
Targeting the Unfolded Protein Response as a Disease-Modifying Pathway in Dementia

 , ,
, , {kind=link}
{kind=link}
Abstract
:1. Introduction
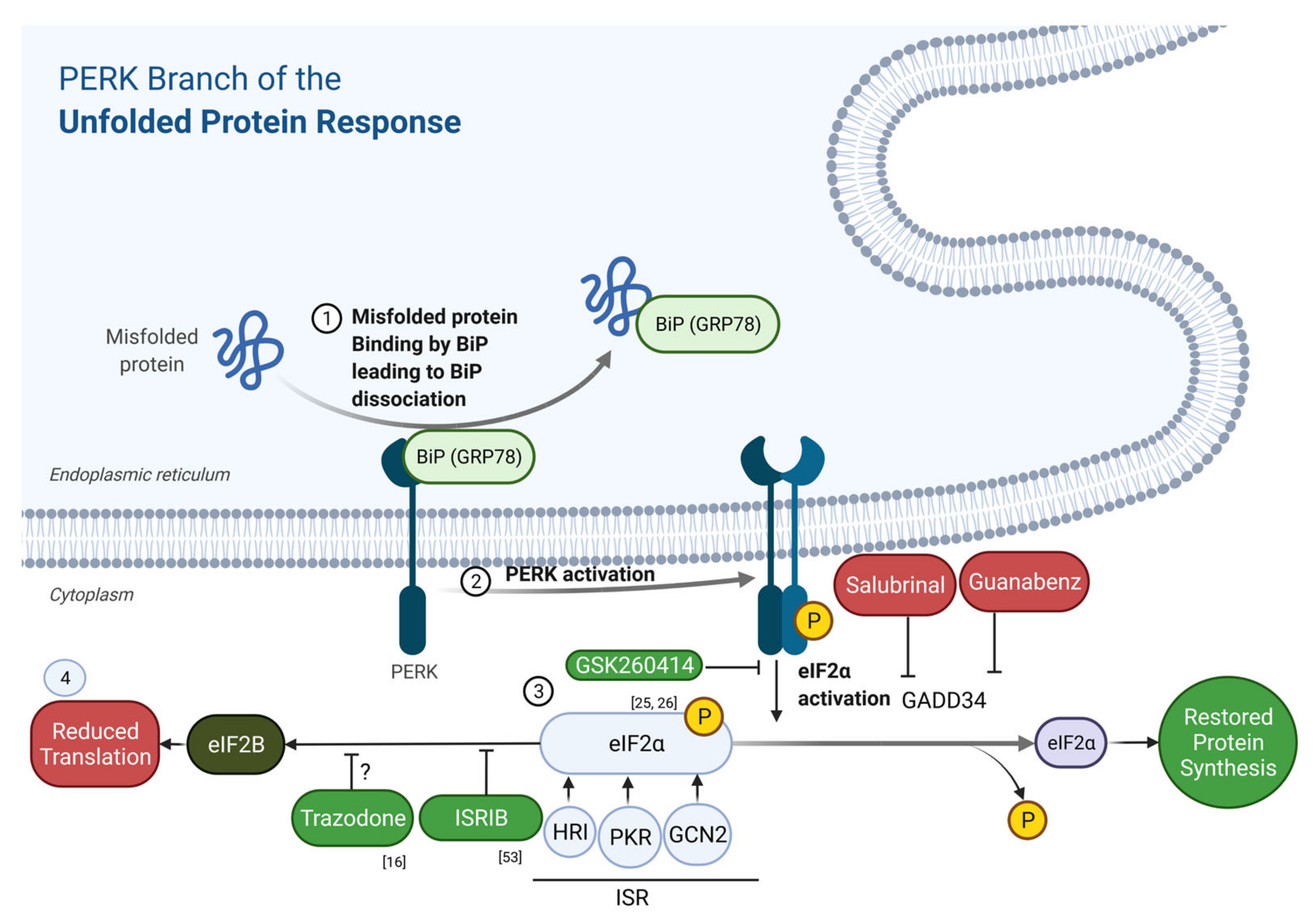
2. The Unfolded Protein Response
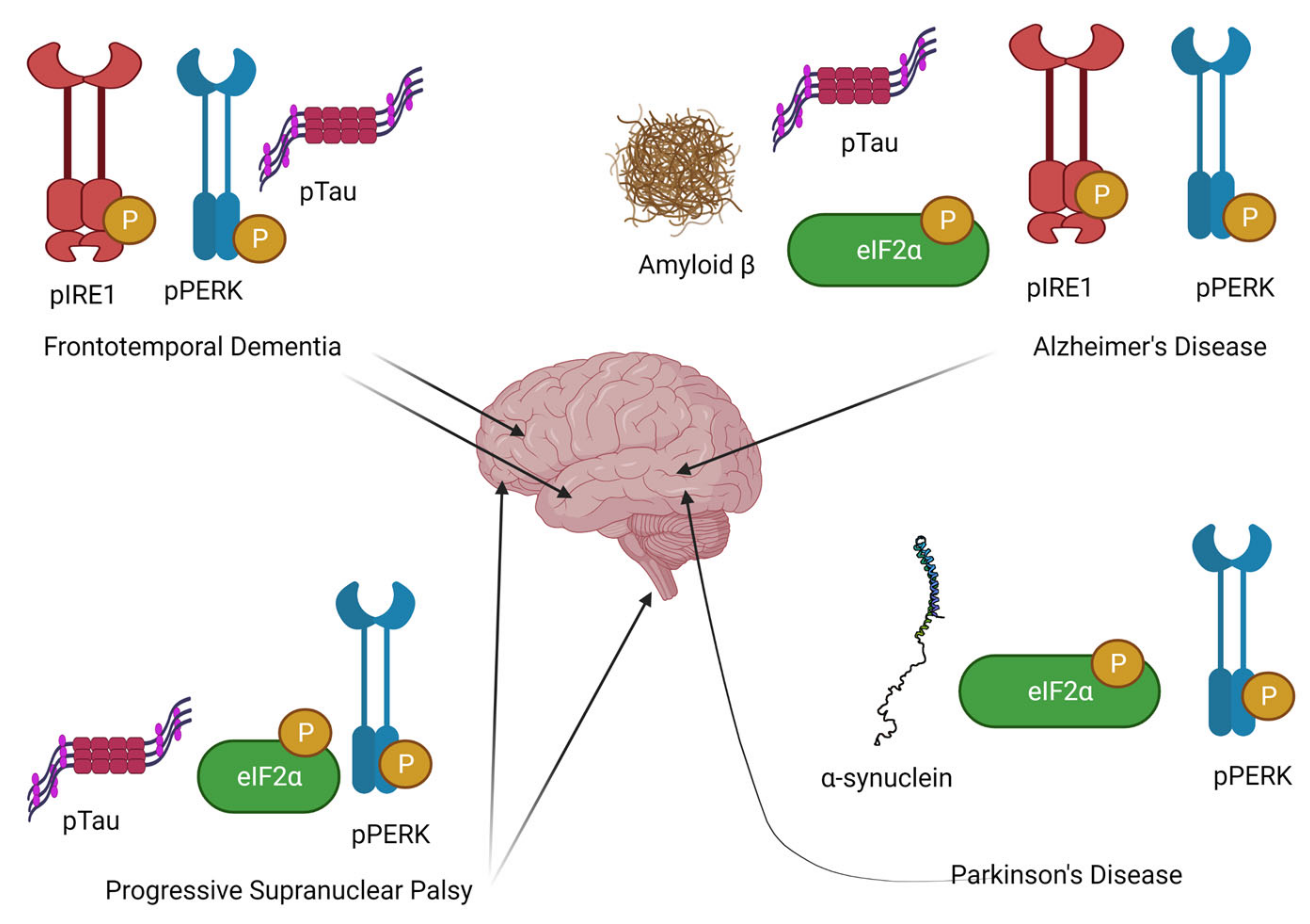
3. Evidence for UPR as a Key Pathogenic Mechanism in Neurodegeneration


4. Drugs Targeting UPR and ISR
5. Epidemiological Data from Observational Studies on Trazodone Use in Dementia
6. Clinical Trial Data on Trazodone Use in Older Adults, including Individuals with Dementia
7. Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Total Deaths in the UK in 2020 and Deaths from Heart Attacks, Heart Disease, Cancer, and Alzheimer’s and Dementia, 2016 to 2020—Office for National Statistics. Available online: https://www.ons.gov.uk/aboutus/transparencyandgovernance/freedomofinformationfoi/totaldeathsintheukin2020anddeathsfromheartattacksheartdiseasecancerandalzheimersanddementia2016to2020 (accessed on 23 December 2021).
- Deaths Due to Dementia|Dementia Statistics Hub. Available online: https://www.dementiastatistics.org/statistics/deaths-due-to-dementia/ (accessed on 23 June 2021).
- Ritchie, K.; Kildea, D. Is Senile Dementia “Age-Related” or “Ageing-Related”?--Evidence from Meta-Analysis of Dementia Prevalence in the Oldest Old. Lancet 1995, 346, 931–934. [Google Scholar] [CrossRef]
- Ferri, C.P.; Prince, M.; Brayne, C. Global prevalence of dementia: A Delphi consensus study. Lancet 2005, 366, 2112–2117. [Google Scholar] [CrossRef]
- 2020 Alzheimer’s Disease Facts and Figures. Alzheimer’s Dement. 2020, 16, 391–460. [CrossRef]
- Herrmann, N.; Chau, S.A.; Kircanski, I.; Lanctôt, K.L. Current and Emerging Drug Treatment Options for Alzheimers Disease: A Systematic Review. Drugs 2011, 71, 2031–2065. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, S.; Bouchard, R.; Lamontagne, A.; Bailey, P.; Bergman, H.; Ratner, J.; Tesfaye, Y.; Saint-Martin, M.; Bacher, Y.; Carrier, L.; et al. Tetrahydroaminoacridine-Lecithin Combination Treatment in Patients with Intermediate-Stage Alzheimer’s Disease. Results of a Canadian Double-Blind, Crossover, Multicenter Study. N. Engl. J. Med. 1990, 322, 1272–1276. [Google Scholar] [CrossRef] [PubMed]
- Zissimopoulos, J.; Crimmins, E.; St.clair, P. The Value of Delaying Alzheimer’s Disease Onset. Forum Heal. Econ. Policy 2015, 18, 25–39. [Google Scholar] [CrossRef]
- Dementia UK Report|Alzheimer’s Society. Available online: https://www.alzheimers.org.uk/about-us/policy-and-influencing/dementia-uk-report#:~:text=Thetotalcostofdementia,servicesforstatefundedcare (accessed on 23 June 2021).
- Ravikumar, B.; Futter, M.; Jahreiss, L.; Korolchuk, V.I.; Lichtenberg, M.; Luo, S.; Massey, D.C.O.; Menzies, F.M.; Narayanan, U.; Renna, M.; et al. Mammalian Macroautophagy at a Glance. J. Cell Sci. 2009. [Google Scholar] [CrossRef] [Green Version]
- Halliday, M.; Hughes, D.; Mallucci, G.R. Fine-Tuning PERK Signaling for Neuroprotection. J. Neurochem. 2017, 142, 812–826. [Google Scholar] [CrossRef]
- Glass, C.K.; Saijou, K.; Winner, B. Mechanisms Underlying Inflammation in Neurodegeneration. Cell 2010, 140, 918–934. [Google Scholar] [CrossRef] [Green Version]
- Yaribeygi, H.; Panahi, Y.; Javadi, B.; Sahebkar, A. The Underlying Role of Oxidative Stress in Neurodegeneration: A Mechanistic Review. CNS Neurol. Disord. Drug Targets 2018, 17, 207–215. [Google Scholar] [CrossRef]
- Mullard, A. NLRP3 Inhibitors Stoke Anti-Inflammatory Ambitions. Nat. Rev. Drug Discov. 2019, 18, 405–408. [Google Scholar] [CrossRef] [PubMed]
- Underwood, B.R.; Green-Thompson, Z.W.; Pugh, P.J.; Lazic, S.E.; Mason, S.L.; Griffin, J.; Jones, P.S.; Rowe, J.B.; Rubinsztein, D.C.; Barker, R.A. An Open-Label Study to Assess the Feasibility and Tolerability of Rilmenidine for the Treatment of Huntington’s Disease. J. Neurol. 2017, 264, 2457–2463. [Google Scholar] [CrossRef] [Green Version]
- Halliday, M.; Radford, H.; Zents, K.A.M.M.; Molloy, C.; Moreno, J.A.; Verity, N.C.; Smith, E.; Ortori, C.A.; Barrett, D.A.; Bushell, M.; et al. Repurposed Drugs Targeting EIF2α-P-Mediated Translational Repression Prevent Neurodegeneration in Mice. Brain 2017, 140, 1768–1783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Bertolotti, A.; Zhang, Y.; Hendershot, L.M. Dynamic Interaction of BiP and ER Stress Transducers in the Unfolded-Protein Response. Nat. Cell Biol. 2000, 2, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Fawcett, T.W.; Martindale, J.L.; Guyton, K.Z.; Hai, T.; Holbrook, N.J. Complexes Containing Activating Transcription Factor (ATF)/CAMP-Responsive-Element-Binding Protein (CREB) Interact with the CCAAT/Enhancer-Binding Protein (C/EBP)–ATF Composite Site to Regulate Gadd153 Expression during the Stress Response. Biochem. J. 1999, 339, 135–141. [Google Scholar] [CrossRef]
- Harding, H.; Zhang, Y.; Ron, D. Protein Translation and Folding Are Coupled by an Endoplasmic-Reticulum-Resident Kinase. Nature 1999, 397, 271–274. [Google Scholar] [CrossRef] [PubMed]
- Costa-Mattioli, M.; Walter, P. The Integrated Stress Response: From Mechanism to Disease. Science 2020, 368, eaat5314. [Google Scholar] [CrossRef] [PubMed]
- Kashiwagi, K.; Takahashi, M.; Nishimoto, M.; Hiyama, T.B.; Higo, T.; Umehara, T.; Sakamoto, K.; Ito, T.; Yokoyama, S. Crystal Structure of Eukaryotic Translation Initiation Factor 2B. Nature 2016, 531, 122–125. [Google Scholar] [CrossRef]
- Ma, Y.; Hendershot, L.M. Delineation of a Negative Feedback Regulatory Loop That Controls Protein Translation during Endoplasmic Reticulum Stress. J. Biol. Chem. 2003, 278, 34864–34873. [Google Scholar] [CrossRef] [Green Version]
- Moreno, J.A.; Radford, H.; Peretti, D.; Steinert, J.R.; Verity, N.; Martin, M.G.; Halliday, M.; Morgan, J.; Dinsdale, D.; Ortori, C.A.; et al. Sustained Translational Repression by EIF2α-P Mediates Prion Neurodegeneration. Nature 2012, 485, 507–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno, J.A.; Halliday, M.; Molloy, C.; Radford, H.; Verity, N.; Axten, J.M.; Ortori, C.A.; Willis, A.E.; Fischer, P.M.; Barrett, D.A.; et al. Oral Treatment Targeting the Unfolded Protein Response Prevents Neurodegeneration and Clinical Disease in Prion-Infected Mice. Sci. Transl. Med. 2013, 5, 206ra138. [Google Scholar] [CrossRef] [PubMed]
- Radford, H.; Moreno, J.A.; Verity, N.; Halliday, M.; Mallucci, G.R. PERK Inhibition Prevents Tau-Mediated Neurodegeneration in a Mouse Model of Frontotemporal Dementia. Acta Neuropathol. 2015, 130, 633–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LaFerla, F.M.; Green, K.N.; Oddo, S. Intracellular Amyloid-Beta in Alzheimer’s Disease. Nat. Rev. Neurosci. 2007, 8, 499–509. [Google Scholar] [CrossRef]
- Del Prete, D.; Suski, J.M.; Oulès, B.; Debayle, D.; Gay, A.S.; Lacas-Gervais, S.; Bussiere, R.; Bauer, C.; Pinton, P.; Paterlini-Bréchot, P.; et al. Localization and Processing of the Amyloid-β Protein Precursor in Mitochondria-Associated Membranes. J. Alzheimers. Dis. 2017, 55, 1549–1570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoozemans, J.J.M.; Veerhuis, R.; Van Haastert, E.S.; Rozemuller, J.M.; Baas, F.; Eikelenboom, P.; Scheper, W. The Unfolded Protein Response Is Activated in Alzheimer’s Disease. Acta Neuropathol. 2005, 110, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Stutzbach, L.D.; Xie, S.X.; Naj, A.C.; Albin, R.; Gilman, S.; Lee, V.M.Y.; Trojanowski, J.Q.; Devlin, B.; Schellenberg, G.D. The Unfolded Protein Response Is Activated in Disease-Affected Brain Regions in Progressive Supranuclear Palsy and Alzheimer’s Disease. Acta Neuropathol. Commun. 2013, 1, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Ito, D.; Fujisawa, T.; Iida, H.; Suzuki, N. Characterization of Seipin/BSCL2, a Protein Associated with Spastic Paraplegia 17. Neurobiol. Dis. 2008, 31, 266–277. [Google Scholar] [CrossRef]
- Hoozemans, J.J.M.; Van Haastert, E.S.; Nijholt, D.A.T.; Rozemuller, A.J.M.; Eikelenboom, P.; Scheper, W. The Unfolded Protein Response Is Activated in Pretangle Neurons in Alzheimer’s Disease Hippocampus. Am. J. Pathol. 2009, 174, 1241–1251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoozemans, J.J.M.; Van Haastert, E.S.; Nijholt, D.A.T.; Rozemuller, A.J.M.; Scheper, W. Activation of the Unfolded Protein Response Is an Early Event in Alzheimer’s and Parkinson’s Disease. Neurodegener. Dis. 2012, 10, 212–215. [Google Scholar] [CrossRef] [PubMed]
- Murray, H.C.; Dieriks, B.V.; Swanson, M.E.V.; Anekal, P.V.; Turner, C.; Faull, R.L.M.; Belluscio, L.; Koretsky, A.; Curtis, M.A. The Unfolded Protein Response Is Activated in the Olfactory System in Alzheimer’s Disease. Acta Neuropathol. Commun. 2020, 8, 109. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Saxena, S. ER Stress and the Unfolded Protein Response in Neurodegeneration. Nat. Rev. Neurol. 2017, 13, 477–491. [Google Scholar] [CrossRef] [PubMed]
- Duran-Aniotz, C.; Cornejo, V.H.; Espinoza, S.; Ardiles, Á.O.; Medinas, D.B.; Salazar, C.; Foley, A.; Gajardo, I.; Thielen, P.; Iwawaki, T.; et al. IRE1 Signaling Exacerbates Alzheimer’s Disease Pathogenesis. Acta Neuropathol. 2017, 134, 489–506. [Google Scholar] [CrossRef]
- Abisambra, J.F.; Jinwal, U.K.; Blair, L.J.; O’Leary, J.C.; Li, Q.; Brady, S.; Wang, L.; Guidi, C.E.; Zhang, B.; Nordhues, B.A.; et al. Tau Accumulation Activates the Unfolded Protein Response by Impairing Endoplasmic Reticulum-Associated Degradation. J. Neurosci. 2013, 33, 9498–9507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devi, L.; Ohno, M. PERK Mediates EIF2α Phosphorylation Responsible for BACE1 Elevation, CREB Dysfunction and Neurodegeneration in a Mouse Model of Alzheimer’s Disease. Neurobiol. Aging 2014, 35, 2272–2281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saxena, S.; Cabuy, E.; Caroni, P. A Role for Motoneuron Subtype-Selective ER Stress in Disease Manifestations of FALS Mice. Nat. Neurosci. 2009, 12, 627–636. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.J.; Khatiwada, S.; Cui, Y.; Reineke, L.C.; Dooling, S.W.; Kim, J.J.; Li, W.; Walter, P.; Costa-Mattioli, M. Activation of the ISR Mediates the Behavioral and Neurophysiological Abnormalities in Down Syndrome. Science 2019, 366, 843–849. [Google Scholar] [CrossRef] [PubMed]
- Koren, S.A.; Hamm, M.J.; Cloyd, R.; Fontaine, S.N.; Chishti, E.; Lanzillotta, C.; Rodriguez-Rivera, J.; Ingram, A.; Bell, M.; Galvis-Escobar, S.M.; et al. Broad Kinase Inhibition Mitigates Early Neuronal Dysfunction in Tauopathy. Int. J. Mol. Sci. 2021, 22, 1186. [Google Scholar] [CrossRef] [PubMed]
- Hughes, D.; Mallucci, G.R. The Unfolded Protein Response in Neurodegenerative Disorders—Therapeutic Modulation of the PERK Pathway. FEBS J. 2019, 286, 342–355. [Google Scholar] [CrossRef] [PubMed]
- Smith, H.L.; Freeman, O.J.; Butcher, A.J.; Holmqvist, S.; Humoud, I.; Schätzl, T.; Hughes, D.T.; Verity, N.C.; Swinden, D.P.; Hayes, J. Astrocyte Unfolded Protein Response Induces a Specific Reactivity State That Causes Non-Cell-Autonomous Neuronal Degeneration. Neuron 2020, 105, 855–866.e5. [Google Scholar] [CrossRef] [Green Version]
- Moradi Majd, R.; Mayeli, M.; Rahmani, F. Pathogenesis and Promising Therapeutics of Alzheimer Disease through EIF2α Pathway and Correspondent Kinases. Metab. Brain Dis. 2020, 35, 1241–1250. [Google Scholar] [CrossRef] [PubMed]
- Costa-Mattioli, M.; Gobert, D.; Stern, E.; Gamache, K.; Colina, R.; Cuello, C.; Sossin, W.; Kaufman, R.; Pelletier, J.; Rosenblum, K.; et al. EIF2α Phosphorylation Bidirectionally Regulates the Switch from Short- to Long-Term Synaptic Plasticity and Memory. Cell 2007, 129, 195–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, P.J.; Huang, W.; Kalikulov, D.; Yoo, J.W.; Placzek, A.N.; Stoica, L.; Zhou, H.; Bell, J.C.; Friedlander, M.J.; Krnjević, K.; et al. Suppression of PKR Promotes Network Excitability and Enhanced Cognition by Interferon-γ-Mediated Disinhibition. Cell 2011, 147, 1384–1396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, T.; Trinh, M.A.; Wexler, A.J.; Bourbon, C.; Gatti, E.; Pierre, P.; Cavener, D.R.; Klann, E. Suppression of EIF2α Kinases Alleviates Alzheimer’s Disease-Related Plasticity and Memory Deficits. Nat. Neurosci. 2013, 16, 1299–1305. [Google Scholar] [CrossRef] [Green Version]
- Mercado, G.; Castillo, V.; Soto, P.; López, N.; Axten, J.M.; Sardi, S.P.; Hoozemans, J.J.M.; Hetz, C. Targeting PERK Signaling with the Small Molecule GSK2606414 Prevents Neurodegeneration in a Model of Parkinson’s Disease. Neurobiol. Dis. 2018, 112, 136–148. [Google Scholar] [CrossRef] [PubMed]
- Sidrauski, C.; Acosta-Alvear, D.; Khoutorsky, A.; Vedantham, P.; Hearn, B.R.; Li, H.; Gamache, K.; Gallagher, C.M.; Ang, K.K.; Wilson, C.; et al. Pharmacological Brake-Release of mRNA Translation Enhances Cognitive Memory. Elife 2013, 2, e00498. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.L.; LeBon, L. The Small Molecule ISRIB Rescues the Stability and Activity of Vanishing White Matter Disease EIF2B Mutant Complexes. Elife 2018, 7, e32733. [Google Scholar] [CrossRef] [PubMed]
- Westergard, T.; McAcoy, K.; Russell, K. Repeat-Associated Non-AUG Translation in C9orf72-ALS/FTD Is Driven by Neuronal Excitation and Stress. EMBO Mol. Med. 2019, 11, e9423. [Google Scholar] [CrossRef] [PubMed]
- Chou, A.; Krukowski, K.; Jopson, T. Inhibition of the Integrated Stress Response Reverses Cognitive Deficits after Traumatic Brain Injury. Proc. Natl. Acad. Sci. USA 2017, 114, E6420–E6426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halliday, M.; Radford, H.; Sekine, Y.; Moreno, J.; Verity, N.; Le Quesne, J.; Ortori, C.A.; Barrett, D.A.; Fromont, C.; Fischer, P.M.; et al. Partial Restoration of Protein Synthesis Rates by the Small Molecule ISRIB Prevents Neurodegeneration without Pancreatic Toxicity. Cell Death Dis. 2015, 6, e1672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliveira, M.M.; Lourenco, M.V.; Longo, F.; Kasica, N.P.; Yang, W.; Ureta, G.; Ferreira, D.D.P.; Mendonça, P.H.J.; Bernales, S.; Ma, T.; et al. Correction of EIF2-Dependent Defects in Brain Protein Synthesis, Synaptic Plasticity, and Memory in Mouse Models of Alzheimer’s Disease. Sci. Signal. 2021, 14, eabc5429. [Google Scholar] [CrossRef] [PubMed]
- McCleery, J.; Sharpley, A.L. Pharmacotherapies for Sleep Disturbances in Dementia. Cochrane Database Syst. Rev. 2020. [Google Scholar] [CrossRef] [PubMed]
- Akbari, V.; Ghobadi, S.; Mohammadi, S.; Khodarahmi, R. The Antidepressant Drug; Trazodone Inhibits Tau Amyloidogenesis: Prospects for Prophylaxis and Treatment of AD. Arch. Biochem. Biophys. 2020, 679, 108218. [Google Scholar] [CrossRef] [PubMed]
- La, A.L.; Walsh, C.M.; Neylan, T.C.; Vossel, K.A.; Yaffe, K.; Krystal, A.D.; Miller, B.L.; Karageorgiou, E. Long-Term Trazodone Use and Cognition: A Potential Therapeutic Role for Slow-Wave Sleep Enhancers. J. Alzheimer’s Dis. 2019, 67, 911–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, H.Q.; Ren, M.; Jiang, H.Z.; Wang, J.; Zhang, J.; Yin, X.; Wang, S.Y.; Qi, Y.; Wang, X.D.; Feng, H.L. Guanabenz Delays the Onset of Disease Symptoms, Extends Lifespan, Improves Motor Performance and Attenuates Motor Neuron Loss in the SOD1 G93A Mouse Model of Amyotrophic Lateral Sclerosis. Neuroscience 2014, 277, 132–138. [Google Scholar] [CrossRef]
- Wang, M.D.; Gomes, J.; Cashman, N.R.; Little, J.; Krewski, D. Intermediate CAG Repeat Expansion in the ATXN2 Gene Is a Unique Genetic Risk Factor for ALS−A Systematic Review and Meta-Analysis of Observational Studies. PLoS ONE 2014, 9, e105534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, M.M.; Svendsen, C.N. Astrocytes Show Reduced Support of Motor Neurons with Aging That Is Accelerated in a Rodent Model of ALS. Neurobiol. Aging 2015, 36, 1130–1139. [Google Scholar] [CrossRef] [PubMed]
- Watt, J.A.; Gomes, T.; Bronskill, S.E.; Huang, A.; Austin, P.C.; Ho, J.M.; Straus, S.E. Comparative Risk of Harm Associated with Trazodone or Atypical Antipsychotic Use in Older Adults with Dementia: A Retrospective Cohort Study. CMAJ 2018, 190, E1376–E1383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bronskill, S.E.; Campitelli, M.A.; Iaboni, A.; Herrmann, N.; Guan, J.; Maclagan, L.C.; Watt, J.; Rochon, P.A.; Morris, A.M.; Jeffs, L.; et al. Low-Dose Trazodone, Benzodiazepines, and Fall-Related Injuries in Nursing Homes: A Matched-Cohort Study. J. Am. Geriatr. Soc. 2018, 66, 1963–1971. [Google Scholar] [CrossRef] [PubMed]
- Tellone, V.; Rosignoli, M.T.; Picollo, R.; Dragone, P.; Del Vecchio, A.; Comandini, A.; Radicioni, M.; Leuratti, C.; Calisti, F. Effect of 3 Single Doses of Trazodone on QTc Interval in Healthy Subjects. J. Clin. Pharmacol. 2020, 60, 1483–1495. [Google Scholar] [CrossRef]
- Armstrong, S.E.M.; Brown, H.K.; Shorey, C.; Madan, R.; Szabuniewicz, C.; Koh, S.; Crépeau-Gendron, G.; Mah, L. No Association between Trazodone and Corrected-QT Prolongation in Older Adults. J. Clin. Psychopharmacol. 2019, 39, 528–530. [Google Scholar] [CrossRef] [PubMed]
- Viramontes, T.S.; Truong, H.; Linnebur, S.A. Antidepressant-Induced Hyponatremia in Older Adults. Consult. Pharm. 2016, 31, 139–150. [Google Scholar] [CrossRef]
- Brauer, R.; Lau, W.C.Y.; Hayes, J.F.; Man, K.K.C.; Osborn, D.P.J.; Howard, R.; Kim, J.; Wong, I.C.K. Trazodone Use and Risk of Dementia: A Population-Based Cohort Study. PLoS Med. 2019, 16, e1002728. [Google Scholar] [CrossRef]
- Sommerlad, A.; Werbeloff, N.; Perera, G.; Smith, T.; Costello, H.; Mueller, C.; Kormilitzin, A.; Broadbent, M.; Nevado-Holgado, A.; Lovestone, S.; et al. Effect of Trazodone on Cognitive Decline in People with Dementia: Cohort Study Using UK Routinely Collected Data. Int. J. Geriatr. Psychiatry 2021. [Google Scholar] [CrossRef] [PubMed]
- Pirker-Kees, A.; Dal-Bianco, P.; Schmidt, R. Effects of Psychotropic Medication on Cognition, Caregiver Burden, and Neuropsychiatric Symptoms in Alzheimer’s Disease over 12 Months: Results from a Prospective Registry of Dementia in Austria (PRODEM). J. Alzheimer’s Dis. 2019, 71, 623–630. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, S.; Tsunematsu, T. Association between Sleep, Alzheimer’s, and Parkinson’s Disease. Biology 2021, 10, 1127. [Google Scholar] [CrossRef]
- Charernboon, T.; Phanasathit, M. Prevalence of Neuropsychiatric Symptoms in Alzheimer’s Disease: A Cross-Sectional Descriptive Study in Thailand. J. Med. Assoc. Thail. 2014, 97, 560. [Google Scholar]
- Fagiolini, A.; Comandini, A.; Dell’Osso, M.C.; Kasper, S. Rediscovering Trazodone for the Treatment of Major Depressive Disorder. CNS Drugs 2012, 26, 1033–1049. [Google Scholar] [CrossRef]
- Reding, M.J.; Orto, L.A.; Winter, S.W.; Fortuna, I.M.; Ponte, P.; Mcdowell, F.H. Antidepressant Therapy After Stroke: A Double-Blind Trial. Arch. Neurol. 1986, 43, 763–765. [Google Scholar] [CrossRef]
- Tammiala-Salonen, T.; Forssell, H.; Tammiala-Salonen, T.; Forsseii, H.; Forsseil, H. Trazodone in Burning Mouth Pain: A Placebo-Controlled, Double-Blind Study. J. Prosthet. Dent. 1999, 82, 578. [Google Scholar] [CrossRef]
- Kaynak, H.; Kaynak, D.; Gözükirmizi, E.; Guilleminault, C. The Effects of Trazodone on Sleep in Patients Treated with Stimulant Antidepressants. Sleep Med. 2004, 5, 15–20. [Google Scholar] [CrossRef]
- Meinhardt, W.; Schmitz, P.I.M.; Kropman, R.F.; De la Fuente, R.B.; Lycklama a Nijeholt, A.A.B.; Zwartendijk, J. Trazodone, a Double Blind Trial for Treatment of Erectile Dysfunction. Int. J. Impot. Res. 1997, 9, 163–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stryjer, R.; Rosenzcwaig, S.; Bar, F.; Ulman, A.M.; Weizman, A.; Spivak, B. Trazodone for the Treatment of Neuroleptic-Induced Acute Akathisia: A Placebo-Controlled, Double-Blind, Crossover Study. Clin. Neuropharmacol. 2010, 33, 219–222. [Google Scholar] [CrossRef] [PubMed]
- Battistella, P.A.; Ruffilli, R.; Cernetti, R.; Pettenazzo, A.; Baldin, L.; Bertoli, S.; Zacchello, F. A Placebo-Controlled Crossover Trial Using Trazodone in Pediatric Migraine. Headache J. Head Face Pain 1993, 33, 36–39. [Google Scholar] [CrossRef] [PubMed]
- De Wit, S.; Cremers, L.; Hirsch, D.; Zulian, C.; Clumeck, N.; Kormoss, N. Efficacy and Safety of Trazodone versus Clorazepate in the Treatment of HIV-Positive Subjects with Adjustment Disorders: A Pilot Study. J. Int. Med. Res. 1999, 27, 223–232. [Google Scholar] [CrossRef] [Green Version]
- Hudson, J.I.; Pope, H.G.; Keck, P.E.; McElroy, S.L. Treatment of Bulimia Nervosa with Trazodone: Short-Term Response and Long-Term Follow-Up. Clin. Neuropharmacol. 1989, 12, S38–S46. [Google Scholar] [CrossRef]
- Clouse, R.E.; Lustman, P.J.; Eckert, T.C.; Ferney, D.M.; Griffith, L.S. Low-Dose Trazodone for Symptomatic Patients With Esophageal Contraction Abnormalities: A Double-Blind, Placebo-Controlled Trial. Gastroenterology 1987, 92, 1027–1036. [Google Scholar] [CrossRef]
- Pigott, T.A.; L’Heureux, F.; Rubenstein, C.S.; Bernstein, S.E.; Hill, J.L.; Murphy, D.L. A Double-Blind, Placebo Controlled Study of Trazodone in Patients with Obsessive-Compulsive Disorder. J. Clin. Psychopharmacol. 1992, 12, 156–162. [Google Scholar] [CrossRef]
- Rickels, K.; Downing, R.; Schweizer, E.; Hassman, H. Antidepressants for the Treatment of Generalized Anxiety Disorder: A Placebo-Controlled Comparison of Imipramine, Trazodone, and Diazepam. Arch. Gen. Psychiatry 1993, 50, 884–895. [Google Scholar] [CrossRef]
- Le Bon, O.; Murphy, J.R.; Staner, L.; Hoffmann, G.; Kormoss, N.; Kentos, M.; Dupont, P.; Lion, K.; Pelc, I.; Verbanck, P. Double-Blind, Placebo-Controlled Study of the Efficacy of Trazodone in Alcohol Post-Withdrawal Syndrome: Polysomnographic and Clinical Evaluations. J. Clin. Psychopharmacol. 2003, 23, 377–383. [Google Scholar] [CrossRef]
- Bossini, L.; Coluccia, A.; Casolaro, I.; Benbow, J.; Amodeo, G.; De Giorgi, R.; Fagiolini, A. Off-Label Trazodone Prescription: Evidence, Benefits and Risks. Curr. Pharm. Des. 2015, 21, 3343–3351. [Google Scholar] [CrossRef] [PubMed]
- Sultzer, D.L.; Gray, K.F.; Gunay, I.; Wheatley, M.V.; Mahler, M.E. Does Behavioral Improvement with Haloperidol or Trazodone Treatment Depend on Psychosis or Mood Symptoms in Patients with Dementia? J. Am. Geriatr. Soc. 2001, 49, 1294–1300. [Google Scholar] [CrossRef] [PubMed]
- Camargos, E.F.; Louzada, L.L.; Quintas, J.L.; Naves, J.O.S.; Louzada, F.M.; Nóbrega, O.T. Trazodone Improves Sleep Parameters in Alzheimer Disease Patients: A Randomized, Double-Blind, and Placebo-Controlled Study. Am. J. Geriatr. Psychiatry 2014, 22, 1565–1574. [Google Scholar] [CrossRef] [PubMed]
- Teri, L.; Logsdon, R.G.; Peskind, E.; Raskind, M.; Weiner, M.F.; Tractenberg, R.E.; Foster, N.L.; Schneider, L.S.; Sano, M.; Whitehouse, P.; et al. Treatment of Agitation in AD: A Randomized, Placebo-Controlled Clinical Trial. Neurology 2000, 55, 1271–1278. [Google Scholar] [CrossRef] [PubMed]
- Lebert, F.; Stekke, W.; Hasenbroekx, C.; Pasquier, F. Frontotemporal dementia: A randomized, controlled trial with trazodone. Dement. Geriatr. Cogn. Disord. 2004, 17, 355–359. [Google Scholar] [CrossRef] [PubMed]
- Gonçalo, A.M.G.; Vieira-Coelho, M.A. The Effects of Trazodone on Human Cognition: A Systematic Review. Eur. J. Clin. Pharmacol. 2021, 77, 1623–1637. [Google Scholar] [CrossRef]
- Revised Guideline on Clinical Studies for Alzheimer’s Disease Medicines|European Medicines Agency. Available online: https://www.ema.europa.eu/en/news/revised-guideline-clinical-studies-alzheimers-disease-medicines (accessed on 23 December 2021).
- Trial Drugs|MND-SMART. Available online: https://mnd-smart.org/about/trial-drugs (accessed on 17 November 2021).
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sidhom, E.; O’Brien, J.T.; Butcher, A.J.; Smith, H.L.; Mallucci, G.R.; Underwood, B.R. Targeting the Unfolded Protein Response as a Disease-Modifying Pathway in Dementia. Int. J. Mol. Sci. 2022, 23, 2021. https://doi.org/10.3390/ijms23042021
Sidhom E, O’Brien JT, Butcher AJ, Smith HL, Mallucci GR, Underwood BR. Targeting the Unfolded Protein Response as a Disease-Modifying Pathway in Dementia. International Journal of Molecular Sciences. 2022; 23(4):2021. https://doi.org/10.3390/ijms23042021
Chicago/Turabian StyleSidhom, Emad, John T. O’Brien, Adrian J. Butcher, Heather L. Smith, Giovanna R. Mallucci, and Benjamin R. Underwood. 2022. "Targeting the Unfolded Protein Response as a Disease-Modifying Pathway in Dementia" International Journal of Molecular Sciences 23, no. 4: 2021. https://doi.org/10.3390/ijms23042021