
Secondary Structure of Influenza A Virus Genomic Segment 8 RNA Folded in a Cellular Environment

 , , ,
, , ,  ,
, 
Abstract
:1. Introduction
2. Results
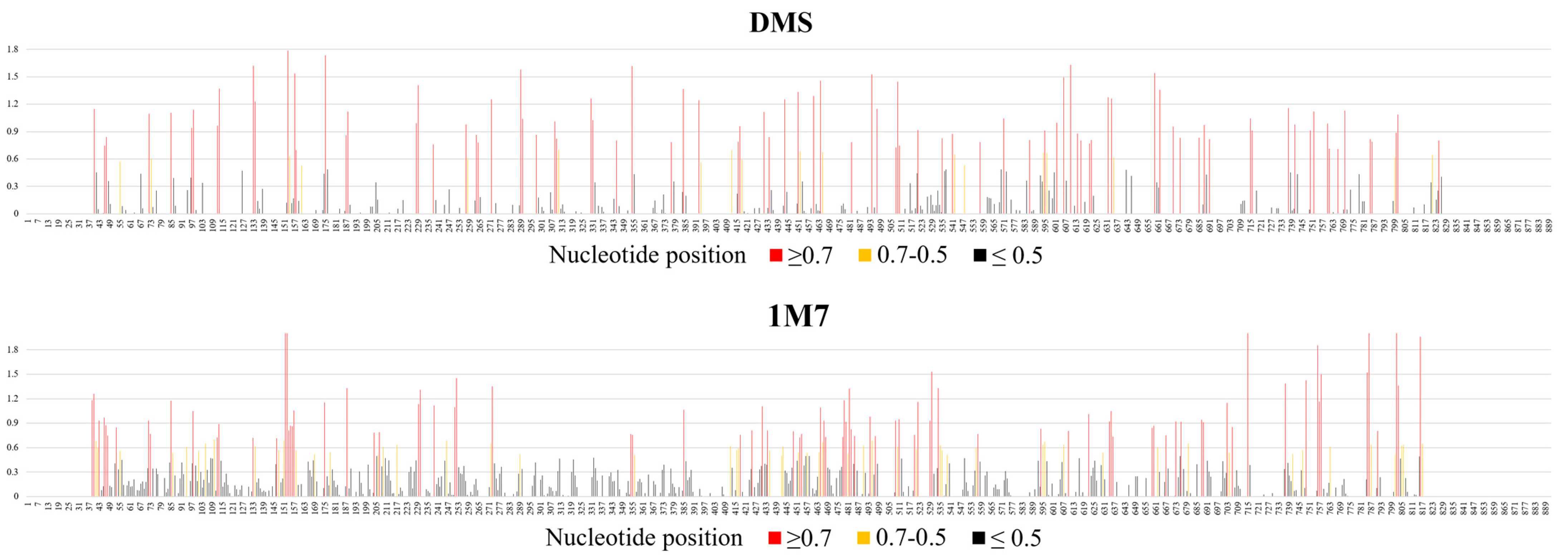
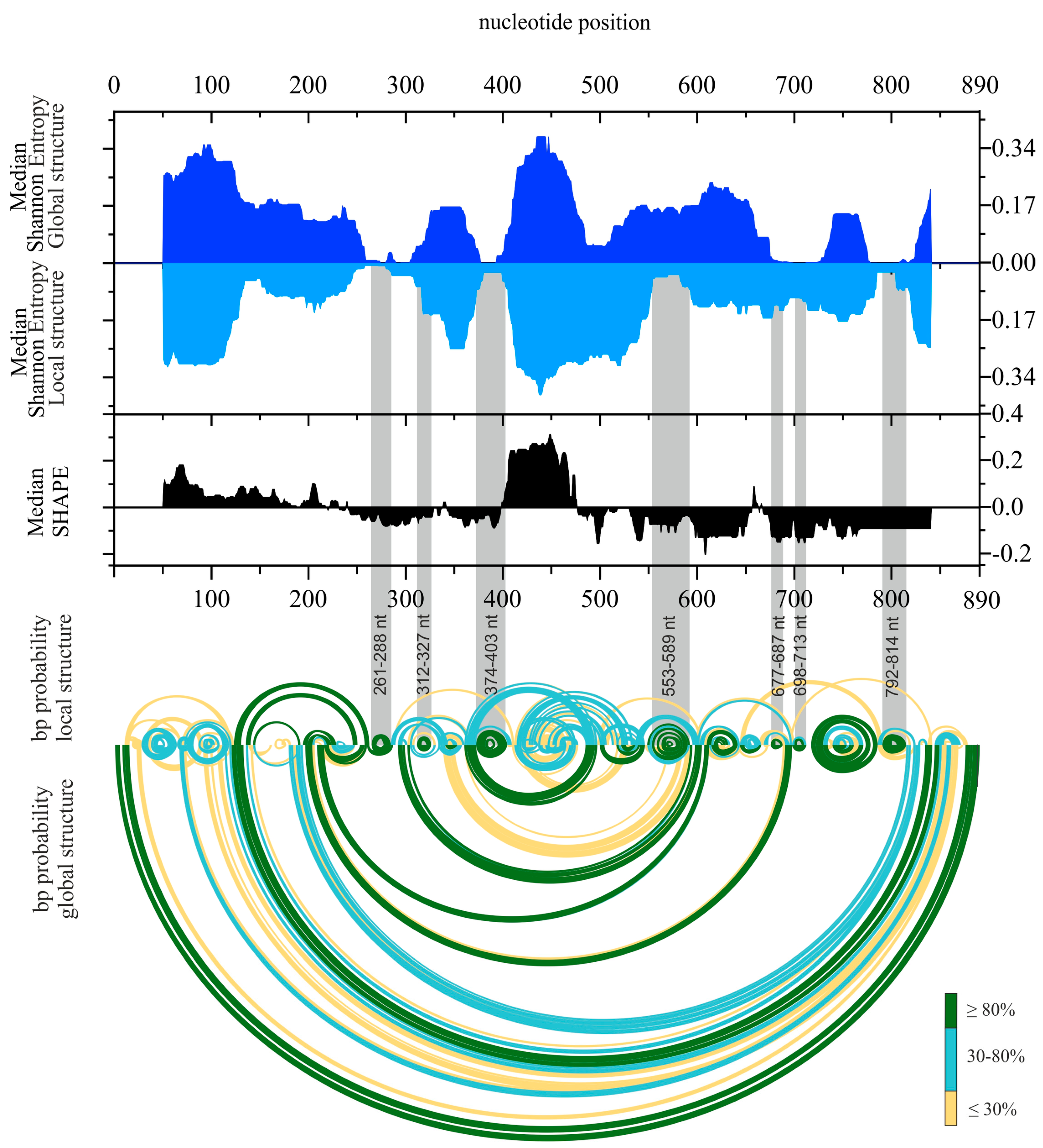
2.1. Chemical Mapping of vRNA8 in Cell Lysates
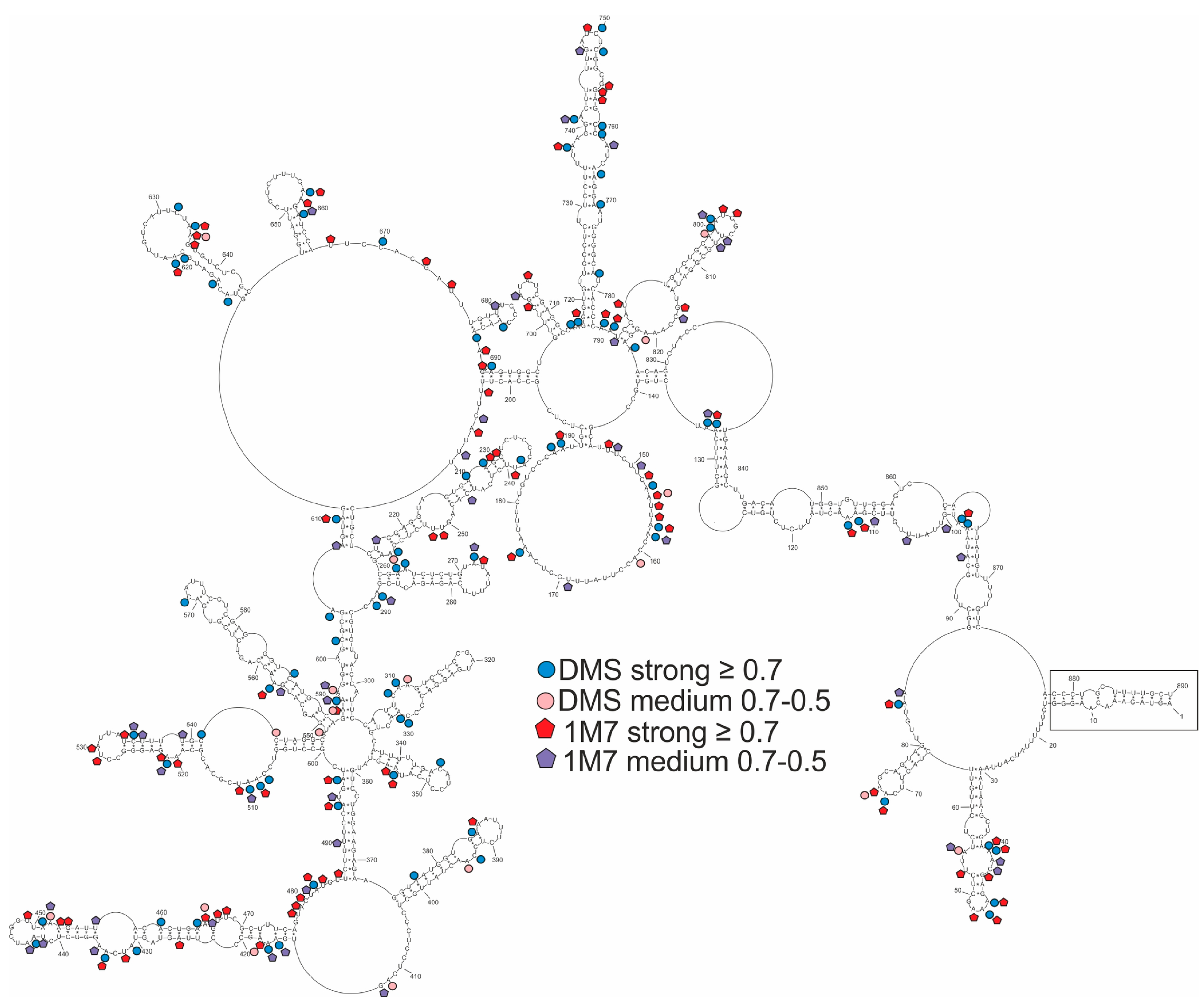
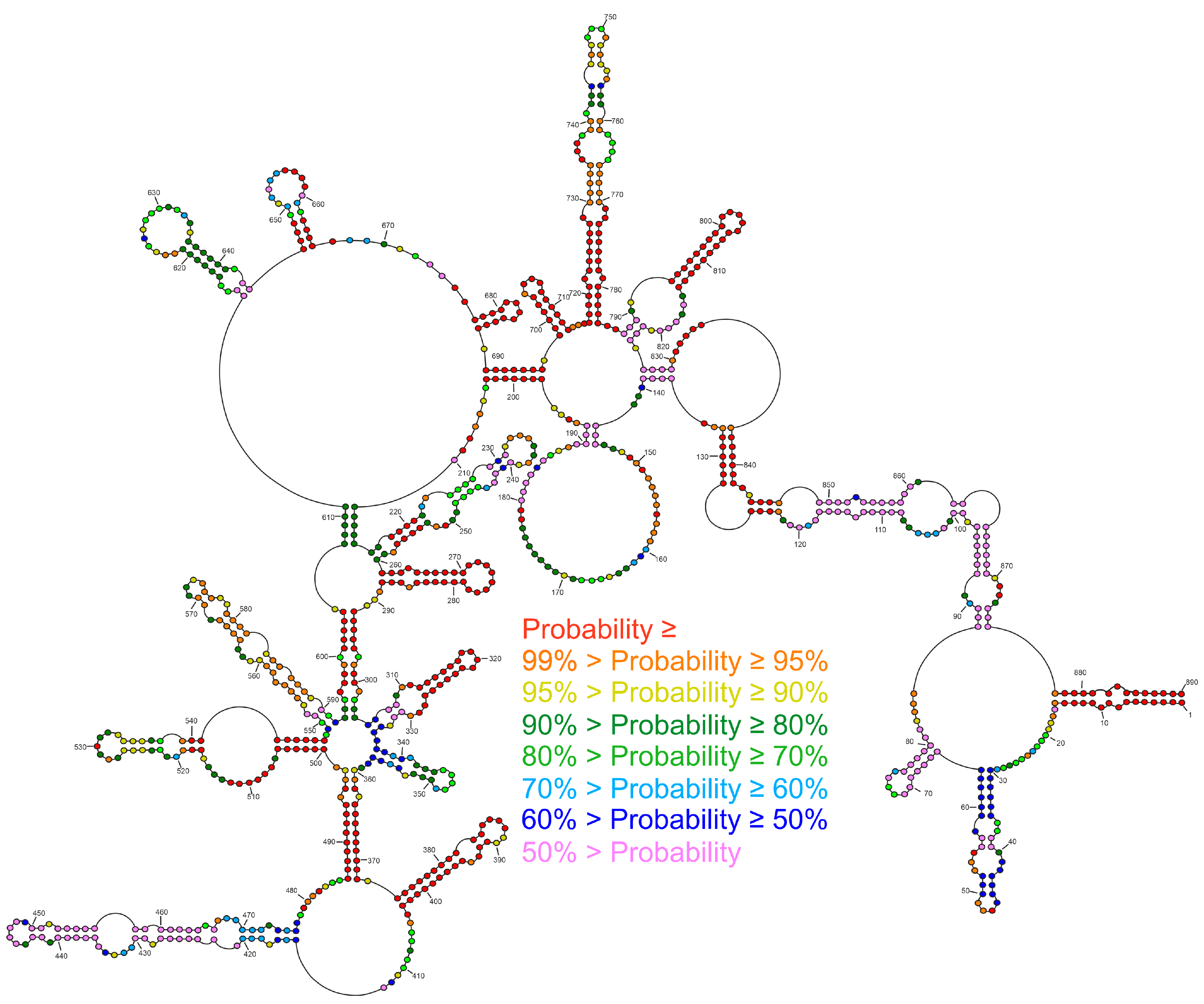
2.2. Secondary Structure of Segment 8 vRNA in Cell Lysate
2.3. Well-Defined Structural Motifs of vRNA8 in Cell Lysate
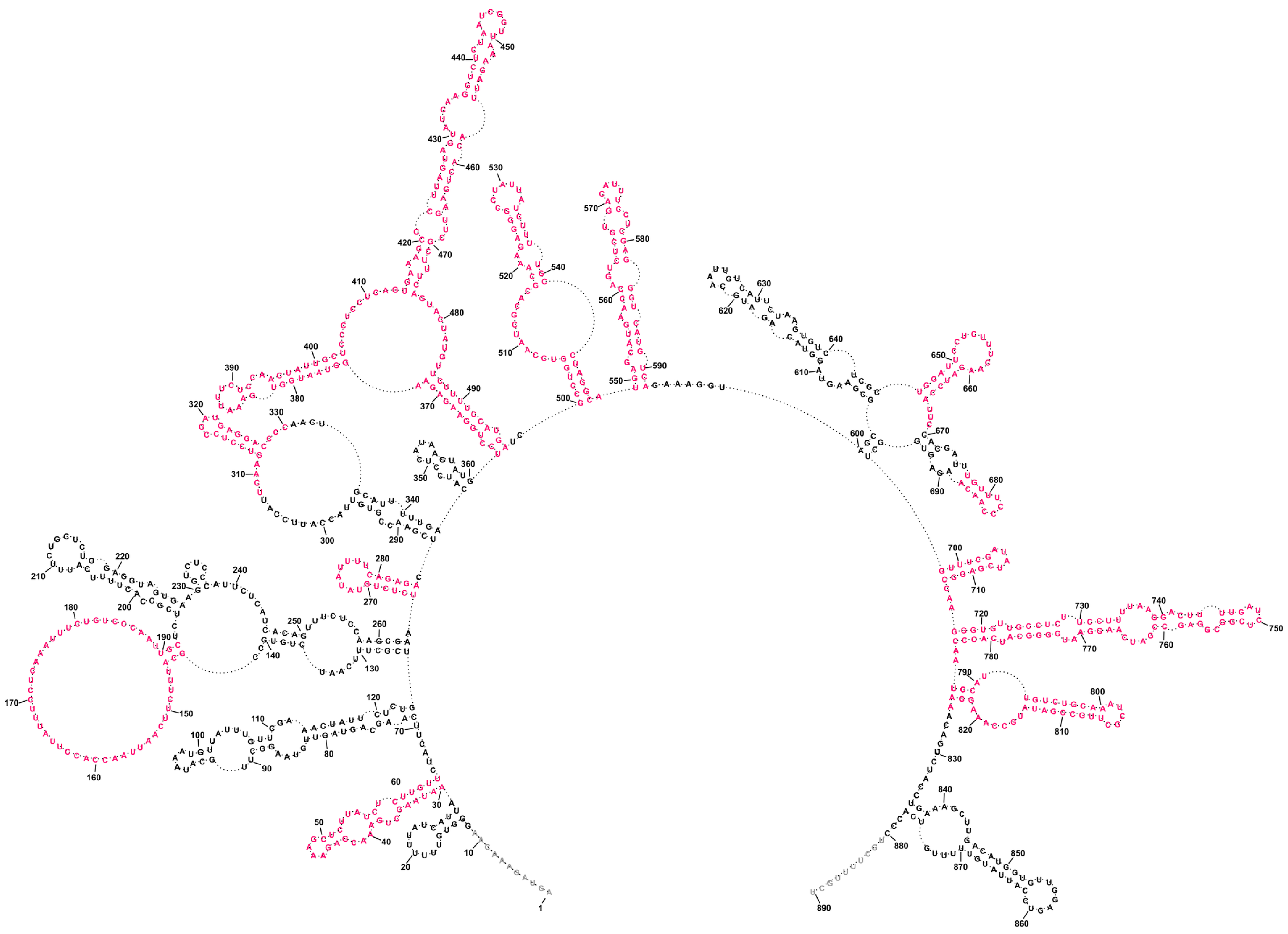
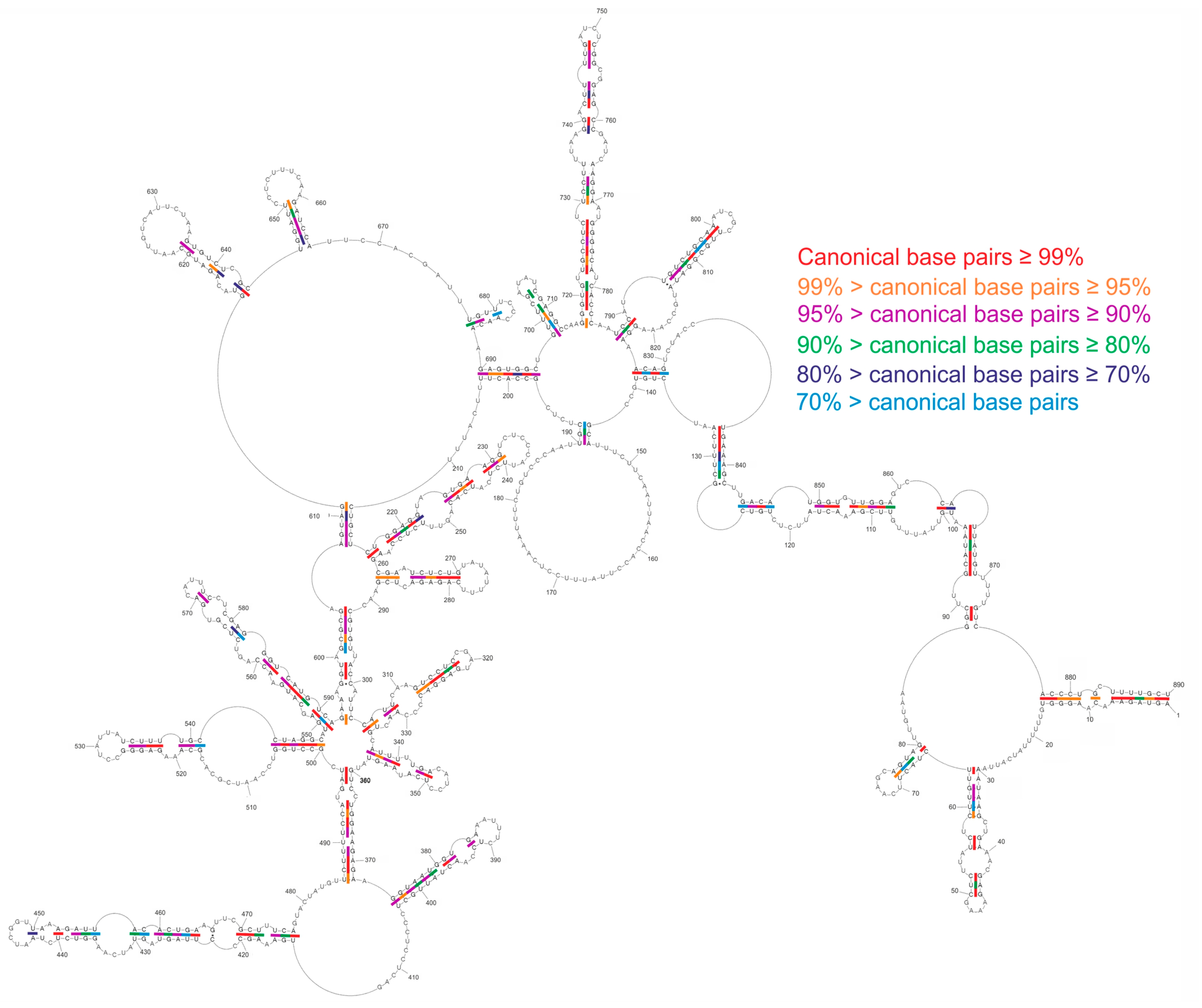
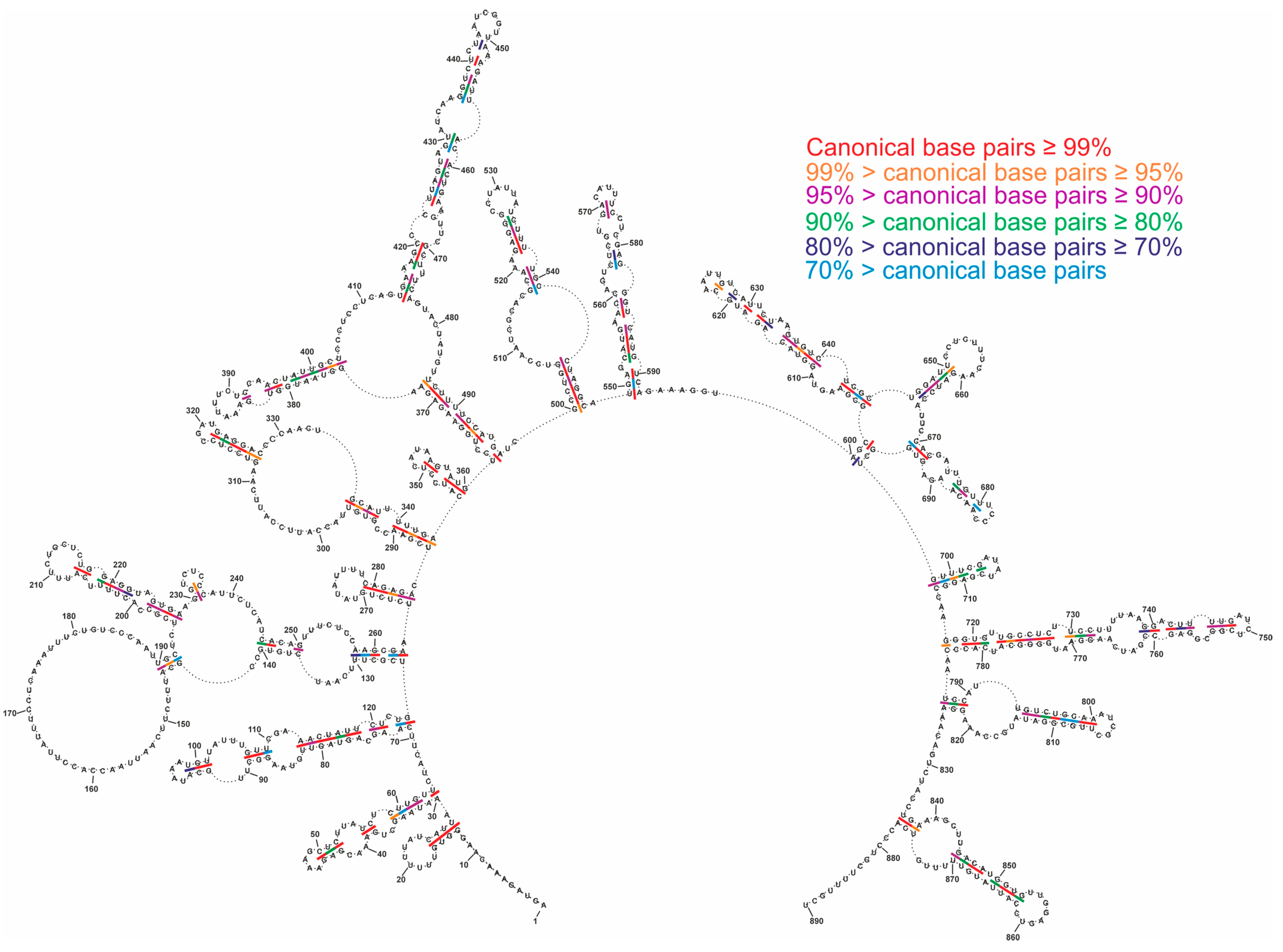
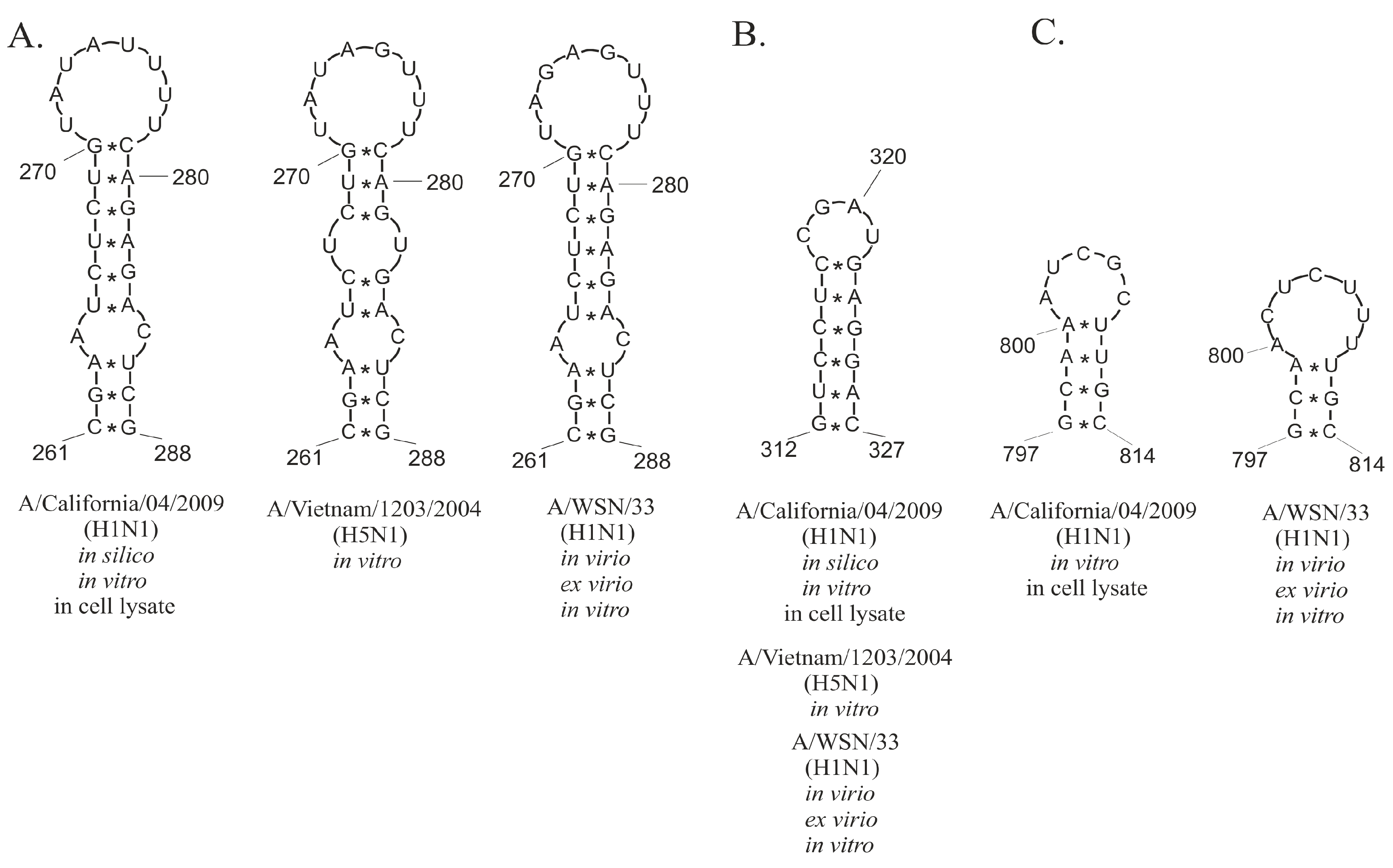
2.4. Conservation of Predicted vRNA8 Structure in Cell Lysate throughout Influenza A Strains
3. Discussion
3.1. Structural Probing of vRNA8 in Cellular Environment Showed Difference in RNA Accessibility to Chemical Reagents
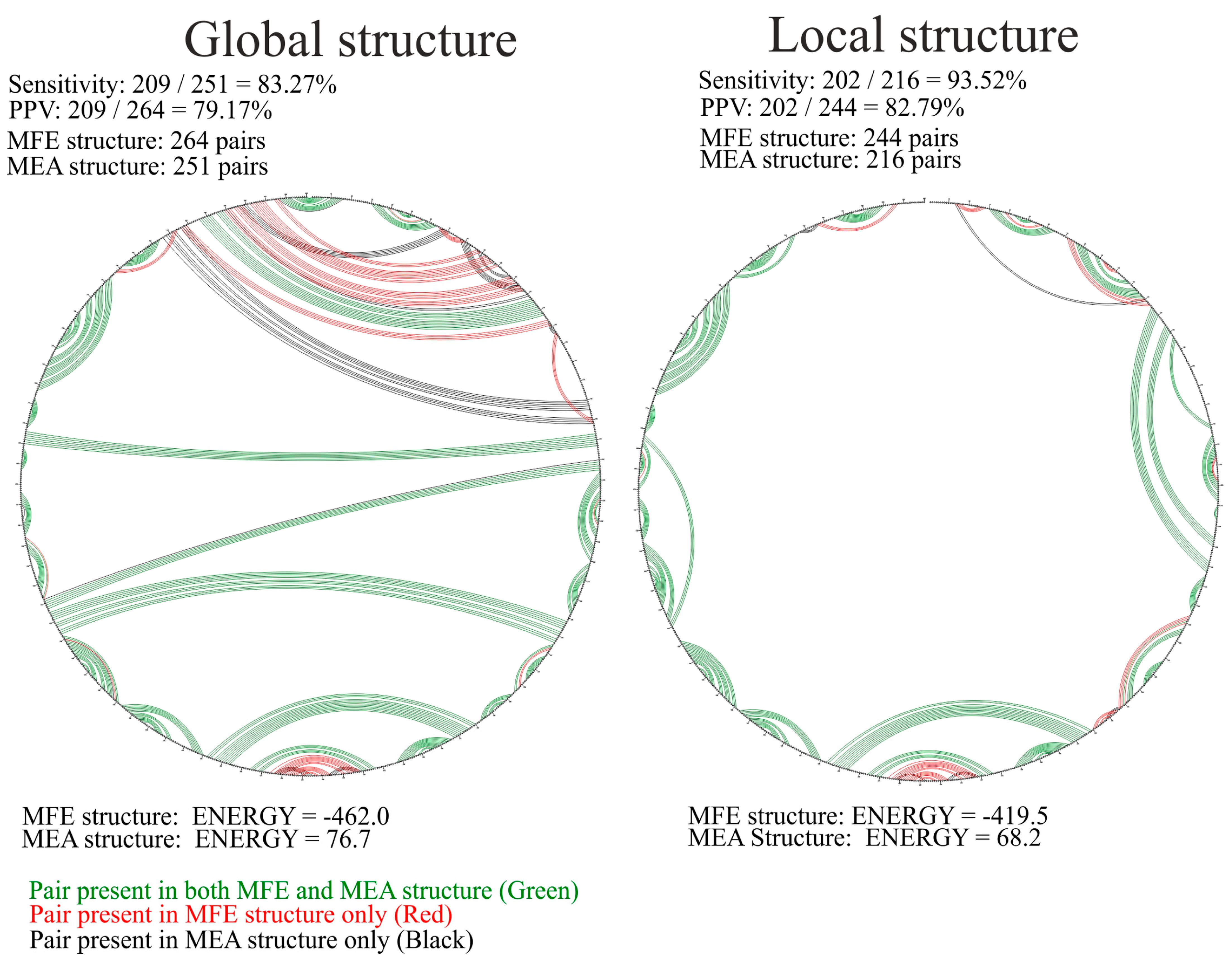
3.2. Different RNA Structure Prediction Approaches Identified Structural Motifs with High Fidelity
3.3. Several Well-Defined and Conserved for IAV Structural Motifs of vRNA8 Were Identified
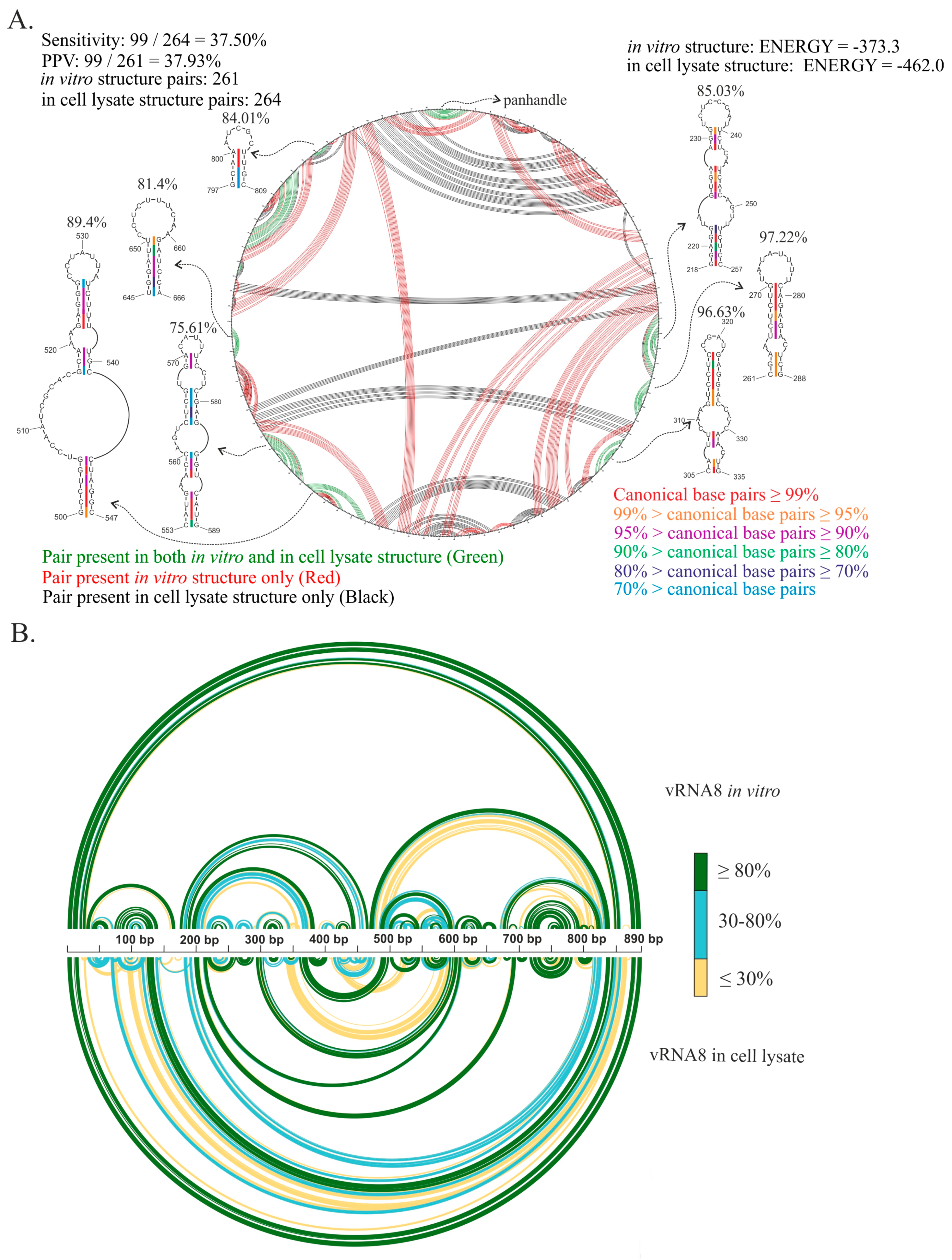
3.4. Comparison of vRNA8 Structure in Cell Lysates and In Vitro
3.5. Motifs of the Highest Base-Pairing Probability Are Present in NP-Enriched and NP-Depleted Regions of vRNA in vRNP Complexes
3.6. vRNA8 Structure Accessibility in a Cellular Environment Could Be Useful to Design Antivirals
4. Materials and Methods
4.1. Cell Culture and Virus Propagation
4.2. Preparation of the Cellular-Viral Lysates
4.3. In Vitro Transcription of vRNA8
4.4. Primers Synthesis and Fluorescent Labeling of the Primers
4.5. Chemical Probing
4.6. Primer Extension by Reverse Transcription
4.7. DNA Sequencing Ladders
4.8. Real-Time PCR
4.9. Western Blot
4.10. Chemical Mapping Data Analysis
4.11. Secondary Structure Prediction and Visualization
4.12. Bioinformatic Analysis of Structure Conservation
4.13. Calculations of Shannon entropy
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Su, S.; Fu, X.; Li, G.; Kerlin, F.; Veit, M. Novel Influenza D virus: Epidemiology, pathology, evolution and biological characteristics. Virulence 2017, 8, 1580–1591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mostafa, A.; Abdelwhab, E.M.; Mettenleiter, T.C.; Pleschka, S. Zoonotic potential of influenza A viruses: A comprehensive overview. Viruses 2018, 10, 497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health Organisation. Global Influenza Strategy Summary 2019–2030 Influenza; World Health Organisation: Geneva, Switzerland, 2019; pp. 1–2. [Google Scholar]
- Francis, M.E.; King, M.L.; Kelvin, A.A. Back to the future for influenza preimmunity—Looking back at influenza virus history to infer the outcome of future infections. Viruses 2019, 11, 122. [Google Scholar] [CrossRef] [Green Version]
- Duwe, S. Influenza viruses-antiviral therapy and resistance. GMS Infect. Dis. 2017, 5, Doc04. [Google Scholar] [CrossRef] [PubMed]
- Houser, K.; Subbarao, K. Influenza vaccines: Challenges and solutions. Cell Host Microbe 2015, 17, 295–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- States, U. CDC Estimates of 2009 H1N1 Influenza Cases, Hospitalizations and Deaths in the United States, April–December 12, 2009. Emerg. Infect. Dis. 2010, 2009, 1–7. [Google Scholar]
- Kim, H.; Webster, R.G.; Webby, R.J. Influenza Virus: Dealing with a Drifting and Shifting Pathogen. Viral Immunol. 2018, 31, 174–183. [Google Scholar] [CrossRef]
- Shao, W.; Li, X.; Goraya, M.U.; Wang, S.; Chen, J.L. Evolution of influenza a virus by mutation and re-assortment. Int. J. Mol. Sci. 2017, 18, 1650. [Google Scholar] [CrossRef] [Green Version]
- Balgi, A.D.; Wang, J.; Cheng, D.Y.H.; Ma, C.; Pfeifer, T.A.; Shimizu, Y.; Anderson, H.J.; Pinto, L.H.; Lamb, R.A.; DeGrado, W.F.; et al. Inhibitors of the Influenza A Virus M2 Proton Channel Discovered Using a High-Throughput Yeast Growth Restoration Assay. PLoS ONE 2013, 8, e55271. [Google Scholar] [CrossRef]
- Hayden, F.G. Newer influenza antivirals, biotherapeutics and combinations. Influenza Other Respir. Viruses 2012, 7, 63–75. [Google Scholar] [CrossRef]
- Masuda, H.; Suzuki, H.; Oshitani, H.; Saito, R.; Kawasaki, S.; Nishikawa, M.; Satoh, H. Incidence of amantadine-resistant influenza A viruses in sentinel surveillance sites and nursing homes in Niigata, Japan. Microbiol. Immunol. 2000, 44, 833–839. [Google Scholar] [CrossRef] [PubMed]
- Saito, R.; Sakai, T.; Sato, I.; Sano, Y.; Oshitani, H.; Sato, M.; Suzuki, H. Frequency of amantadine-resistant influenza A viruses during two seasons featuring cocirculation of H1N1 and H3N2. J. Clin. Microbiol. 2003, 41, 2164–2165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Young, J.S.; Jeung, H.P.; Lee, J.H.; Yun, H.B.; Song, M.S.; Oh, T.K.; Han, H.S.; Pascua, P.N.Q.; Choi, Y.K. Emergence of amantadine-resistant H3N2 avian influenza A virus in South Korea. J. Clin. Microbiol. 2008, 46, 3788–3790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaraket, H.; Saito, R.; Suzuki, Y.; Baranovich, T.; Dapat, C.; Caperig-Dapat, I.; Suzuki, H. Genetic makeup of amantadine-resistant and oseltamivir-resistant human influenza A/H1N1 viruses. J. Clin. Microbiol. 2010, 48, 1085–1092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gultyaev, A.P.; Fouchier, R.A.M.; Olsthoorn, R.C.L. Influenza virus RNA structure: Unique and common features. Int. Rev. Immunol. 2010, 29, 533–556. [Google Scholar] [CrossRef]
- Ferhadian, D.; Contrant, M.; Printz-Schweigert, A.; Smyth, R.P.; Paillart, J.C.; Marquet, R. Structural and functional motifs in influenza virus RNAs. Front. Microbiol. 2018, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Jiang, T.; Nogales, A.; Baker, S.F.; Martinez-Sobrido, L.; Turner, D.H. Mutations designed by ensemble defect to misfold conserved RNA structures of influenza A segments 7 and 8 affect splicing and attenuate viral replication in cell culture. PLoS ONE 2016, 11, e0156906. [Google Scholar] [CrossRef] [Green Version]
- Priore, S.F.; Kierzek, E.; Kierzek, R.; Baman, J.R.; Moss, W.N.; Dela-Moss, L.I.; Turner, D.H. Secondary structure of a conserved domain in the intron of influenza A NS1 mRNA. PLoS ONE 2013, 8, e70615. [Google Scholar] [CrossRef]
- Piasecka, J.; Jarmolowicz, A.; Kierzek, E. Organization of the Influenza A Virus Genomic RNA in the Viral Replication Cycle—Structure, Interactions, and Implications for the Emergence of New Strains. Pathogens 2020, 9, 951. [Google Scholar] [CrossRef]
- Tomescu, A.I.; Robb, N.C.; Hengrung, N.; Fodor, E.; Kapanidis, A.N. Single-molecule FRET reveals a corkscrew RNA structure for the polymerase-bound influenza virus promoter. Proc. Natl. Acad. Sci. USA 2014, 111, E3335–E3342. [Google Scholar] [CrossRef] [Green Version]
- Coloma, R.; Arranz, R.; De La Rosa-Trevín, J.M.; Sorzano, C.O.S.; Munier, S.; Carlero, D.; Naffakh, N.; Ortín, J.; Martín-Benito, J. Structural insights into influenza A virus ribonucleoproteins reveal a processive helical track as transcription mechanism. Nat. Microbiol. 2020, 5, 727–734. [Google Scholar] [CrossRef] [PubMed]
- Pflug, A.; Guilligay, D.; Reich, S.; Cusack, S. Structure of influenza A polymerase bound to the viral RNA promoter. Nature 2014, 516, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Fodor, E.; Brownlee, G.G.; Seong, B.L. Mutational analysis of the RNA-fork model of the influenza A virus vRNA promoter in vivo. J. Gen. Virol. 1997, 78, 353–357. [Google Scholar] [CrossRef] [PubMed]
- Le Sage, V.; Kanarek, J.P.; Snyder, D.J.; Cooper, V.S.; Lakdawala, S.S.; Lee, N. Mapping of Influenza Virus RNA-RNA Interactions Reveals a Flexible Network. Cell Rep. 2020, 31, 107823. [Google Scholar] [CrossRef]
- Wandzik, J.M.; Kouba, T.; Karuppasamy, M.; Pflug, A.; Drncova, P.; Provaznik, J.; Azevedo, N.; Cusack, S. A structure-based model for the complete transcription cycle of influenza polymerase. Cell 2020, 181, 877–893. [Google Scholar] [CrossRef]
- Noble, E.; Mathews, D.H.; Chen, J.L.; Turner, D.H.; Takimoto, T.; Kim, B. Biophysical Analysis of Influenza A Virus RNA Promoter at Physiological Temperatures. J. Biol. Chem. 2011, 286, 22965–22970. [Google Scholar] [CrossRef] [Green Version]
- Crescenzo-Chaigne, B.; Barbezange, C.; van der Werf, S. The Panhandle Formed by Influenza A and C Virus NS Non-Coding Regions Determines NS Segment Expression. PLoS ONE 2013, 8, e81550. [Google Scholar]
- Dadonaite, B.; Gilbertson, B.; Knight, M.L.; Trifkovic, S.; Rockman, S.; Laederach, A.; Brown, L.E.; Fodor, E.; Bauer, D.L.V. The structure of the influenza A virus genome. Nat. Microbiol. 2019, 4, 1781–1789. [Google Scholar] [CrossRef]
- Lee, M.-K.; Kim, H.-E.; Park, E.-B.; Lee, J.; Kim, K.-H.; Lim, K.; Yum, S.; Lee, Y.-H.; Kang, S.-J.; Lee, J.-H.; et al. Structural features of influenza A virus panhandle RNA enabling the activation of RIG-I independently of 5′-triphosphate. Nucleic Acids Res. 2016, 44, 8407–8416. [Google Scholar] [CrossRef]
- Michalak, P.; Soszynska-Jozwiak, M.; Biala, E.; Moss, W.N.W.N.; Kesy, J.; Szutkowska, B.; Lenartowicz, E.; Kierzek, R.; Kierzek, E. Secondary structure of the segment 5 genomic RNA of influenza A virus and its application for designing antisense oligonucleotides. Sci. Rep. 2019, 9, 3801. [Google Scholar] [CrossRef] [Green Version]
- Ruszkowska, A.; Lenartowicz, E.; Moss, W.N.; Kierzek, R.; Kierzek, E. Secondary structure model of the naked segment 7 influenza A virus genomic RNA. Biochem. J. 2016, 473, 4327–4348. [Google Scholar] [CrossRef] [PubMed]
- Lenartowicz, E.; Kesy, J.; Ruszkowska, A.; Soszynska-Jozwiak, M.; Michalak, P.; Moss, W.N.; Turner, D.H.; Kierzek, R.; Kierzek, E. Self-folding of naked segment 8 genomic RNA of influenza a virus. PLoS ONE 2016, 11, e0148281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenartowicz, E.; Nogales, A.; Kierzek, E.; Kierzek, R.; Martínez-Sobrido, L.; Turner, D.H. Antisense Oligonucleotides Targeting Influenza A Segment 8 Genomic RNA Inhibit Viral Replication. Nucleic Acid Ther. 2016, 26, 277–285. [Google Scholar] [CrossRef] [Green Version]
- Kierzek, E. Binding of short oligonucleotides to RNA: Studies of the binding of common RNA structural motifs to isoenergetic microarrays. Biochemistry 2009, 48, 11344–11356. [Google Scholar] [CrossRef] [PubMed]
- Nakano, M.; Sugita, Y.; Kodera, N.; Miyamoto, S.; Muramoto, Y.; Wolf, M.; Noda, T. Ultrastructure of influenza virus ribonucleoprotein complexes during viral RNA synthesis. Commun. Biol. 2021, 4, 858. [Google Scholar] [CrossRef]
- Arranz, R.; Coloma, R.; Chichón, F.J.; Conesa, J.J.; Carrascosa, J.L.; Valpuesta, J.M.; Ortín, J.; Martín-Benito, J. The structure of native influenza virion ribonucleoproteins. Science 2012, 338, 1634–1637. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.; Le Sage, V.; Nanni, A.V.; Snyder, D.J.; Cooper, V.S.; Lakdawala, S.S. Genome-wide analysis of influenza viral RNA and nucleoprotein association. Nucleic Acids Res. 2017, 45, 8968–8977. [Google Scholar] [CrossRef] [Green Version]
- Le Sage, V.; Nanni, A.V.; Bhagwat, A.R.; Snyder, D.J.; Cooper, V.S.; Lakdawala, S.S.; Lee, N. Non-uniform and non-random binding of nucleoprotein to influenza A and B viral RNA. Viruses 2018, 10, 522. [Google Scholar] [CrossRef] [Green Version]
- Moeller, A.; Kirchdoerfer, R.N.; Potter, C.S.; Carragher, B.; Wilson, I.A. Organization of the influenza virus replication machinery. Science 2012, 338, 1631–1634. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Gu, M.; Zheng, Q.; Gao, R.; Liu, X. Packaging signal of influenza A virus. Virol. J. 2021, 18, 1–10. [Google Scholar] [CrossRef]
- Williams, G.D.; Townsend, D.; Wylie, K.M.; Kim, P.J.; Amarasinghe, G.K.; Kutluay, S.B.; Boon, A.C.M. Nucleotide resolution mapping of influenza A virus nucleoprotein-RNA interactions reveals RNA features required for replication. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iselin, L.; Palmalux, N.; Kamel, W.; Simmonds, P.; Mohammed, S.; Castello, A. Uncovering viral RNA–host cell interactions on a proteome-wide scale. Trends Biochem. Sci. 2022, 47, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Marc, D. Influenza virus non-structural protein NS1: Interferon antagonism and beyond. J. Gen. Virol. 2014, 95, 2594–2611. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Zheng, M.; Wang, P.; Mok, B.W.Y.; Liu, S.; Lau, S.Y.; Chen, P.; Liu, Y.C.; Liu, H.; Chen, Y.; et al. An NS-segment exonic splicing enhancer regulates influenza A virus replication in mammalian cells. Nat. Commun. 2017, 8, 14751. [Google Scholar] [CrossRef] [Green Version]
- Kuo, R.L.; Zhao, C.; Malur, M.; Krug, R.M. Influenza IAV strains that circulate in humans differ in the ability of their NS1 proteins to block the activation of IRF3 and interferon-β transcription. Virology 2010, 408, 146–158. [Google Scholar] [CrossRef] [PubMed]
- Hale, B.G.; Randall, R.E.; Ortin, J.; Jackson, D. The multifunctional NS1 protein of influenza A viruses. J. Gen. Virol. 2008, 89, 2359–2376. [Google Scholar] [CrossRef]
- Lu, Z.J.; Gloor, J.W.; Mathews, D.H. Improved RNA secondary structure prediction by maximizing expected pair accuracy. RNA 2009, 15, 1805–1813. [Google Scholar] [CrossRef] [Green Version]
- Hajiaghayi, M.; Condon, A.; Hoos, H.H. Analysis of energy-based algorithms for RNA secondary structure prediction. BMC Bioinform. 2012, 13, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Soszynska-Jozwiak, M.; Pszczola, M.; Piasecka, J.; Peterson, J.M.; Moss, W.N.; Taras-Goslinska, K.; Kierzek, R.; Kierzek, E. Universal and strain specific structure features of segment 8 genomic RNA of influenza A virus–application of 4-thiouridine photocrosslinking. J. Biol. Chem. 2021, 297, 101245. [Google Scholar] [CrossRef]
- Leamy, K.A.; Assmann, S.M.; Mathews, D.H.; Bevilacqua, P.C. Bridging the gap between in vitro and in vivo RNA folding. Q. Rev. Biophys. 2016, 49, e10. [Google Scholar] [CrossRef] [Green Version]
- Bae, S.H.; Cheong, H.K.; Lee, J.H.; Cheong, C.; Kainosho, M.; Choi, B.S. Structural features of an influenza virus promoter and their implications for viral RNA synthesis. Proc. Natl. Acad. Sci. USA 2001, 98, 10602–10607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, G.; Zhou, Y. Cytoplasm and Beyond: Dynamic Innate Immune Sensing of Influenza A Virus by RIG-I. J. Virol. 2019, 93, e02299-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathews, D.H. RNA secondary structure analysis using RNAstructure. Curr. Protoc. Bioinform. 2014, 46, 12.6.1–12.6.14. [Google Scholar] [CrossRef] [Green Version]
- Mathews, D.H. Using the RNAstructure Software Package to Predict Conserved RNA Structures. Curr. Protoc. Bioinform. 2014, 46, 12–22. [Google Scholar] [CrossRef]
- Hutchinson, E.C.; von Kirchbach, J.C.; Gog, J.R.; Digard, P. Genome packaging in influenza A virus. J. Gen. Virol. 2010, 91, 313–328. [Google Scholar] [CrossRef] [PubMed]
- Dias, N.; Stein, C.A. Antisense oligonucleotides: Basic concepts and mechanisms. Mol. Cancer Ther. 2002, 1, 347–355. [Google Scholar]
- Hussain, M.; Galvin, H.D.; Haw, T.Y.; Nutsford, A.N.; Husain, M. Drug resistance in influenza A virus: The epidemiology and management. Infect. Drug Resist. 2017, 10, 121–134. [Google Scholar] [CrossRef] [Green Version]
- Tarn, W.Y.; Cheng, Y.; Ko, S.H.; Huang, L.M. Antisense oligonucleotide-based therapy of viral infections. Pharmaceutics 2021, 13, 2015. [Google Scholar] [CrossRef]
- Vickers, T.A.; Wyatt, J.R.; Freier, S.M. Effects of RNA secondary structure on cellular antisense activity. Nucleic Acids Res. 2000, 28, 1340–1347. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.; Liu, Y.; Chemparathy, A.; Pande, T.; La Russa, M.; Qi, L.S. A comprehensive analysis and resource to use CRISPR-Cas13 for broad-spectrum targeting of RNA viruses. Cell Rep. Med. 2021, 2, 100245. [Google Scholar] [CrossRef]
- Freije, C.A.; Myhrvold, C.; Boehm, C.K.; Lin, A.E.; Welch, N.L.; Carter, A.; Metsky, H.C.; Luo, C.Y.; Abudayyeh, O.O.; Gootenberg, J.S.; et al. Programmable Inhibition and Detection of RNA Viruses Using Cas13. Mol. Cell 2019, 76, 826–837.e11. [Google Scholar] [CrossRef] [Green Version]
- Abudayyeh, O.O.; Gootenberg, J.S.; Konermann, S.; Joung, J.; Slaymaker, I.M.; Cox, D.B.T.; Shmakov, S.; Makarova, K.S.; Semenova, E.; Minakhin, L.; et al. C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science 2016, 353, aaf5573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbott, T.R.; Dhamdhere, G.; Liu, Y.; Lin, X.; Goudy, L.; Zeng, L.; Chemparathy, A.; Chmura, S.; Heaton, N.S.; Debs, R.; et al. Development of CRISPR as an Antiviral Strategy to Combat SARS-CoV-2 and Influenza. Cell 2020, 181, 865–876.e12. [Google Scholar] [CrossRef]
- Wessels, H.-H.; Méndez-Mancilla, A.; Guo, X.; Legut, M.; Daniloski, Z.; Sanjana, N.E. Massively parallel Cas13 screens reveal principles for guide RNA design. Nat. Biotechnol. 2020, 38, 722–727. [Google Scholar] [CrossRef] [PubMed]
- Kesy, J.; Patil, K.M.K.M.; Kumar, S.R.; Shu, Z.; Yong, H.Y.H.Y.; Zimmermann, L.; Ong, A.A.L.A.A.L.; Toh, D.-F.K.D.F.K.; Krishna, M.S.M.S.; Yang, L.; et al. A Short Chemically Modified dsRNA-Binding PNA (dbPNA) Inhibits Influenza Viral Replication by Targeting Viral RNA Panhandle Structure. Bioconjugate Chem. 2019, 30, 931–943. [Google Scholar] [CrossRef] [PubMed]
- Meyer, S.M.; Williams, C.C.; Akahori, Y.; Tanaka, T.; Aikawa, H.; Tong, Y.; Childs-Disney, J.L.; Disney, M.D. Small molecule recognition of disease-relevant RNA structures. Chem. Soc. Rev. 2020, 49, 7167–7199. [Google Scholar] [CrossRef]
- Umuhire Juru, A.; Hargrove, A.E. Frameworks for targeting RNA with small molecules. J. Biol. Chem. 2021, 296, 100191. [Google Scholar] [CrossRef]
- Warner, K.D.; Hajdin, C.E.; Weeks, K.M. Principles for targeting RNA with drug-like small molecules. Nat. Rev. Drug Discov. 2018, 17, 547–558. [Google Scholar] [CrossRef]
- Klimov, A.; Balish, A.; Veguilla, V.; Sun, H.; Schiffer, J.; Lu, X. Influenza Virus Titration, Antigenic Characterization, and Serological Methods for Antibody Detection. In Influenza Virus: Methods and Protocols; Kawaoka, Y., Neumann, G., Eds.; Humana Press: Totowa, NJ, USA, 2012; pp. 25–51. [Google Scholar] [CrossRef]
- Shatzkes, K.; Teferedegne, B.; Murata, H. A simple, inexpensive method for preparing cell lysates suitable for downstream reverse transcription quantitative PCR. Sci. Rep. 2014, 4, 1–7. [Google Scholar] [CrossRef]
- Kierzek, E.; Kierzek, R. The synthesis of oligoribonucleotides containing N6-alkyladenosines and 2-methylthio-N6-alkyladenosines via post-synthetic modification of precursor oligomers. Nucleic Acids Res. 2003, 31, 4461–4471. [Google Scholar] [CrossRef] [Green Version]
- Kierzek, E.; Kierzek, R. The thermodynamic stability of RNA duplexes and hairpins containing N6-alkyladenosines and 2-methylthio-N6-alkyladenosines. Nucleic Acids Res. 2003, 31, 4472–4480. [Google Scholar] [CrossRef] [PubMed]
- Fratczak, A.; Kierzek, R.; Kierzek, E. LNA-modified primers drastically improve hybridization to target RNA and reverse transcription. Biochemistry 2009, 48, 514–516. [Google Scholar] [CrossRef]
- Soszynska-Jozwiak, M.; Michalak, P.; Moss, W.N.W.N.; Kierzek, R.; Kesy, J.; Kierzek, E. Influenza virus segment 5 (+)RNA-Secondary structure and new targets for antiviral strategies. Sci. Rep. 2017, 7, 1–15. [Google Scholar] [CrossRef]
- Beutner, G.L.; Kuethe, J.T.; Yasuda, N. A practical method for preparation of 4-hydroxyquinolinone esters. J. Org. Chem. 2007, 72, 7058–7061. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, E.; Watanabe, T.; Fujii, K.; Goto, H.; Watanabe, S.; Noda, T.; Kawaoka, Y. Strand-specific real-time RT-PCR for distinguishing influenza vRNA, cRNA, and mRNA. J. Virol. Methods 2011, 173, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vester, D.; Lagoda, A.; Hoffmann, D.; Seitz, C.; Heldt, S.; Bettenbrock, K.; Genzel, Y.; Reichl, U. Real-time RT-qPCR assay for the analysis of human influenza A virus transcription and replication dynamics. J. Virol. Methods 2010, 168, 63–71. [Google Scholar] [CrossRef]
- Vasa, S.M.; Guex, N.; Wilkinson, K.A.; Weeks, K.M.; Giddings, M.C. ShapeFinder: A software system for high-throughput quantitative analysis of nucleic acid reactivity information resolved by capillary electrophoresis. RNA 2008, 14, 1979–1990. [Google Scholar] [CrossRef] [Green Version]
- Low, J.T.; Weeks, K.M. SHAPE-directed RNA secondary structure prediction. Methods 2010, 52, 150–158. [Google Scholar] [CrossRef] [Green Version]
- Reuter, J.S.; Mathews, D.H. RNAstructure: Software for RNA secondary structure prediction and analysis. BMC Bioinformatics 2010, 11, 129. [Google Scholar] [CrossRef] [Green Version]
- Mathews, D.H.; Disney, M.D.; Childs, J.L.; Schroeder, S.J.; Zuker, M.; Turner, D.H. Incorporating chemical modification constraints into a dynamic programming algorithm for prediction of RNA secondary structure. Proc. Natl. Acad. Sci. USA 2004, 101, 7287–7292. [Google Scholar] [CrossRef] [Green Version]
- Turner, D.H.; Mathews, D.H. The nearest neighbor parameter database for predicting stability of nucleic acid secondary structure. Nucleic Acids Res. 2009, 38, 2009–2011. [Google Scholar] [CrossRef]
- Byun, Y.; Han, K. PseudoViewer: Web application and web service for visualizing RNA pseudoknots and secondary structures. Nucleic Acids Res. 2006, 34, 416–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative Genome Viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huynen, M.; Gutell, R.; Konings, D. Assessing the reliability of RNA folding using statistical mechanics. J. Mol. Biol. 1997, 267, 1104–1112. [Google Scholar] [CrossRef] [Green Version]
- Mathews, D.H. Using an RNA secondary structure partition function to determine confidence in base pairs predicted by free energy minimization. RNA 2004, 10, 1178–1190. [Google Scholar] [CrossRef] [PubMed] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Global MFE vRNA8 Structure | Local MFE vRNA8 Structure | ||
|---|---|---|---|
| motif | conservation of base pairs | motif | conservation of base pairs |
| 1–15/890–877 | 98.13% | 13–16/28–25 | 99.6% |
| 216–231/260–240 | 95% | 265–270/284–279 | 97.02% |
| 261–270/288–279 including 265–270/284–279 | 97.22% | 286–317/344–322 including 312–317/327–322 | 97.28% 96.09% |
| 305–317/335–322 including 312–317/327–322 | 96.63% 96.09% | 345–350/360–355 | 99.81% |
| 719–728/782–773 | 96.53% | 719–728/782–773 | 96.53% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szutkowska, B.; Wieczorek, K.; Kierzek, R.; Zmora, P.; Peterson, J.M.; Moss, W.N.; Mathews, D.H.; Kierzek, E. Secondary Structure of Influenza A Virus Genomic Segment 8 RNA Folded in a Cellular Environment. Int. J. Mol. Sci. 2022, 23, 2452. https://doi.org/10.3390/ijms23052452
Szutkowska B, Wieczorek K, Kierzek R, Zmora P, Peterson JM, Moss WN, Mathews DH, Kierzek E. Secondary Structure of Influenza A Virus Genomic Segment 8 RNA Folded in a Cellular Environment. International Journal of Molecular Sciences. 2022; 23(5):2452. https://doi.org/10.3390/ijms23052452
Chicago/Turabian StyleSzutkowska, Barbara, Klaudia Wieczorek, Ryszard Kierzek, Pawel Zmora, Jake M. Peterson, Walter N. Moss, David H. Mathews, and Elzbieta Kierzek. 2022. "Secondary Structure of Influenza A Virus Genomic Segment 8 RNA Folded in a Cellular Environment" International Journal of Molecular Sciences 23, no. 5: 2452. https://doi.org/10.3390/ijms23052452