Coordinators, N.R. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2018, 46, D8–D13. [Google Scholar] [CrossRef] [Green Version]
- Bayer, P.E.; Edwards, D.; Batley, J. Bias in resistance gene prediction due to repeat masking. Nat. Plants 2018, 4, 762–765. [Google Scholar] [CrossRef]
- Sherman, R.M.; Salzberg, S.L. Pan-genomics in the human genome era. Nat. Rev. Genet. 2020, 21, 243–254. [Google Scholar] [CrossRef]
- Sirén, J.; Garrison, E.; Novak, A.M.; Paten, B.; Durbin, R. Haplotype-aware graph indexes. Bioinformatics 2020, 36, 400–407. [Google Scholar] [CrossRef]
- Ou, L.; Li, D.; Lv, J.; Chen, W.; Zhang, Z.; Li, X.; Yang, B.; Zhou, S.; Yang, S.; Li, W.; et al. Pan-genome of cultivated pepper (Capsicum) and its use in gene presence–absence variation analyses. New Phytol. 2018, 220, 360–363. [Google Scholar] [CrossRef] [Green Version]
- Jiao, W.-B.; Schneeberger, K. Chromosome-level assemblies of multiple Arabidopsis genomes reveal hotspots of rearrangements with altered evolutionary dynamics. Nat. Commun. 2020, 11, 989. [Google Scholar] [CrossRef] [Green Version]
- Gordon, S.P.; Contreras-Moreira, B.; Levy, J.J.; Djamei, A.; Czedik-Eysenberg, A.; Tartaglio, V.S.; Session, A.; Martin, J.; Cartwright, A.; Katz, A.; et al. Gradual polyploid genome evolution revealed by pan-genomic analysis of Brachypodium hybridum and its diploid progenitors. Nat. Commun. 2020, 11, 3670. [Google Scholar] [CrossRef]
- Hübner, S.; Bercovich, N.; Todesco, M.; Mandel, J.R.; Odenheimer, J.; Ziegler, E.; Lee, J.S.; Baute, G.J.; Owens, G.L.; Grassa, C.J.; et al. Sunflower pan-genome analysis shows that hybridization altered gene content and disease resistance. Nat. Plants 2019, 5, 54–62. [Google Scholar] [CrossRef]
- Sun, X.; Jiao, C.; Schwaninger, H.; Chao, C.T.; Ma, Y.; Duan, N.; Khan, A.; Ban, S.; Xu, K.; Cheng, L.; et al. Phased diploid genome assemblies and pan-genomes provide insights into the genetic history of apple domestication. Nat. Genet. 2020, 52, 1423–1432. [Google Scholar] [CrossRef]
- Zhou, P.; Silverstein, K.A.T.; Ramaraj, T.; Guhlin, J.; Denny, R.; Liu, J.; Farmer, A.D.; Steele, K.P.; Stupar, R.M.; Miller, J.R.; et al. Exploring structural variation and gene family architecture with De Novo assemblies of 15 Medicago genomes. BMC Genom. 2017, 18, 261. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Zhu, W.; Diao, S.; Wu, X.; Lu, J.; Ding, C.; Su, X. The poplar pangenome provides insights into the evolutionary history of the genus. Commun. Biol. 2019, 2, 215. [Google Scholar] [CrossRef]
- Okada, R.; Kiyota, E.; Moriyama, H.; Toshiyuki, F.; Valverde, R.A. A new endornavirus species infecting Malabar spinach (Basella alba L.). Arch. Virol. 2014, 159, 807–809. [Google Scholar] [CrossRef]
- Wang, X.; Larrea-Sarmiento, A.; Borth, W.B.; Barone, R.; Olmedo-Velarde, A.; Melzer, M.J.; Suzuki, J.Y.; Wall, M.M.; Hu, J.S. First report of Basella alba naturally infected with basella rugose mosaic virus in Hawaii. Plant Dis. 2020, 104, 2296. [Google Scholar] [CrossRef] [Green Version]
- Silva, L.; Techio, V.; Resende, L.; TBraz, G.; Resende, K.; Samartini, C. Unconventional vegetables collected in Brazil: Chromosome number and description of nuclear DNA content ARTICLE. Crop Breed. Appl. Biotechnol. 2017, 17, 320–326. [Google Scholar] [CrossRef] [Green Version]
- Tripathi, P.; Shahin, L.; Sangra, A.; Bajaj, R.; Arun, A.; Berrios, J.A.N. Current Status and future prospects for select underutilized medicinally valuable plants of puerto rico: A case study. In Medicinal Plants: From Farm to Pharmacy; Joshee, N., Dhekney, S.A., Parajuli, P., Eds.; Springer International Publishing: Cham, Switzerland, 2019; pp. 81–110. [Google Scholar]
- Paz, F.S.; Pinto, C.E.; de Brito, R.M.; Imperatriz-Fonseca, V.L.; Giannini, T.C. Edible fruit plant species in the amazon forest rely mostly on bees and beetles as pollinators. J. Econ. Entomol. 2021, 114, 710–722. [Google Scholar] [CrossRef] [PubMed]
- Lysak, M.A.; Cheung, K.; Kitschke, M.; Bures, P. Ancestral chromosomal blocks are triplicated in Brassiceae species with varying chromosome number and genome size. Plant Physiol. 2007, 145, 402–410. [Google Scholar] [CrossRef] [PubMed]
- Grande, F.; Rizzuti, B.; Occhiuzzi, M.A.; Ioele, G.; Casacchia, T.; Gelmini, F.; Guzzi, R.; Garofalo, A.; Statti, G. Identification by molecular docking of homoisoflavones from Leopoldia comosa as ligands of estrogen receptors. Molecules 2018, 23, 894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boulfia, M.; Lamchouri, F.; Senhaji, S.; Lachkar, N.; Bouabid, K.; Toufik, H. Mineral content, chemical analysis, in vitro antidiabetic and antioxidant activities, and antibacterial power of aqueous and organic extracts of moroccan Leopoldia comosa (L.) parl. bulbs. Evid.-Based Complementary Altern. Med. 2021, 2021, 9932291. [Google Scholar] [CrossRef]
- Maroyi, A. Contribution of Schinziophyton rautanenii to sustainable diets, livelihood needs and environmental sustainability in Southern Africa. Sustainability 2018, 10, 581. [Google Scholar] [CrossRef] [Green Version]
- Frankova, A.; Manourova, A.; Kotikova, Z.; Vejvodova, K.; Drabek, O.; Riljakova, B.; Famera, O.; Ngula, M.; Ndiyoi, M.; Polesny, Z.; et al. The chemical composition of oils and cakes of Ochna serrulata (Ochnaceae) and other underutilized traditional oil trees from Western Zambia. Molecules 2021, 26, 5210. [Google Scholar] [CrossRef]
- Brunt, A.; Phillips, S.U.E.; Jones, R.; Kenten, R. Viruses detected in Ullucus tuberosus (Basellaceae) from Peru and Bolivia. Ann. Appl. Biol. 2008, 101, 65–71. [Google Scholar] [CrossRef]
- Fox, A.; Fowkes, A.; Skelton, A.; Harju, V.; Buxton-Kirk, A.; Kelly, M.; Forde, S.; Pufal, H.; Conyers, C.; Ward, R.; et al. Using high throughput sequencing in support of a plant health outbreak reveals novel viruses in Ullucus tuberosus (Basellaceae). Plant Pathol. 2019, 68, 576–587. [Google Scholar]
- Dholakia, H.; Mehta, D.; Joshi, M.; Delvadiya, I. Molecular characterization of Indian bean (Lablab purpureus L.) genotypes. J. Pharmacogn. Phytochem. 2019, 8, 455–463. [Google Scholar]
- Wang, J.; Chen, X.; Chu, S.; You, Y.; Chi, Y.; Wang, R.; Yang, X.; Hayat, K.; Zhang, D.; Zhou, P. Comparative cytology combined with transcriptomic and metabolomic analyses of Solanum nigrum L. in response to Cd toxicity. J. Hazard. Mater. 2022, 423, 127168. [Google Scholar] [CrossRef]
- Xu, J.; Sun, J.; Du, L.; Liu, X. Comparative transcriptome analysis of cadmium responses in Solanum nigrum and Solanum torvum. New Phytol. 2012, 196, 110–124. [Google Scholar] [CrossRef]
- Khan, A.R.; Park, C.E.; Park, G.-S.; Seo, Y.-J.; So, J.-H.; Shin, J.-H. The whole chloroplast genome sequence of black nightshade plant (Solanum nigrum). Mitochondrial DNA Part A 2017, 28, 169–170. [Google Scholar] [CrossRef]
- Cho, K.-S.; Park, T.-H. Complete chloroplast genome sequence of Solanum nigrum and development of markers for the discrimination of S. nigrum. Hortic. Environ. Biotechnol. 2016, 57, 69–78. [Google Scholar] [CrossRef]
- Leu, Y.-L.; Hwang, T.-L.; Kuo, P.-C.; Liou, K.-P.; Huang, B.-S.; Chen, G.-F. Constituents from Vigna vexillata and their anti-inflammatory activity. Int. J. Mol. Sci. 2012, 13, 9754–9768. [Google Scholar] [CrossRef] [Green Version]
- Dachapak, S.; Tomooka, N.; Somta, P.; Naito, K.; Kaga, A.; Srinives, P. QTL analysis of domestication syndrome in zombi pea (Vigna vexillata), an underutilized legume crop. PLoS ONE 2018, 13, e0200116. [Google Scholar] [CrossRef] [Green Version]
- Marubodee, R.; Ogiso-Tanaka, E.; Isemura, T.; Chankaew, S.; Kaga, A.; Naito, K.; Ehara, H.; Tomooka, N. Construction of an SSR and RAD-marker based molecular linkage map of Vigna vexillata (L.) A. Rich. PLoS ONE 2015, 10, e0138942. [Google Scholar] [CrossRef]
- Damayanti, F.; Lawn, R.J.; Bielig, L.M. Genetic compatibility among domesticated and wild accessions of the tropical tuberous legume Vigna vexillata (L.) A. Rich. Crop Pasture Sci. 2010, 61, 785–797. [Google Scholar] [CrossRef]
- Lulin, H.; Xiao, Y.; Pei, S.; Wen, T.; Shangqin, H. The first illumina-based De Novo transcriptome sequencing and analysis of safflower flowers. PLoS ONE 2012, 7, e38653. [Google Scholar] [CrossRef]
- Chen, J.; Tang, X.; Ren, C.; Wei, B.; Wu, Y.; Wu, Q.; Pei, J. Full-length transcriptome sequences and the identification of putative genes for flavonoid biosynthesis in safflower. BMC Genom. 2018, 19, 548. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.H.; Liao, R.; Dong, X.; Qin, R.; Liu, H. Complete chloroplast genome sequence of Carthamus tinctorius L. from PacBio Sequel Platform. Mitochondrial DNA B Resour. 2019, 4, 2635–2636. [Google Scholar] [CrossRef] [Green Version]
- Couvreur, T.L.P.; Hahn, W.J.; Granville, J.J.d.; Pham, J.L.; Ludeña, B.; Pintaud, J.C. Phylogenetic relationships of the cultivated neotropical palm Bactris gasipaes (Arecaceae) with its wild relatives inferred from chloroplast and nuclear DNA polymorphisms. Syst. Bot. 2007, 32, 519–530. [Google Scholar] [CrossRef]
- Bazzo, B.R.; de Carvalho, L.M.; Carazzolle, M.F.; Pereira, G.A.G.; Colombo, C.A. Development of novel EST-SSR markers in the macaúba palm (Acrocomia aculeata) using transcriptome sequencing and cross-species transferability in Arecaceae species. BMC Plant Biol. 2018, 18, 276. [Google Scholar] [CrossRef]
- Xiao, Y. Efficient isolation of high quality RNA from tropical palms for RNA-seq analysis. Plant Omics 2012, 5, 584–589. [Google Scholar]
- Santos da Silva, R.; Roland Clement, C.; Balsanelli, E.; de Baura, V.A.; Maltempi de Souza, E.; Pacheco de Freitas Fraga, H.; do Nascimento Vieira, L. The plastome sequence of Bactris gasipaes and evolutionary analysis in tribe Cocoseae (Arecaceae). PLoS ONE 2021, 16, e0256373. [Google Scholar] [CrossRef] [PubMed]
- Levi, A.; Simmons, A.M.; Massey, L.; Coffey, J.; Wechter, W.P.; Jarret, R.L.; Tadmor, Y.; Nimmakayala, P.; Reddy, U.K. Genetic diversity in the desert watermelon Citrullus colocynthis and its relationship with Citrullus species as determined by high-frequency oligonucleotides-targeting active gene markers. J. Am. Soc. Hortic. Sci. 2017, 142, 47–56. [Google Scholar] [CrossRef]
- Wang, Z.; Hu, H.; Goertzen, L.R.; McElroy, J.S.; Dane, F. Analysis of the Citrullus colocynthis transcriptome during water deficit stress. PLoS ONE 2014, 9, e104657. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Zhao, S.; Sun, H.; Wang, X.; Wu, S.; Lin, T.; Ren, Y.; Gao, L.; Deng, Y.; Zhang, J.; et al. Resequencing of 414 cultivated and wild watermelon accessions identifies selection for fruit quality traits. Nat. Genet. 2019, 51, 1616–1623. [Google Scholar] [CrossRef]
- Gao, P.; Xu, W.; Yan, T.; Zhang, C.; Lv, X.; He, Y. Application of near-infrared hyperspectral imaging with machine learning methods to identify geographical origins of dry narrow-leaved oleaster (Elaeagnus angustifolia) fruits. Foods 2019, 8, 620. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Zhu, J.; Yang, X.; Wu, H.; Wei, Q.; Wei, H.; Zhang, H. Growth performance, organ-level ionic relations and organic osmoregulation of Elaeagnus angustifolia in response to salt stress. PLoS ONE 2018, 13, e0191552. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.; Li, J.P.; Yuan, F.; Yang, Z.; Wang, B.S.; Chen, M. Transcriptome profiling of genes involved in photosynthesis in Elaeagnus angustifolia L. under salt stress. Photosynthetica 2018, 56, 998–1009. [Google Scholar] [CrossRef]
- Liu, X.; Chen, C.; Liu, Y.; Liu, Y.; Zhao, Y.; Chen, M. The presence of moderate salt can increase tolerance of Elaeagnus angustifolia seedlings to waterlogging stress. Plant Signal. Behav. 2020, 15, 1743518. [Google Scholar] [CrossRef]
- Yemataw, Z.; Muzemil, S.; Ambachew, D.; Tripathi, L.; Tesfaye, K.; Chala, A.; Farbos, A.; O’Neill, P.; Moore, K.; Grant, M.; et al. Genome sequence data from 17 accessions of Ensete ventricosum, a staple food crop for millions in Ethiopia. Data Brief 2018, 18, 285–293. [Google Scholar] [CrossRef]
- Harrison, J.; Moore, K.A.; Paszkiewicz, K.; Jones, T.; Grant, M.R.; Ambacheew, D.; Muzemil, S.; Studholme, D.J. A draft genome sequence for Ensete ventricosum, the drought-tolerant “tree against hunger”. Agronomy 2014, 4, 13–33. [Google Scholar] [CrossRef] [Green Version]
- Biswas, M.K.; Darbar, J.N.; Borrell, J.S.; Bagchi, M.; Biswas, D.; Nuraga, G.W.; Demissew, S.; Wilkin, P.; Schwarzacher, T.; Heslop-Harrison, J.S. The landscape of microsatellites in the enset (Ensete ventricosum) genome and web-based marker resource development. Sci. Rep. 2020, 10, 15312. [Google Scholar] [CrossRef]
- Price, E.J.; Drapal, M.; Perez-Fons, L.; Amah, D.; Bhattacharjee, R.; Heider, B.; Rouard, M.; Swennen, R.; Becerra Lopez-Lavalle, L.A.; Fraser, P.D. Metabolite database for root, tuber, and banana crops to facilitate modern breeding in understudied crops. Plant J. 2020, 101, 1258–1268. [Google Scholar] [CrossRef]
- Ferreira, L.T.; Venancio, V.P.; Kawano, T.; Abrão, L.C.C.; Tavella, T.A.; Almeida, L.D.; Pires, G.S.; Bilsland, E.; Sunnerhagen, P.; Azevedo, L.; et al. Chemical genomic profiling unveils the in vitro and in vivo antiplasmodial mechanism of açaí (Euterpe oleracea Mart.) polyphenols. ACS Omega 2019, 4, 15628–15635. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, L.C.; de Oliveira, M.d.S.P.; Davide, L.C.; Torres, G.A. Karyotype and genome size in Euterpe Mart. (Arecaceae) species. Comp Cytogenet. 2016, 10, 17–25. [Google Scholar]
- Kron, K.A.; Powell, E.A.; Luteyn, J.L. Phylogenetic relationships within the blueberry tribe (Vaccinieae, Ericaceae) based on sequence data from MATK and nuclear ribosomal ITS regions, with comments on the placement of Satyria. Am. J. Bot. 2002, 89, 327–336. [Google Scholar] [CrossRef]
- Llivisaca, S.; Manzano, P.; Ruales, J.; Flores, J.; Mendoza, J.; Peralta, E.; Cevallos-Cevallos, J.M. Chemical, antimicrobial, and molecular characterization of mortiño (Vaccinium floribundum Kunth) fruits and leaves. Food Sci. Nutr. 2018, 6, 934–942. [Google Scholar] [CrossRef]
- Ligarreto, G.A.; Patiño Mdel, P.; Magnitskiy, S.V. Phenotypic plasticity of Vaccinium meridionale (Ericaceae) in wild populations of mountain forests in Colombia. Rev. Biol. Trop. 2011, 59, 569–583. [Google Scholar]
- Arango-Varela, S.S.; Luzardo-Ocampo, I.; Reyes-Dieck, C.; Yahia, E.M.; Maldonado-Celis, M.E. Antiproliferative potential of Andean Berry (Vaccinium meridionale Swartz) juice in combination with Aspirin in human SW480 colon adenocarcinoma cells. J. Food Biochem. 2021, 45, e13760. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tay Fernandez, C.G.; Nestor, B.J.; Danilevicz, M.F.; Gill, M.; Petereit, J.; Bayer, P.E.; Finnegan, P.M.; Batley, J.; Edwards, D. Pangenomes as a Resource to Accelerate Breeding of Under-Utilised Crop Species. Int. J. Mol. Sci. 2022, 23, 2671. https://doi.org/10.3390/ijms23052671
Tay Fernandez CG, Nestor BJ, Danilevicz MF, Gill M, Petereit J, Bayer PE, Finnegan PM, Batley J, Edwards D. Pangenomes as a Resource to Accelerate Breeding of Under-Utilised Crop Species. International Journal of Molecular Sciences. 2022; 23(5):2671. https://doi.org/10.3390/ijms23052671
Chicago/Turabian StyleTay Fernandez, Cassandria Geraldine, Benjamin John Nestor, Monica Furaste Danilevicz, Mitchell Gill, Jakob Petereit, Philipp Emanuel Bayer, Patrick Michael Finnegan, Jacqueline Batley, and David Edwards. 2022. "Pangenomes as a Resource to Accelerate Breeding of Under-Utilised Crop Species" International Journal of Molecular Sciences 23, no. 5: 2671. https://doi.org/10.3390/ijms23052671