
Polymodal Control of TMEM16x Channels and Scramblases


Department of Pharmacology, University of Oxford, Mansfield Road, Oxford OX1 3QT, UK
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2022, 23(3), 1580; https://doi.org/10.3390/ijms23031580
Submission received: 5 December 2021
/
Revised: 20 January 2022
/
Accepted: 20 January 2022
/
Published: 29 January 2022
(This article belongs to the Special Issue Ca2+-Activated Chloride Channels and Phospholipid Scramblases)
Abstract
:The TMEM16A/anoctamin-1 calcium-activated chloride channel (CaCC) contributes to a range of vital functions, such as the control of vascular tone and epithelial ion transport. The channel is a founding member of a family of 10 proteins (TMEM16x) with varied functions; some members (i.e., TMEM16A and TMEM16B) serve as CaCCs, while others are lipid scramblases, combine channel and scramblase function, or perform additional cellular roles. TMEM16x proteins are typically activated by agonist-induced Ca2+ release evoked by Gq-protein-coupled receptor (GqPCR) activation; thus, TMEM16x proteins link Ca2+-signalling with cell electrical activity and/or lipid transport. Recent studies demonstrate that a range of other cellular factors—including plasmalemmal lipids, pH, hypoxia, ATP and auxiliary proteins—also control the activity of the TMEM16A channel and its paralogues, suggesting that the TMEM16x proteins are effectively polymodal sensors of cellular homeostasis. Here, we review the molecular pathophysiology, structural biology, and mechanisms of regulation of TMEM16x proteins by multiple cellular factors.
Keywords:
Ca2+-activated Cl− channels; anoctamin; TMEM16x; scramblases; lipids; gating; Ca2+ signalling; SARS-CoV-21. Introduction to TMEM16x Physiology
The TMEM16x eukaryotic protein family (HUGO gene nomenclature: Anoctamin) is composed of 10 paralogues in mammals that share high sequence homology while forming a functionally diverse group of proteins. TMEM16x proteins may (1) form Ca2+-activated Cl− channels (CaCCs) (TMEM16A and B) [1]; (2) function as lipid scramblases, which facilitate bidirectional movement of lipids across the cell membranes, possibly in combination with non-selective ion channel activity (TMEM16D, E, F, K and J) [2,3,4,5,6,7,8]; or (3) play additional cellular roles (TMEM16C, G, H) [4,9,10]. Unlike the mammalian TMEM16x family, lower eukaryotes generally have fewer than 10 TMEM16x paralogues [11,12,13].
The TMEM16A and B channels have the highest (~60%) sequence homology within the family [12,14,15,16,17], and present similar electrophysiological properties, including comparable degrees of selectivity and permeability to a range of anions, and sensitivity to intracellular Ca2+ [1,18,19,20,21,22,23]. TMEM16A and B channels also share some pharmacological properties; for example, they are modulated in a complex manner by antracene-9-carboxilic acid (A9C) [24,25], and are inhibited by other commonly used Cl− channel blockers—such as 4,4′-diisothiocyano-2,2′-stilbenedisulfonic acid (DIDS) and niflumic acid (NFA)—to a similar extent [22,26,27]. In contrast, a recently identified drug—2-(4-chloro-2-methylphenoxy)-acetic acid 2-[(2-methoxyphenyl) methylene] hydrazide (Ani9)—selectively inhibited TMEM16A, with no significant effect on TMEM16B [28]. The pharmacology of TMEM16x scramblases remains poorly explored, although niclosamide, a therapeutic anthelminthic drug, reportedly inhibits both TMEM16F ion and lipid transport [29], and also inhibits TMEM16A ionic currents [30].
TMEM16x proteins differ in their expression profiles across tissues. TMEM16F and K, for example, are almost ubiquitously expressed in mammalian cells [31]. Other members have more restricted expression profiles and, consequently, play more specific cellular roles. Aspects of the cellular pathophysiology of TMEM16A and some of its paralogues have recently been reviewed [15,32,33,34,35,36,37,38,39,40,41,42]; thus, here we provide a succinct overview of this topic.
TMEM16A is primarily involved in transepithelial Cl− transport [23,43,44,45], and in the regulation of smooth muscle tone [46,47,48,49,50], while TMEM16B participates in the modulation of sensory processes such as olfaction and vision [51,52,53,54,55], as well as the control of the excitability of neuronal and glial cells [45,56,57,58]. The TMEM16D plasmalemmal scramblase [42,59,60] also operates as a non-selective cation channel [6], and can be localised in intracellular membranes during heterologous expression [36]. TMEM16D is primarily expressed in the brain [61] and in endocrine glands, including the adrenal zona glomerulosa, where it may stimulate aldosterone secretion and, thus, contribute to renal control of mean arterial pressure [62]. TMEM16D has been linked to various neuronal disorders [63], such as schizophrenia [64], Alzheimer’s disease [65], and anxiety disorders [66]. TMEM16E and F combine channel and scramblase functions, and have established links to human disease [3,67,68,69,70]. Gain- and loss-of-function mutations in the human TMEM16E gene are linked to the bone disease gnathodiaphyseal dysplasia (GDD) [3,68] and muscular dystrophy (MD) [67,68,71], respectively. Mutations in the TMEM6F gene are associated with Scott syndrome, a bleeding disorder caused by defective phospholipid scrambling in platelets [59,60,69,70].
Some TMEM16x (TMEM16C, G, and H) proteins play cellular roles other than serving as ion channels or lipid scramblases. TMEM16C interacts with the Na+-activated K+ (SLACK) channel, increasing the single-channel activity and sodium sensitivity of the SLACK channel [9]. Mutations in the TMEM16C gene may lead to autosomal-dominant craniocervical dystonia in human subjects [72]. TMEM16C is also associated with scramblase activity [59], and an early report suggested that it may also have intracellular localisation [36]. TMEM16G is highly expressed in both prostate cancer and normal prostate tissues; it is a proposed candidate for both diagnosis and immunotherapy of prostate cancer [4,73], but the detailed cellular roles of TMEM16G are not fully defined. In some [31,74]—but not all [36,59,60]—published reports, TMEM16G was found to mediate non-selective currents in heterologous systems (FTR and HEK-293 cells), and it was also suggested that it may localise in the endoplasmic reticulum (ER) [36]. TMEM16G also mediated lipid scrambling in a cell line in which the TMEM16F gene was deleted [59,60]. TMEM16G may also interact with proteins upregulated during cancer progression [75], including the cellular vesicles staphylococcal nuclease and tudor domain-containing 1 (SND1), the heat shock protein family A (Hsp70) member 1A (HSPA1A), the adaptor-related protein complex 2 subunit beta 1 (AP2B1), and the coatomer protein complex subunit gamma 2 (COPG2); however, the functional significance of these interactions remains undefined [76]. TMEM16H participates in the formation of junctions between the ER and the cell membrane; this process may favour the interaction of proteins and receptors involved in Ca2+ release from intracellular stores such as the stromal interaction molecule 1 (STIM1), the inositol 1,4,5-triphosphate (IP3) receptor, and the sarco/endoplasmic reticulum calcium ATPase 2 (SERCA2) [10].
While some TMEM16x proteins (such as TMEM16A, B and F) are located in the plasma membrane, TMEM16E, G, H, J, and K are expressed primarily or exclusively in the membranes of intracellular compartments, including the ER; however, there are some conflicting results regarding TMEM16x cellular localisation [2,3,4,10,31,36,74,77,78,79,80,81]. TMEM16K is one of the most studied of the intercellular TMEM16x proteins. TMEM16K is an ER-resident lipid scramblase with non-specific ion channel activity and a dependence on Ca2+ and short-chain lipids for optimal activity. The ER membrane, unlike the plasma membrane, has symmetrical lipid distribution. Since many lipids are synthesised on the cytoplasmic side of the ER, scramblases play an important role in the formation of the ER membrane’s symmetrical lipid distribution. TMEM16K has been associated with several cellular phenomena, including spindle formation [82], Ca2+ signalling [83], volume regulation [77], and apoptosis [82,83]. The notion that TMEM16K truncations and missense variants lead to autosomal recessive spinocerebellar ataxia type 10 (SCAR10) suggests that incorrect lipid distribution in the ER participates in the pathophysiology of this disease [84,85]. TMEM16J is another TMEM16x with reported intracellular localisation [31,80,81]; however, heterologous expression in HEK293T cells promotes plasma membrane expression, where the TMEM16J works as a cation channel activated by a cAMP-dependent protein kinase A (PKA) [5]. TMEM16J is overexpressed in pancreatic cancer cells [86], in gastrointestinal cancer [87], and in oesophageal squamous-cell carcinoma (ESCC) [81], and is associated with tumour progression [81,86,87].
2. TMEM16x Splice Variants
Some TMEM16x genes undergo alternative splicing (Figure 1), which leads to the generation of TMEM16x proteins with different functional properties (e.g., sensitivity to Ca2+) [7,43,54,55,88,89,90,91,92,93,94,95,96,97,98,99]. The alternatively spliced exons in TMEM16A are termed a, b, c, d, and exon 0 (Figure 1). The segment a is under the control of an alternative promoter, and codes for an N-terminal cytoplasmic region. The segment b distally follows segment a in the N-terminus. The segment c is located in the first intracellular loop, and codes for a segment of only four amino acids (EAVK). The segment d distally follows segment c in the first intracellular loop. An alternatively spliced exon, termed exon 0, located upstream of segment a in the N-terminus, has also been reported [96]. Finally, another variant has been identified in human interstitial cells of Cajal that lacks exons 1 and 2, as well as part of exon 3, but no specific nomenclature has been attributed to this variant [95,96,97]. The presence of an alternative start codon at position 117 in the primary sequence of abcTMEM16A has been reported. The resulting N-terminal-truncated TMEM16A variant has a restricted tissue expression profile, having only been detected in human testes [98].
Splicing variants have also been reported for TMEM16B, and involve exon 4—within the cytoplasmic N-terminus—and exon 14, which encodes for four amino acids (EAVK) in the first predicted intracellular loop. Alternative starting exons give rise to TMEM16B variants with N-termini of different length (termed A (long) and B (short)). When these two isoforms also lack exon 4 they are termed AΔ4 or BΔ4, respectively [54,55,91]. Four alternative spliced variants of TMEM16F have been identified [7]. Variant V1 contains 10 transmembrane (TM) segments, and both the N- and C-termini are cytosolic [7]. Variant V2 has an alternative starting exon that gives rise to a shorter N-terminus segment [7]. Variant V3 [7] (also termed V4 in available databases [7]) contains an alternative 3′-terminal exon that results in a TMEM16F isoform with a longer C-terminus portion exposed to the extracellular environment. Variant V5 contains an additional in-frame coding exon in the N-terminus [7,92].
3. The Factors Controlling Ion and Lipid Transport in TMEM16x Proteins
TMEM16x proteins are activated in response to an increase in intracellular Ca2+ concentration ([Ca2+]i), typically evoked in response to activation of Gq-protein-coupled receptors (GqPCRs). In addition, the TMEM16A channel and other TMEM16x proteins are controlled by a series of other cellular factors, including pH, anions, hypoxia, ATP, heat, auxiliary proteins and lipid. In virtue of these varied mechanisms of regulation, the TMEM16x proteins effectively constitute polymodal sensors of cellular homeostasis. Here, we review the various cellular mechanisms of regulation of TMEM16x proteins.
Biophysics of Ion and Lipid Transport
The amplitude of a whole-cell ion channel current (I) is determined by the product of the number of channels in the membrane (N), the channel-open probability (Po), and the single-channel current (i) as I = NPoi. The magnitude of the single-channel current (i.e., the current flowing through the open pore) is determined by the ionic electrochemical gradient and the chemical characteristics of the permeation pathway (pore conductance). The fraction of time the channel spends in the open state defines the Po.
For the TMEM16x channels, the Po is primarily regulated by changes in [Ca2+]i and voltage (Vm), since binding of Ca2+ to a specific high-affinity site, which is favoured at depolarised Vm, leads to channel opening (see Section 4.1). Other factors, such as pH, ATP, heat etc., also modulate TMEM16x channel gating. N, the number of channels in the membrane, is the result of gene expression and protein trafficking, processes that are dynamically regulated and can be altered in disease. The regulation of N typically occurs in minutes to days, while the channel response to changes in Ca2+ and Vm is much more rapid. Furthermore, TMEM16x splice variants vary in their functional properties; thus, regulation of TMEM16x splicing offers an additional means of modulation of whole-cell current properties (kinetics, Ca2+ sensitivity). The equation above implies that permeation (i) and gating (Po) are separate factors. However, this is an approximation, since for the TMEM16x channels the permeating anion may also influence channel gating and apparent Ca2+ sensitivity. For example, activation of TMEM16A and B is facilitated by anions with high permeability, or by an increase in extracellular Cl− [19,100], suggesting that the conductive pore and Ca2+-dependent gating are coupled. It has been proposed that bulkier anions favour the channel-open state by increasing the mean open time via a mechanism similar to the ‘foot in the door’ produced by quaternary ammonium ions on K+ channels [101,102,103].
A conceptually similar equation to the one described above for the TMEM16x current could be used to describe TMEM16x scramblase function. For scramblases, the equivalent of i is given by the lipid transport occurring through a single TMEM16x scramblase protein per unit of time, Po is the fraction of time the scramblase spends in the ‘lipid-conductive’ mode, and N is the number of TMEM16x scramblases expressed on the membrane. The cellular factors (reviewed below) that control TMEM16x primarily affect the Po; however, it has been reported that ATP may modulate i of TMEM16A via phosphorylation of a serine in the third intracellular loop [104]. Some structural domains in TMEM16A and B that control their expression on the plasma membrane (which influences N) have also been identified [18].
4. Overview of the Structure–Function Relationship in TMEM16x Proteins
The structures of the mammalian TMEM16A channel [105,106,107,108] and fungal [109,110,111,112] or mammalian TMEM16x lipid scramblases [2,113,114] have been experimentally resolved via X-ray crystallography and/or cryo-electron (cryo-EM) microscopy. Here, we offer a concise summary of the relationship between structure and function in TMEM16x proteins. Residues are defined by the International Union of Pure and Applied Chemistry (IUPAC) one-letter codes, and their position in the primary sequence is given alongside the splice variant utilised in each cited study (when this was specified in the original publications).
The X-ray structure of the Nectria haematococca TMEM16 (nhTMEM16)—a scramblase with non-selective channel activity—demonstrated a dimer arranged in a bi-lobal ‘butterfly’ fashion, with each subunit presenting 10 TM α-helices [109] (Figure 1 and Figure 2). Each monomer has a hydrophilic, membrane-spanning groove that provides a pathway for lipid headgroups to move across membranes. This lipid-scrambling mechanism has been reproduced in silico via molecular dynamics (MD) [115,116]. The structure of another fungal TMEM16 protein—the Aspergillus fumigatus afTMEM16 [110]—in conjunction with the structures of nhTMEM16 [111], murine TMEM16F [113,114], and human TMEM16K [2], demonstrated a range of conformations that these scramblases can assume. These involve movements of helices near the lipid-scrambling pathway enabling a range of activation states, potentially triggered by diverse stimuli such as membrane thickness, lipid composition, post-translational modification, or binding of cofactors.
Lipid scramblase activity critically depends on both the membrane lipid composition and membrane thickness. For example, lipid scrambling by TMEM16K is greatly augmented in the presence of shorter chain lipids, which form thinner bilayers, similarly to the ER membrane [2,79]. Continuous scrambling occurs in the ER membranes to help to redistribute lipids that are synthesised on the cytoplasmic side of the ER membrane. In contrast, the plasma membrane has a highly asymmetrical lipid distribution, which is dissipated under specific conditions, such as apoptosis. Thus, plasmalemmal scramblases, such as TMEM16F, need to be tightly regulated [7,8,60,70]. The dependence of TMEM16K on membrane thickness might provide a safety mechanism to preclude this protein from dissipating the plasma membrane asymmetry in case of aberrant trafficking leading to TMEM16K insertion to non-ER membranes [2].
Like the TMEM16x scramblases, the TMEM16A channel is a homodimer, and each monomer forms an independent pore [117,118] (Figure 2 and Figure 3). Experimentally determined structures of murine TMEM16A revealed two transmembrane α-helices (TM4 and TM6), effectively blocking the top of what constitutes the scramblase groove in scramblase homologues [105,107,108] (Figure 1). This results in the formation of a protein-enclosed ion-conductive pore in TMEM16A that is for the most part shielded from the membrane, but which might be partly accessible to lipids on its intracellular side [105,107,108], where the detachment of TM4 and TM6 forms a funnel-shaped vestibule that has portions directly exposed to the cytoplasm and the lipid bilayer. The shape of this ion-conductive pore resembles the shape of an hourglass. The hydrophilic membrane-exposed cavity in TMEM16x scramblases has evolved in TMEM16x channels to form an aqueous pore mostly shielded from the membrane lipids (Figure 1). The intracellular region connecting TM4 and TM5 plays a role in lipid scrambling in TMEM16F (termed the ‘scramblase domain’) [8]. Transfer of the TMEM16F or TMEM16E scrambling domains to TMEM16A confers scrambling activity to TMEM16A [8,119,120].
4.1. Gating Mechanisms
Each TMEM16A monomer has a principal high-affinity Ca2+-binding pocket for two Ca2+ ions; binding of Ca2+ at this site triggers channel opening [105,107] (Figure 1, Figure 2 and Figure 3). Each pore possesses a steric gate constituted by an intracellular portion of the sixth transmembrane α-helix (TM6) [107,121]. A hinge-point formed by glycine at position 640 (in aTMEM16A) enables a conformational rearrangement of this gate in response to Ca2+ binding [107,121] (Figure 3). Alanine substitution of I637 or Q645 (in aTMEM16A) stabilises the TM6 gate in the open state, which results in channel opening in the absence of intracellular Ca2+ at positive Vm [121,122,123]. The principal Ca2+-binding pocket encompasses a series of negatively charged residues [123], namely, E650, E698, E701, E730, and D734 (in aTMEM16A); except for E698, these residues are highly conserved in the various TMEM16x paralogues and homologues [124,125,126,127,128,129]. The proximity of these residues to the permeation pathway means that the vacant Ca2+-binding pocket provides an electrostatic barrier to anion permeation (termed the ‘electrostatic gate’). Binding of Ca2+ at this site screens the negative charge density of the Ca2+-binding pocket, thus producing attenuation of the electrostatic gate [123].
The existence of a low-affinity Ca2+-binding site in an intracellular domain of the channel has also been suggested [117,118], but the residues forming this site and its functional role are not fully defined. The recently solved TMEM16K [2] and TMEM16F [113,114] structures indicate a potential additional binding site for Ca2+ at the interface between the two TMEM16x monomers, and involving residues in TM2 and TM10 (Figure 1). This site is also found in TMEM16A, and mutations of key residues (i.e., E425, K428, D879, and D884 in aTMEM16A) reduce the channel activation in response to [Ca2+]i elevation [130]. Furthermore, an EF-hand-like region is also present at the N-terminus of TMEM16A and B [131] (Figure 1). Electrophysiology experiments demonstrated that TMEM16F proteins engineered to harbour this EF-hand-like domain have enhanced Ca2+ sensitivity [132]. In addition, the intracellular loop between TM2 and TM3 encompasses a segment composed of four glutamates in succession, adjacent to the spliced segment c of TMEM16A (exon 14 in TMEM16B) (Figure 1). This region is known to modulate Ca2+ sensitivity, and may bind Ca2+ directly in both TMEM16A and B channels [100,133,134].
For the TMEM16x scramblases such as human TMEM16K, afTMEM16, and nhTMEM16, Ca2+ binding triggers widening of the outer region of the groove to enable lipid scrambling [2,110,135]. An analogous gating movement is observed in the outer pore of the TMEM16A channel (Figure 3) [106,122,136]. This gating structure is composed of hydrophobic residues in TM4 and TM6, such as I550, I551, and I641 in acTMEM16A [106,136,137], and V539 and I636 in aTMEM16A [122,137] (Figure 2). Thus, TMEM16x channels and scramblases may share a common Ca2+-dependent mechanism that widens the outer pore/scrambling groove. The observation that single-point pore mutations confer scramblase activity to TMEM16A [138] is consistent with the idea that the lipid- and ion-permeation pathways in TMEM16x proteins may share a similar overall structural arrangement. These single-point mutations are (1) substitution of V543 in acTMEM16A into a threonine or serine (i.e., the nhTMEM16 and TMEM16F residues found in the position corresponding to V543 of TMEM16A), or (2) substitution of K588 in acTMEM16A into asparagine (i.e., the equivalent residue found in nhTMEM16) [138].
Intracellular Ca2+ is mandatory for TMEM16F activity [60,113,128], while afTMEM16 [139], nhTMEM16 [109] and, to a lesser degree, human TMEM16K [2], show some constitutive scrambling activity even in the absence of intracellular Ca2+. This basal activity might be ascribed to a high degree of mobility for TM3 and TM4 in fungal scramblases, and multiple flexible residues (such as P332 and G339 for nhTMEM16) may be involved [110,111]. The possibility that scramblase activity in the absence of Ca2+ is due to the presence of other cofactors cannot be ruled out [3,8,138].
4.2. Ion and Lipid Conduction Pathways
The ion conduction pathway in TMEM16A consists of a funnel-shaped intracellular vestibule that narrows to a tight pore at the extracellular part of the membrane [105,108]. Permeating ions need to shed their hydration shell as they pass through the narrow section of the pore; thus, the anion selectivity of TMEM16A follows a lyotropic sequence [1,17,140]. The relative anion permeability of TMEM16A measured under bi-ionic conditions is SCN− > NO3− > I− > Br− > Cl− > F− > gluconate [1,17,18,141]. Similar anion selectivity and permeability sequences have also been reported for TMEM16B and F [18,19,22,54,142,143,144,145].
The TMEM16A pore is amphiphilic and contains charged, polar, and apolar residues. Anion permeation in TMEM16A requires Cl− binding to a series of positively charged residues within the pore, including K584, R617, and K641 (in aTMEM16A) [108,117,118,122,146]. Mutations of pore residues R573 and K540 in TMEM16B (corresponding to R617 and K584 in aTMEM16A, respectively) lead to alterations in the ion selectivity and Ca2+ sensitivity of the TMEM16B channel, emphasising that TMEM16A and B channels share similar pore structures and permeation mechanisms [18,21].
TMEM16F channels are poorly selective for anions and cations. Whole-cell recordings of TMEM16F currents in a heterologous system (HEK-293 cells) demonstrated a slightly higher permeability to Cl− than to Na+ [142,144,147,148,149,150,151,152]; however, recordings obtained in excised inside-out patches (HEK-293 cells) revealed that TMEM16F was more permeable to cations than to anions [113,128,143]. Thus, the electrophysiological properties of TMEM16F are altered during inside-out recording, possibly because patch excision may cause disruption of the interaction of TMEM16F with the cytoskeleton and/or cytoplasmic factors [153]. The residues lining the pores of TMEM16F are not fully defined. It is noteworthy that K584 in aTMEM16A corresponds to Q559 in TMEM16F. The anionic selectivity of TMEM16A, relative to Na+, is reduced by glutamine substitution of K584, while the cationic selectivity and scramblase activity of TMEM16F are reduced by lysin substitution of Q559 [113,114,128,143,145,154].
As noted above, in TMEM16x scramblases, the TM4 and TM5 form a groove that enables lipid translocation. The region that constitutes the scramblase domain (see Section 4) can accommodate the headgroups of different sized lipids, but the narrow neck (the narrowest point of the hourglass shape) needs to widen in order to enable lipid scrambling [59,60,109,139]. MD simulations of nhTMEM16 demonstrated that lipids interact with charged and polar amino acids in the intracellular (e.g., E352, K353, R505, N378, Q374) and extracellular vestibules (e.g., E318, E313, R432, T333, Y439) [115,135,138]. It has been proposed that these interactions trigger conformational rearrangements that open the narrow neck [135], enabling lipid scrambling through a so-called ‘credit card mechanism’ [155]. Polar uncharged residues (e.g., T381, S382, T340) within the groove pathway are also important for lipid translocation, and mutations of these residues result in impaired lipid trafficking [120,138]. The E529 and K530 residues in V2-TMEM16F (conserved in TMEM16C, D, and E, and corresponding to E352 and K353 of nhTMEM16, respectively) are located deeper in the membrane compared to nhTMEM16, and only K530, but not E529, is essential for PS exposure [120]. Moreover, Y439, which is important for scrambling in fungal homologues, is not conserved in mammalian TMEM16x scramblases. Thus, despite a low sequence similarity between distant family members, such as TMEM16F and nhTMEM16, there is a remarkable conservation of function, indicating that scramblase activity tolerates sequence divergence [156,157].
A series of mechanisms can explain the combined ion channel and scramblase function of some TMEM16x proteins; these include (1) the ‘alternating pore–cavity’ model, which implies that ions permeate through an intermediate, semi-closed conformation of the lipid/ion permeation pathway that is nonconductive to lipids, but permeable to ions [113], and (2) the proteolipidic pore model, which implies that ions and lipids are transported through the fully open cavity lined with lipid headgroups [158]. These propositions have been tested in silico using MD simulations of fungal nhTMEM16, human TMEM16K, and murine TMEM16F. The results of these investigations suggest that the intermediate state of the permeation pathway does not allow the passage of ions and lipids [159]. Thus, Ca2+ binding may promote full opening of the pore/groove, enabling both ion conduction and lipid scrambling [138,159].
5. Mechanisms of Modulation of TMEM16x Proteins’ Function
5.1. Ca2+ and Other Divalent Cations
An increase in [Ca2+]i is the prime stimulus for the activation of TMEM16x proteins (Figure 4, Table 1). The response of native CaCCs to Ca2+ is characterised by a half-maximal effective concentration (EC50) in the range of 0.2–5 µM [14,160]. The Ca2+ response curve for native CaCC currents has a Hill coefficient > 1, which is consistent with the estimated binding of 2–3 Ca2+ required for channel activation [161,162,163], and the recent structural findings showing two bound Ca2+ ions in each TMEM16x monomer [105,107] (Figure 2). The Ca2+ sensitivity of cloned TMEM16A and B channels is in the same range as that of native CaCCs, with EC50 values depending on the clone species, splice variant, and recording conditions (e.g., the configuration of patch-clamp technique, or the Vm at which the recording was made) [17,18,22,54,100]. Intracellular Ca2+ activates TMEM16x channels in a Vm-dependent manner; the Ca2+ sensitivity increases proportionally to the degree of Vm depolarisation [1,18,127,133]. This is likely a direct consequence of the location of the principal Ca2+-binding pocket within the membrane-spanning region of TMEM16x proteins—an arrangement that favours intracellular Ca2+ binding at depolarised Vm [105]. As a result, the current versus Vm relationship for either TMEM16A or B channels is outwardly rectifying at low Ca2+ (<~1 μM), but becomes almost linear at high Ca2+, when Ca2+ binding occurs at depolarised and hyperpolarised Vm [18,22,164]. The possibility of Vm-dependent conformational changes taking place after Ca2+ binding has also been suggested [106,157].
TMEM16A currents can be activated in the absence of intracellular Ca2+ at highly depolarised Vm (>~120 mV) [25,100] (Figure 4, Table 1). Unlike other Vm-dependent channels—such as Vm-gated Na+ (Nav), Ca2+ (Cav), and K+ (Kv) channels—the TMEM16x proteins lack any specific Vm-sensing domains, comprising a series of positively charged residues. Consistently, no gating currents (which reflect the movement of the voltage sensors of Vm-gated channels) have been detected in TMEM16A channels during recordings in 0 [Ca2+]i and in the presence of blockers to prevent the ionic currents [165]. The intrinsic Vm dependence in TMEM16A channels may be the result of allosteric activation of the channel by extracellular Cl− [165]. TMEM16x channels also lack fast inactivation, which is typical of some Vm-gated channels such as Nav, Cav, and some Kv. However, prolonged exposure to high [Ca2+]i induces TMEM16x channel desensitisation [21,22,54,105,129,141,145,164] via a mechanism that might involve PIP2 depletion [164,166,167,168].
TMEM16F has an EC50 for Ca2+ of ~1 µM for scrambling activity [113]. The reported Ca2+ sensitivity of the TMEM16F channel function varies dramatically depending on the patch-clamp configuration used, with the reported EC50 equalling ~5–30 µM or 10–100 µM during inside-out [113,128,143,145] or whole-cell [132,142,144,145] recordings, respectively. These differences might be due to loss of cellular factors (e.g., PIP2) during patch excision [164] and/or disruption of interaction of TMEM16F with the cytoskeleton [169]. In addition, during recordings in the inside-out patch-clamp configuration, the TMEM16F current is rapidly activated as soon as the patch is excised and exposed to a solution supplemented with Ca2+ [128,143,145,170], while during whole-cell recordings the TMEM16F current gradually increases to reach a steady-state level within minutes [8,142,145]. In contrast, large heterologous TMEM16A and B currents are detected as soon as the whole-cell configuration is established [1,18,22,43,54,133].
A range of divalent cations may lead to activation of TMEM16x channels, but with different apparent EC50 (Ca2+ > Sr2+ ≈ Ni2+ > Ba2+) [54,100,141,171] (Figure 4, Table 1). The Hill coefficients of the dose–response curves for Ca2+, Sr2+, and Ba2+ vary from ~2.5 for Ca2+ to ~1.5 for Sr2+ and Ba2+, possibly indicating different degrees of cooperativity for these ligands [141]. These divalent cations may bind to the principal Ca2+-binding pocket; thus, in the presence of saturating [Ca2+]i, the addition of Sr2+, Ni2+, and Ba2+ produces no further current activation [171]. Intracellular divalent cations may also produce surface charge screening on the headgroups of phospholipids in the vicinity of the permeation pathways of TMEM16x channels, thus affecting the electrostatic potential in the proximity of the pore, which may influence ion permeation [170,172].
Other divalent cations such as Co2+, Zn2+, and Mg2+ (Table 1) are equivalent to competitive antagonists that lack the capacity to induce opening of the TM6 steric gate but compete with Ca2+ binding to the principal Ca2+-binding pocket, thereby producing a rightward shift in the current versus [Ca2+]i relationship for TMEM16A [141,171,173]. Binding of divalent (e.g., Mg2+) or trivalent (Gd3+) (Table 1) cations to the principal Ca2+-binding pocket also produces some attenuation of the electrostatic gate and increases the conductance of the TMEM16A channel [122,123].
5.2. Calmodulin
The possibility that TMEM16x sensitivity to intracellular Ca2+ is modulated by the Ca2+-binding messenger protein calmodulin (CaM), which is ubiquitously expressed in eukaryotic cells, has been explored extensively, but the outcome of this research effort is still inconclusive (Figure 4, Table 1). The following observations support the proposition that CaM is not mandatory for the response of TMEM16x channels to intracellular Ca2+: (1) CaM had no effect on TMEM16A and B currents when applied during inside-out patch-clamp recordings [22,54,174]; (2) heterologous expression of mutant forms of CaM with reduced Ca2+ sensitivity did not affect the amplitude of TMEM16A [174,175] or B [175] currents; (3) co-immunoprecipitation experiments revealed weak association between TMEM16A and CaM [174,176,177]; (4) low concentrations of intracellular Ba2+ (which does not bind to CaM [178]) activated the TMEM16A current [174]; (5) purified TMEM16A proteins reconstituted into liposomes elicited Ca2+-activated currents [177]; and (6) heterologous TMEM16F currents were not affected by pharmacological inhibition of Ca2+/CaM-dependent protein kinase II (CaMKII) [144].
In spite of the observations described above, a series of putative CaM-binding sites on TMEM16x proteins were identified. Bioinformatics analysis led to the identification of putative CaM-binding domains (CaM-BD1 and CaM-BD2) in TMEM16A [179] (Figure 1); however, there was no experimental evidence that CaM can bind to CaM-BD2 [179]. In addition, the CaM-BD1 sequence is in part encompassed within the splice segment b. The notion that TMEM16A channels lacking segment b mediate CaCC currents [46,48,180] suggests that CaM binding at this site is not mandatory for TMEM16A channel activity. Furthermore, splice segment b is associated with reduced Ca2+ sensitivity in TMEM16A [88].
An additional putative CaM-binding site, termed regulatory calmodulin-binding motif (RCBM), was identified in the N-terminus of TMEM16A and B channels, and Ca2+-CaM was shown to bind to isolated RCBM [175] (Figure 1). More recently, Jung et al. [181] reported two CaM-binding motifs (CBM1 and CBM2), with CBM1 partly overlapping with the CaM-BD1 described in [179] (Figure 1). Application of CaM to excised patches enhanced the HCO3− permeability at high [Ca2+]i in the acTMEM16A splice variant [181], although another study showed that in equimolar Cl− conditions exogenous CaM did not alter TMEM16A anion permeability [174].
Native CaCC currents in arterial smooth muscle cells are inhibited by CaMKII; thus, CaCC current activation can be evoked by (1) treatment with generic inhibitors of phosphorylation (synthetic AMP-PNP), (2) omission of ATP in the intracellular solution, or (3) gene silencing with siRNA directed against CaMKII [161,182,183,184] (Figure 4, Table 1); the former two types of treatment also reduce heterologous TMEM16A current rundown in HEK-293 cells [74,179,185]. Dephosphorylation of native CaCCs in isolated rabbit pulmonary arterial smooth muscle cells (PASMCs) may lead to an increase in current density. Dephosphorylation was induced via intracellular dialysis of a solution deprived of ATP and supplemented with KN-93 (a CaMKII inhibitor) during whole-cell patch-clamp recordings [186]. In isolated PASMCs, CaMKII affected CaCCs’ gating in response to depolarising Vm steps, and produced a rightward shift in the Vm dependency of the current [182]. Noise analysis of heterologous TMEM16A currents revealed that single-channel current (i) was significantly reduced by CaMKII phosphorylation, while the maximum open probability (Po) and channel number (N) were unaffected [104].
CaMKII (γ isoform) may physically interact with heterologous acTMEM16A channels and catalyse phosphorylation of (1) S673 in proximity to the principal Ca2+-binding pocket, and (2) S471, in the first intracellular loop [104,183,185]. Phosphorylation of these residues leads to current inhibition [104,183,185]. In contrast to the role of CaMKII in arterial smooth muscle cells and heterologous expression systems, CaMKII (β isoform) may enhance the surface expression and TMEM16A channel activity in a glioblastoma cell line and promote cell migration and invasion [187].
5.3. Intra- and Extracellular pH
Acidification of intracellular pH (pHi) below 6 favours protonation of residues (e.g., N650, E654, E702, E705, E734, and D738 in acTMEM16A) that form the principal Ca2+-binding pocket of TMEM16A [188,189]. Thus, H+ effectively competes with Ca2+ for the same site, leading to apparent current inhibition (Figure 4, Table 1), as illustrated by the observations that (1) at saturating [Ca2+]i the inhibitory effect of intracellular H+ ceased [189], and (2) amino acid substitution of the residues forming the principal Ca2+-binding pocket led to reduction in the sensitivity of the TMEM16A channel to intracellular H+ (N650A/E654Q (mild effect), E702Q/E705Q (strong effect), and E734Q/D738N (strong effect)) [188]. The inhibition of TMEM16A by intracellular H+ may constitute a protective feedback mechanism in some epithelial cells [188]. In pancreatic acinar cells, the HCO3− efflux through TMEM16A channels contributes to pH balance in the lumen of the acini in face of the acidification caused by the release of zymogen granules within the acinar lumen [190,191]; in turn, the HCO3− efflux leads to intracellular acidification and TMEM16A inhibition in acinar cells [191], constituting a negative feedback loop that prevents excessive loss of HCO3− in the acinar cells [188].
Another study suggested that intracellular H+ may effectively work as a low efficacy agonist, which may contribute to the presence of a TMEM16A current at highly depolarised Vm in the absence of intracellular Ca2+ (see also, Section 5.1) [192]. This is because titration of the acidic residues in the principal Ca2+-binding pocket of TMEM16A is favoured at depolarised Vm; the resulting protonation of the residues of the Ca2+-binding pocket may trigger channel opening and, possibly, attenuation of the electrostatic gate [192]. Indeed, the substitution of the acidic residues of the principal Ca2+-binding pocket with glutamines, mimicking a permanent protonation, allowed the Vm gating at physiological pHi, even with 0 [Ca2+]i [192].
The TMEM16F protein is also affected by changes in pHi. Liang and Yang [189] reported that pHi < 7 reduced both TMEM16F channel and scramblase activities, while pHi > 8 potentiated these functions. The activation of the TMEM16F channel at low pHi was detected at [Ca2+]i < 15 μM, while at higher [Ca2+]i the effect of low pHi became less prominent, suggesting that intracellular Ca2+ and H+ compete for the same site [189]. Substitution of E667 with glutamine within the principal Ca2+-binding pocket of TMEM16F shifted the peak of [Ca2+]i, which reduced TMEM16F pHi sensitivity from 15 μM to 3 mM [189]. In contrast, alanine substitution of D859 and E395 within the auxiliary Ca2+-binding site located on TM2-TM10 did not affect the sensitivity of TMEM16F to intracellular H+, suggesting no role for this site in controlling pHi sensitivity [189]. The pHi-mediated modulation of TMEM16F channel and scramblase functions may be relevant in the pathophysiology of cancer, since dysregulated pH is a hallmark of tumour progression [193]. TMEM16A and F are upregulated in several cancer types [34,194]. The notion that these TMEM16x members are capable of sensing changes in pHi may provide new insights into the involvement of TMEM16x in carcinogenesis and tumour progression.
The TMEM16A channel is also controlled by extracellular H+ [195] (Figure 4, Table 1). The channel is activated when extracellular pH (pHo) reaches values below 7. A glutamate (E623 in acTMEM16A) that is located in the extracellular loop connecting TM5 and TM6 is crucially involved in this response [195] (Figure 1). Non-stationary noise analysis revealed that titration of E623 by extracellular H+ allosterically increases the Po of the TMEM16A channel, without altering the single-channel conductance, thus enabling activation of TMEM16A at non-saturating [Ca2+]i—possibly through a conformational rearrangement that stabilises the principal Ca2+-binding pocket [195]. The residue is highly conserved in different TMEM16x homologues and paralogues; thus, it may also confer H+ sensitivity in other TMEM16x proteins [195]. Modulation of TMEM16A by extracellular H+ could be important in pathophysiological conditions, since changes in pHo occur during cellular injury, ischaemia, or tumour progression [196] in the brain [197], retina [198,199,200], or epithelial cells [201,202,203,204,205,206,207], where CaCC channels are expressed [17,46,208].
5.4. Intra- and Extracellular ATP
Adenosine triphosphate (ATP) is an important extracellular signalling molecule in several epithelia, and serves as a neurotransmitter in both the peripheral and central nervous systems. Extracellular ATP binds to purinergic P2Y (Gq-protein-coupled receptors) and P2X (non-selective cation channels) receptors, leading to an increase in [Ca2+]i that may promote activation of TMEM16x scramblases [209] and/or TMEM16x channels in airway epithelial cells [210,211,212,213,214], colonic epithelial cells [215], murine taste cells [216], supporting cells of the murine olfactory epithelium [217], and arterial smooth muscle cells [180].
Intracellular ATP may also modulate TMEM16A activity, as exemplified by the observation that the inclusion of apyrase, an ATP-cleaving enzyme, in the pipette solution during whole-cell patch-clamp recordings dampened heterologous abcTMEM16A and acTMEM16A channel activity [179]. Intracellular ATP may indirectly induce TMEM16A current activation by serving as a substrate for the synthesis of PIP2 and/or CaMKII-mediated phosphorylation of serine residues in TMEM16A [104,218,219]. Other TMEM16x proteins are also influenced by intracellular ATP (Figure 4, Table 1). Intracellular MgATP, but not Na2ATP, prevented TMEM16F current rundown in inside-out patches, mimicking the activating effect of PIP2 [164]. Whole-cell TMEM16F current rundown could also be prevented by MgATP-promoted cytoskeletal actin polymerisation [169]. Somewhat counterintuitively, disruption of the actin cytoskeleton with cytochalasin-D [169] or as a result of patch excision [145,169] hastened the activation kinetics of the TMEM16F current, while actin-filament-stabilising agents inhibited TMEM16F activity [169].
Reduced cytoplasmic concentration of ATP during altered metabolic states—for example, during cell hypoxia and ischaemia—has the potential to reduce the extrusion of Ca2+ from the cytoplasm (e.g., via the plasma membrane Ca2+-ATPase) and, thus, lead to an increase in [Ca2+]i and the activation of TMEM16x. It is plausible that this mechanism may constitute a relevant aspect of cellular responses to ischemia, and participate for example in arteriolar and pericyte contraction that may occur during cerebral ischaemia, given that the TMEM16A channel forms a crucial depolarising force in contractile vascular cells [220,221].
5.5. Hypoxia and Reactive Oxygen Species (ROS)
Tissue hypoxia is associated with conditions such as stroke, angina pectoris, myocardial infarction, heart failure, and peripheral artery disease. Hypoxia has been shown to enhance TMEM16A current density in cultured murine cardiac vascular endothelial cells [222] (Figure 4, Table 1). The underlying mechanism is an increase in TMEM16A expression, including alteration of the repertoire of the splicing variants being expressed [222]. Chronic hypoxic pulmonary hypertension is associated with enhanced TMEM16A current density and increased levels of TMEM16A mRNA and protein expression in murine PASMCs [223]. These effects may lead to vessel contraction and remodelling [223]. TMEM16A is also upregulated in idiopathic forms of pulmonary hypertension in humans [224]. Long-term hypoxia can also enhance TMEM16A expression and current density in epithelia, such as in cultured sinonasal epithelial layers [225].
In an airway-epithelium-derived cell line, hypoxia-induced augmentation of reactive oxygen species (ROS)—and the consequent peroxidation of plasma membrane lipids—increased TMEM16F activity [152]. This resulted in PS (phosphatidylserine) exposure, inflammatory cell death, and apoptosis [226] (Figure 4, Table 1). Hypoxia, oxidative stress, and lipid peroxidation may also activate TMEM16A during conditions such as polycystic kidney disease [227]. The underlying mechanism likely involves store-operated Ca2+ entry triggered by oxidative stress [227]. Lipid peroxidation induced by ROS enhances heterologous TMEM16A and F currents and activates TMEM16F-mediated phospholipid scrambling in HEK-293 cells [228].
5.6. Heat
The TMEM16x channel activity is highly sensitive to heat [228,229,230,231] (Figure 4, Table 1). The thermal sensitivity of TMEM16A becomes prominent at temperatures > ~37 °C, with an apparent temperature coefficient (Q10) of ~20. The temperature threshold for TMEM16A activation is lowered as [Ca2+]i is increased [230]. At temperatures > 44 °C, small heterologous TMEM16A currents can be detected even at 0 [Ca2+]i [230]. The underlying biophysical mechanism of TMEM16A heat activation is not fully defined; it may involve protein domains serving as specific thermal sensors—as suggested for other strongly temperature-sensitive channels, such as TRP channels [232,233,234]—or it may arise from the difference in heat capacity between open and closed states [235]. The increases in [Ca2+]i and temperature have a synergistic effect on TMEM16A activation; thus, concomitant increases in these variables stimulate the TMEM16A channel more substantially than either factor alone [230]. Furthermore, the heat-mediated activation can be observed even at saturating [Ca2+]i [17,230]. It remains to be established whether heat promotes changes in Po and/or affects i or N.
In virtue of its thermal sensitivity and high expression in dorsal root ganglion (DRG) neurons, the TMEM16A channel plays a key role in nociceptive thermal sensitivity [230,236]. Consistently, DRG neurons from mice in which the Tmem16A gene was deleted (knockout) had reduced heat-activated Cl− currents [230,237]. A role for TMEM16A in nociception was also demonstrated by the observation that the channel is activated in response to bradykinin, which is released at sites of tissue damage and inflammation [237]. Behavioural experiments demonstrated that pharmacological inhibition of TMEM16A (with mefloquine or with small interfering RNA (siRNA)) or Tmem16A knockout impaired the normal response to heat in tail-withdrawal tests [230]. TMEM16B, but not TMEM16D or E, is also temperature sensitive [230,238]. However, a major role for TMEM16B in thermal nociception was excluded because it is expressed in DRG neurons at very low levels compared to TMEM16A [230].
TMEM16F proteins are also markedly heat sensitive, and demonstrate increased Ca2+ sensitivity during whole-cell patch-clamp recordings at 37 °C relative to the activity measured at room temperature [228]. All TMEM16F splice variants are activated at temperatures between 37 and 42 °C in the submicromolar range of [Ca2+]i [231]. However, the TMEM16F current amplitude was not significantly increased by temperatures > 42 °C, in contrast with what was observed for the TMEM16A channel [230,231]. The temperature sensitivity of TMEM16F scramblase function remains not fully defined, having been reported to either decrease [231] or increase [239] at 37 °C relative to measurements of scramblase activity at room temperature.
5.7. Lipids
5.7.1. PIP2
As noted above (see Section 3), TMEM16x proteins are activated by GqPCR stimulation. The associated activation of PLC leads to PIP2 breakdown and the formation of IP3, which promotes an increase in [Ca2+]i and TMEM16x activation. PIP2 binds to cloned and native smooth muscle TMEM16A channels in membrane extracts [240]. Ta et al. [241] investigated whether PIP2 controls the function of TMEM16A and B channels. Cloned acTMEM16A was activated by a water-soluble PIP2 analogue during inside-out patch-clamp recordings. Depletion of endogenous PIP2 with a genetically encoded Danio rerio voltage-sensitive phosphatase (DrVSP) reduced heterologous TMEM16A currents [241]. This effect was attenuated by an inactivating mutation in DrVSP and antagonised by co-expression of a phosphatidylinositol-4-phosphate 5-kinase that catalyses PIP2 formation. In contrast, the TMEM16B channel was inhibited by PIP2 [241]. The effect of PIP2 on TMEM16A channels was Vm-independent, and was especially pronounced in the low micromolar range of [Ca2+]i (<~2 μM). In contrast, the effect of PIP2 on TMEM16B did not differ significantly over a wide range of [Ca2+]i, but was only detectable at highly depolarised Vm (≥50 mV) [241]. PIP2 affected TMEM16A and B current amplitude via modulation of Po, while i and ion selectivity remained unaltered [241]. It was proposed that in vivo PIP2 may modulate TMEM16A under resting conditions, as well as during membrane depolarisation, to constitute a negative feedback on GqPCR-mediated TMEM16A current activation by [Ca2+]i [241]. In contrast, TMEM16B may be modulated only at highly depolarised Vm, which might be reached by some types of excitable cells during action potential firing—especially during pathological conditions associated with elevations of the action potential peak, such as hypernatremia [241].
Other groups also reported that PIP2 activated cloned TMEM16A channels (Figure 4, Table 1) and prevented current rundown during recordings in excised patches [166,167,215]. In Xenopus laevis oocytes, depletion of membrane PIP2 through applications of a PIP2-sequestering agent (neomycin) resulted in inactive TMEM16A channels even in the presence of saturating [Ca2+]i, suggesting that PIP2 and Ca2+ are both mandatory for TMEM16A function [219]. Native CaCC currents in rodent PASMCs, which are mediated by TMEM16A [48], were found to be inhibited by PIP2 [240]. In addition, internal application of water-soluble PIP2 analogues in whole-cell recordings produced little-to-no effect on endogenous TMEM16A currents in HT29 colonic epithelial cells [215]. Thus, the mechanism of PIP2 regulation of native CaCCs in mammalian cells may not be univocal, and may vary depending on cell-type-specific regulatory components.
Regulation of the TMEM16A channel by PIP2 may vary depending on the splice variant, with heterologous acTMEM16A displaying higher channel activity and more pronounced current rundown following PIP2 depletion than aTMEM16A [104]. PIP2 resynthesis, dependent on cytosolic ATP, was essential for acTMEM16A recovery from rundown [218,241]. Mutagenesis and atomistic simulations suggested that CaMKII phosphorylation of S673 (in acTMEM16A) in the third intracellular loop reduced the channel’s sensitivity to PIP2 [104]. TMEM16A splice variants lacking the segment c were less sensitive to S673 phosphorylation [104].
Le et al. [167] identified a putative PIP2-binding site in aTMEM16A composed of six basic residues (three arginine and three lysine) at the cytosolic interface of TM3–TM5. Binding of PIP2 at this site stabilised the open state and prevented current rundown [167] (Figure 1). The ion-conducting pore of TMEM16A therefore consists of two functionally distinct modules: the principal Ca2+-binding pocket module formed by residues in TM6–TM8, and the PIP2-binding site regulatory module involving residues in TM3–TM5 [167] (Figure 1). It was suggested that Ca2+ and PIP2 synergistically promote TMEM16A activation [167]. The possibility that other members of the family may share the dual regulation by Ca2+ and PIP2 observed in TMEM16A could not be excluded [167]. Using unbiased atomistic MD simulations and single-point mutagenesis, Yu et al. [242] identified eight additional putative interaction sites for PIP2 on acTMEM16A. Maximal activation of TMEM16A requires interaction with both PIP2 and Ca2+ [218,242,243].
The experimentally determined structure of the open TMEM16A channel is not yet available. Using atomistic simulations, Jia and Chen [137] obtained a model of the open channel. Binding of PIP2 to TMEM16A was sufficient to induce spontaneous dilation of the pore through a rearrangement of TM3 and TM4, similarly to another open-channel model proposed by a different group [122] (Figure 2). These studies provide insights into the conformational changes triggered by PIP2 binding, as well as the conformation of the open TMEM16A channel.
The TMEM16F channel/scramblase is also activated by PIP2 [164]. TMEM16F’s Ca2+ response is desensitised by a brief exposure to high [Ca2+]i, but subsequent exposure to PIP2 or water-soluble PIP2 analogues restores TMEM16F channel activity [164]. Mutagenetic analysis revealed that electrostatic interactions of PIP2 with a cluster of positively charged amino acids within the N-terminus modulate TMEM16F synergistically with Vm depolarisation to facilitate Ca2+ gating [164] (Figure 1).
5.7.2. Other Lipids
Plasmalemmal lipids can serve as both substrates and modulators of scramblase activity and ion conduction in afTMEM16, nhTMEM16, and mammalian TMEM16x scramblases [156,157] (Table 1). For example, in liposomes composed of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylethanolamine/1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylglycerol (POPE/POPG), afTMEM16 and nhTMEM16 had reduced ion transport rates, which were enhanced in liposomes supplemented with 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylcholine (POPC) [139,244]. Liposomes composed of POPC/POPG led to reduced scrambling activity of nhTMEM16 [111].
The primary structure of TMEM16A encompasses 14 potential cholesterol-binding motifs, but only the site near the extracellular end of TM5 has been shown—via computational docking—to bind cholesterol [166]. Exposure of isolated murine portal vein myocytes to methyl-β-cyclodextrin (M-βCD)—an agent that depletes membrane cholesterol—increased the CaCC current amplitude (Figure 4, Table 1) without affecting other channel properties, such as the sensitivity to NFA and A9C (generic Cl− channel blockers) [245]. Acute application of M-βCD also enhanced THE whole-cell TMEM16A current heterologously expressed in HEK-293T cells [166].
A number of fatty acids, including some dietary polyunsaturated fatty acids (PUFAs), inhibit TMEM16A channel activity (Figure 4, Table 1). These include saturated stearic acid (SA), uncharged methyl stearate (Me-S), monounsaturated oleic acid (OA), polyunsaturated arachidonic acid (AA), polyunsaturated eicosapentaenoic acid (EPA), and polyunsaturated docosahexaenoic acid (DHA) [166]. The underlying mechanism (gating modification or direct occlusion of the pore) remains undefined [166]. The effect of EPA and OA (poly- and monounsaturated long-chain fatty acids, respectively) was detectable only at positive Vm (120 mV), while DHA (a polyunsaturated long-chain fatty acid) exhibited current inhibition even at negative Vm (−100 mV) [166]. The mechanism of Vm dependence of inhibition did not rely on the charge of the lipid headgroup, since uncharged saturated fatty acid analogues of OA also inhibited TMEM16A in a Vm-dependent fashion, albeit to a lesser extent than OA [166]. Thus, the mechanism underlying the Vm dependence of fatty acid inhibition may be secondary to Vm-dependent conformational changes in TMEM16A. The saturated SA produced a much smaller effect in comparison with unsaturated fatty acids with one or more double bonds [166]. The number of double hydrogen bonds in cis geometry in the fatty acid tail is a determinant of fatty acid modulation of several ion channels [246], and may also be a determinant of TMEM16A inhibition.
TMEM16A regulation by lipids may also be important in pathology. Whole-cell TMEM16A currents increased in a murine lung adenocarcinoma cell line (LA795) treated with 10 µg lipopolysaccharide (LPS) (Table 1) for 24 h [247]; this appears to have been due to an increase in TMEM16A mRNA and protein levels after LPS exposure [247]. LPS treatment (10 μg/mL LPS, 24–36 h) induced TMEM16A overexpression in adenocarcinomic human alveolar basal epithelial cells (A549). The underling mechanism remains not fully defined, but TMEM16A activation was associated with reductions in tumour necrosis factor-α (TNF-α) and interleukin 8 (IL-8) secretions, as well as inhibition of the ‘nuclear factor kappa light-chain enhancer of activated B cell’ (NF-κB) pathway [247,248]. TMEM16A expression in rat intestinal epithelial (RAW264.7) cells was increased by exposure to high doses of LPS (10 µg/mL, 12–36 h) [249]. Treatment of monolayers of a rat intestinal epithelial cell line (EEC-6) with low doses of LPS (0.1–1 µg/mL) worsened cell barrier dysfunction, possibly by activating extracellular-signal-regulated kinase1/myosin light-chain kinase (ERK1/MLCK) signalling pathways [249]. However, treatment with higher doses of LPS (10 µg/mL) resulted in a protective effect mediated by TMEM16A activation, and possible subsequent recruitment of the extracellular signal-regulated kinase/B-cell lymphoma-2/Bcl-2-associated x protein (ERK/Bcl-2/Bax) signalling pathways via an undefined mechanism [249]. Collectively, these studies might imply that TMEM16A could play a role in the pathology of LPS-induced inflammation. However, the concentration of LPS used in these studies was significantly higher than the plasma LPS levels detected in human patients during severe sepsis [250]; thus, the pathophysiological significance of the above observations remains unclear.
Phospholipids such as lysophosphatidic acid (LPA) (Figure 4, Table 1) and arachidonic acids (AAs) (Table 1) are also modulators of TMEM16x function. Acutely applied AA inhibited heterologous TMEM16F current during whole-cell patch-clamp recordings [251]. Pharmacological modulation of phospholipase A2 (PLA2) impacted heterologous abcTMEM16A and V1-TMEM16F currents and PS scrambling by V1-TMEM16F in HEK-293 cells [228]. Heterologous TMEM16F current is enhanced by stimulation of PLA2 through generation of plasma membrane LPA, independent of any increase in [Ca2+]i [251]. Lysophospholipids (LPL), such as LPA, may affect cell membrane tension [252] and possibly promote scramblase activity, even in the absence of Ca2+ [251]. LPA reaches a plasma concentration of up to ~10 µM [253,254] during activation of platelets [255,256]. LPA triggers an increase in [Ca2+]i by promoting opening of Ca2+ channels within the membranes of erythrocytes [257]. The [Ca2+]i increase provokes TMEM16F-mediated exposure of PS on the outer surface of the erythrocyte membrane that promotes the thrombogenic process and blood clotting [70,258,259,260,261]. Natural products such as tannic acid (TA) and epigallocatechin-3-gallate (EGCG) inhibit TMEM16F-mediated PS exposure induced by LPA [226,239,261,262]. However, a direct effect of TA and EGCG on TMEM16F scramblase activity has been questioned, since these compounds act as fluorescence quenchers [263]; this results in loss of fluorescence emission of annexin V—a commonly used PS-binding probe to report scrambling activities [263]. The studies above thus suggest that the downstream products of PLA2 may have contrasting effects on TMEM16F activity, with AA and LPL causing inhibition or activation of TMEM16F currents, respectively [228,251].
TMEM16A is activated by bile acid uptake in the apical cholangiocyte membrane of murine, rat, and human biliary epithelium, and may promote increased bile flow during cholestasis through ductular secretion [264]. TMEM16A-mediated Cl− transepithelial secretion is rapidly increased by intracellular dialysis of ursodeoxycholic acid (UDCA) or tauroursodeoxycholic acid (TUDCA) (Table 1) [264]. The extracellular application of both UDCA and TUDCA induced exocytosis, ATP release, and an increase in [Ca2+]i, resulting in the activation of TMEM16A currents [264]. TMEM16A currents were not observed when UDCA and TUDCA were applied together with inhibitors of P2Y and IP3 receptors, such as apyrase, suramin, or 2-aminoethoxydiphenyl borate (2-APB) [264]. Bile acid activates TMEM16A currents via purinergic signalling that mediates an increase in [Ca2+]i through IP3-receptor-dependent Ca2+ release from intracellular stores [264]. Thus, bile acid released by hepatocytes activates TMEM16A currents in the downstream cholangiocytes, which may represent an example of hepatobiliary coupling that links bile acid release with modulation of ductular Cl− transport [264].
6. Other Regulators of TMEM16x Activity
6.1. Ca2+-Activated Chloride Channel Regulators 1 and 2 (CLCA1 and 2)
The Ca2+-activated chloride channel regulators (CLCAs) [265] are a family of secreted self-cleaving metalloproteases that modulate CaCC activity in mammalian cells [266]. CLCAs play important roles in mucus homeostasis [267], and their dysfunction is implicated in pathologies such as asthma and chronic obstructive pulmonary disease (COPD) [268]. Overexpression of CLCAs leads to CaCC activation in a variety of cell types [269,270,271,272], and CLACs were initially proposed as CaCCs’ pore-forming subunits [265].
Secreted CLCA1 modulates TMEM16A channels in a paracrine fashion by increasing TMEM16A surface expression, stabilising the TMEM16A homodimer configuration and promoting channel activity through a physical interaction with TMEM16A [273] (Figure 4, Table 1). CLCAs may also stimulate store-operated entry of Ca2+, and the resulting change in [Ca2+]i may affect TMEM16A activity [274] (Figure 4, Table 1). CLCA2 interacts with the store-operated Ca2+ channel ‘calcium-release-activated calcium channel protein 1′ (ORAI-1) and the ER calcium sensor ‘stromal interaction molecule 1′ (STIM-1) in HEK-293 cells stably expressing human CLCA2 [274]. TMEM16A and CLCA2 are upregulated in some tumours, which may lead to enhancement of TMEM16A currents and tumour progression [274,275,276,277].
A secreted form of CLCA1, the N-terminal CLCA1 (N-CLCA1), encompasses a von Willebrand factor type A (VWA) domain that is involved in the interaction with TMEM16A and activation of TMEM16A currents [273,278]. Injection of N-CLCA1 into the trachea of mice produced intraluminal mucus accumulation in the airways, possibly through increased expression of TMEM16A channels in the apical membranes of airway epithelial cells [279].
6.2. KCNE1
KCNE1 is a single-TM-domain protein that forms an ancillary (β) subunit of the Vm-gated K+ channel KCNQ1 [280,281]. Ávalos Prado et al. [282] suggested that KCNE1 also serves as an auxiliary subunit of the TMEM16A channel, and that it assembles with a 2:2 stoichiometry. In the presence of KCNE1, the TMEM16A channel was found to be active at positive Vm even with 0 [Ca2+]i [282]. It was proposed that KCNE1 modulates TMEM16A activity by favouring TM6 (steric gate) opening [282] (Figure 4, Table 1). A region of 13 residues was identified as the minimal portion of KCNE1 sufficient to produce modulation of the TMEM16A current [282]. Moreover, mutant forms of KCNE1 harbouring single-nucleotide polymorphisms—such as S38G and R32H, which are associated with predisposition to heart failure and long QT syndrome (LQTS) in human subjects—failed to regulate TMEM16A currents [282]. This observation possibly implicates KCNE1 regulation of TMEM16A in the onset of these pathologies. KCNE5, a KCNE1 homologue, also physically interacts with TMEM16A [282]. Unlike TMEM16A, the TMEM16B channel appeared not to be regulated by KCNE1 or KCNE5 [282].
7. Additional Regulatory Mechanisms of TMEM16x Proteins
TMEM16x proteins are subjected to post-translational modifications such as protein phosphorylation (reviewed in [99] for the TMEM16A channel). TMEM16x proteins may also be subjected to SUMOylation (covalent attachment of a small ubiquitin-related modifier (SUMO) to a protein), with SUMOylation sites having been identified in TMEM16B, but not TMEM16A [283]. It is currently unknown whether TMEM16x proteins are subject to RNA editing, a form of post-transcriptional modification that applies to a range of ion channels [284].
8. A Role for TMEM16F in the Pathology of SARS-CoV-2
TMEM16F may also play a role in the pathogenesis of SARS-CoV-2. A characteristic feature of SARS-CoV-2 infection is the formation of pneumocyte syncytia, which involves viral spike protein cleavage by host cell proteases [285,286]. Cells expressing SARS-CoV-2 spike proteins have enhanced Ca2+ oscillations and increased TMEM16F activity, leading to PS externalisation, which promotes syncytia formation [29]. SARS-CoV-2 spike proteins may (1) promote Ca2+ release and, thus, activation of TMEM16F scrambling in infected cells (cis modality), and/or (2) stimulate activation of proteases on neighbouring cells, triggering cell fusion (trans modality) [29]. The involvement of TMEM16F in SARS-CoV-2 spike-induced syncytia is supported by the role of PS exposure in a range of other physiological cell fusion events [287,288,289,290,291]. Niclosamide—a therapeutic anthelmintic drug utilised for the treatment of tapeworm infections—reduces syncytial formation by inhibiting TMEM16F activity, and could be repurposed for the treatment of SARS-CoV-2 [29,292].
9. Conclusions
The TMEM16x family of Ca2+-activated channels and scramblases provides a link between intracellular Ca2+ handling and ion/lipid transport. It is becoming apparent that these proteins respond to a range of additional cellular factors, highlighting their capacity to serve as sensors of cellular homeostasis. Uncovering these intricate mechanisms of regulation will enable a more complete understanding of the TMEM16x physiological roles and aid in the exploitation of these potential pharmacological targets for the treatment of a range of human diseases, including stroke, hypertension, vascular dementia, cystic fibrosis, and cancer.
Author Contributions
Both authors researched the literature, and wrote and edited the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
E.A. is a Blaschko Fellow. Research in P.T.’s lab is supported by the British Heart Foundation (BHF) (PG/19/8/34168) and the Biotechnology and Biological Sciences Research Council (BBSRC) (BB/T007664/1).
Institutional Review Board Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Schroeder, B.C.; Cheng, T.; Jan, Y.N.; Jan, L.Y. Expression Cloning of TMEM16A as a Calcium-Activated Chloride Channel Subunit. Cell 2008, 134, 1019–1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bushell, S.R.; Pike, A.C.W.; Falzone, M.E.; Rorsman, N.J.G.; Ta, C.M.; Corey, R.A.; Newport, T.D.; Christianson, J.C.; Scofano, L.F.; Shintre, C.A.; et al. The Structural Basis of Lipid Scrambling and Inactivation in the Endoplasmic Reticulum Scramblase TMEM16K. Nat. Commun. 2019, 10, 3956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Zanni, E.; Gradogna, A.; Scholz-Starke, J.; Boccaccio, A. Gain of Function of TMEM16E/ANO5 Scrambling Activity Caused by a Mutation Associated with Gnathodiaphyseal dysplasia. Cell. Mol. Life Sci. 2018, 75, 1657–1670. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Wang, D.; Dong, Y.; Gao, X.; Tong, H.; Liu, W.; Zhang, L.; Sun, M. ANO7: Insights into Topology, Function, and Potential Applications as a Biomarker and Immunotherapy Target. Tissue Cell 2021, 72, 101546. [Google Scholar] [CrossRef]
- Kim, H.; Kim, H.; Lee, J.; Lee, B.; Kim, H.-R.; Jung, J.; Lee, M.-O.; Oh, U. Anoctamin 9/TMEM16J Is a Cation Channel Activated by CAMP/PKA Signal. Cell Calcium. 2018, 71, 75–85. [Google Scholar] [CrossRef]
- Reichhart, N.; Schöberl, S.; Keckeis, S.; Alfaar, A.S.; Roubeix, C.; Cordes, M.; Crespo-Garcia, S.; Haeckel, A.; Kociok, N.; Föckler, R.; et al. Anoctamin-4 Is a Bona Fide Ca2+-Dependent Non-Selective Cation Channel. Sci. Rep. 2019, 9, 2257. [Google Scholar] [CrossRef]
- Scudieri, P.; Caci, E.; Venturini, A.; Sondo, E.; Pianigiani, G.; Marchetti, C.; Ravazzolo, R.; Pagani, F.; Galietta, L.J.V. Ion Channel and Lipid Scramblase Activity Associated with Expression of TMEM16F/ANO6 Isoforms. J. Physiol. 2015, 593, 3829–3848. [Google Scholar] [CrossRef] [Green Version]
- Yu, K.; Whitlock, J.M.; Lee, K.; Ortlund, E.A.; Yuan Cui, Y.; Hartzell, H.C. Identification of a Lipid Scrambling Domain in ANO6/TMEM16F. eLife 2015, 4, e06901. [Google Scholar] [CrossRef]
- Huang, F.; Wang, X.; Ostertag, E.M.; Nuwal, T.; Huang, B.; Jan, Y.-N.; Basbaum, A.I.; Jan, L.Y. TMEM16C Facilitates Na+-Activated K+ Currents in Rat Sensory Neurons and Regulates Pain Processing. Nat. Neurosci. 2013, 16, 1284–1290. [Google Scholar] [CrossRef]
- Jha, A.; Chung, W.Y.; Vachel, L.; Maleth, J.; Lake, S.; Zhang, G.; Ahuja, M.; Muallem, S. Anoctamin 8 Tethers Endoplasmic Reticulum and Plasma Membrane for Assembly of Ca2+ Signaling Complexes at the ER/PM Compartment. EMBO J. 2019, 38, e101452. [Google Scholar] [CrossRef] [PubMed]
- Galindo, B.E.; Vacquier, V.D. Phylogeny of the TMEM16 Protein Family: Some Members Are Overexpressed in Cancer. Int. J. Mol. Med. 2005, 16, 919–924. [Google Scholar] [CrossRef] [PubMed]
- Milenkovic, V.M.; Brockmann, M.; Stöhr, H.; Weber, B.H.; Strauss, O. Evolution and Functional Divergence of the Anoctamin Family of Membrane Proteins. BMC Evol. Biol. 2010, 10, 319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelz, T.; Drose, D.R.; Fleck, D.; Henkel, B.; Ackels, T.; Spehr, M.; Neuhaus, E.M. An Ancestral TMEM16 Homolog from Dictyostelium Discoideum Forms a Scramblase. PLoS ONE 2018, 13, e0191219. [Google Scholar] [CrossRef] [Green Version]
- Ferrera, L.; Zegarra-Moran, O.; Galietta, L.J.V. Ca2+-Activated Cl− Channels. Compr. Physiol. 2011, 1, 2155–2174. [Google Scholar] [CrossRef]
- Hartzell, H.C.; Yu, K.; Xiao, Q.; Chien, L.-T.; Qu, Z. Anoctamin/TMEM16 Family Members Are Ca2+-Activated Cl− Channels. J. Physiol. 2009, 587, 2127–2139. [Google Scholar] [CrossRef]
- Huang, F.; Wong, X.; Jan, L.Y. International Union of Basic and Clinical Pharmacology. LXXXV: Calcium-Activated Chloride Channels. Pharmacol. Rev. 2012, 64, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.D.; Cho, H.; Koo, J.Y.; Tak, M.H.; Cho, Y.; Shim, W.-S.; Park, S.P.; Lee, J.; Lee, B.; Kim, B.-M.; et al. TMEM16A Confers Receptor-Activated Calcium-Dependent Chloride Conductance. Nature 2008, 455, 1210–1215. [Google Scholar] [CrossRef]
- Adomaviciene, A.; Smith, K.J.; Garnett, H.; Tammaro, P. Putative Pore-Loops of TMEM16/Anoctamin Channels Affect Channel Density in Cell Membranes. J. Physiol. 2013, 591, 3487–3505. [Google Scholar] [CrossRef]
- Betto, G.; Cherian, O.L.; Pifferi, S.; Cenedese, V.; Boccaccio, A.; Menini, A. Interactions between Permeation and Gating in the TMEM16B/Anoctamin2 Calcium-Activated Chloride Channel. J. Gen. Physiol. 2014, 143, 703–718. [Google Scholar] [CrossRef] [Green Version]
- Cruz-Rangel, S.; Jesús-Pérez, J.J.D.; Contreras-Vite, J.A.; Pérez-Cornejo, P.; Hartzell, H.C.; Arreola, J. Gating Modes of Calcium-Activated Chloride Channels TMEM16A and TMEM16B. J. Physiol. 2015, 593, 5283–5298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pifferi, S. Permeation Mechanisms in the TMEM16B Calcium-Activated Chloride Channels. PLoS ONE 2017, 12, e0169572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pifferi, S.; Dibattista, M.; Menini, A. TMEM16B Induces Chloride Currents Activated by Calcium in Mammalian Cells. Pflüg. Arch. Eur. J. Physiol. 2009, 458, 1023–1038. [Google Scholar] [CrossRef] [PubMed]
- Scudieri, P.; Sondo, E.; Ferrera, L.; Galietta, L.J.V. The Anoctamin Family: TMEM16A and TMEM16B as Calcium-Activated Chloride Channels. Exp. Physiol. 2012, 97, 177–183. [Google Scholar] [CrossRef]
- Cherian, O.L.; Menini, A.; Boccaccio, A. Multiple Effects of Anthracene-9-Carboxylic Acid on the TMEM16B/Anoctamin2 Calcium-Activated Chloride Channel. Biochim. Biophys. Acta BBA Biomembr. 2015, 1848, 1005–1013. [Google Scholar] [CrossRef] [Green Version]
- Ta, C.M.; Adomaviciene, A.; Rorsman, N.J.G.; Garnett, H.; Tammaro, P. Mechanism of Allosteric Activation of TMEM16A/ANO1 Channels by a Commonly Used Chloride Channel Blocker. Br. J. Pharmacol. 2016, 173, 511–528. [Google Scholar] [CrossRef] [Green Version]
- Bradley, E.; Fedigan, S.; Webb, T.; Hollywood, M.A.; Thornbury, K.D.; McHale, N.G.; Sergeant, G.P. Pharmacological Characterization of TMEM16A Currents. Channels 2014, 8, 308–320. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Zhang, H.; Huang, D.; Qi, J.; Xu, J.; Gao, H.; Du, X.; Gamper, N.; Zhang, H. Characterization of the Effects of Cl− Channel Modulators on TMEM16A and Bestrophin-1 Ca2+ Activated Cl− Channels. Pflugers Arch. 2015, 467, 1417–1430. [Google Scholar] [CrossRef]
- Seo, Y.; Lee, H.K.; Park, J.; Jeon, D.; Jo, S.; Jo, M.; Namkung, W. Ani9, A Novel Potent Small-Molecule ANO1 Inhibitor with Negligible Effect on ANO2. PLoS ONE 2016, 11, e0155771. [Google Scholar] [CrossRef] [Green Version]
- Braga, L.; Ali, H.; Secco, I.; Chiavacci, E.; Neves, G.; Goldhill, D.; Penn, R.; Jimenez-Guardeño, J.M.; Ortega-Prieto, A.M.; Bussani, R.; et al. Drugs That Inhibit TMEM16 Proteins Block SARS-CoV-2 Spike-Induced Syncytia. Nature 2021, 594, 88–93. [Google Scholar] [CrossRef]
- Miner, K.; Labitzke, K.; Liu, B.; Wang, P.; Henckels, K.; Gaida, K.; Elliott, R.; Chen, J.J.; Liu, L.; Leith, A.; et al. Drug Repurposing: The Anthelmintics Niclosamide and Nitazoxanide Are Potent TMEM16A Antagonists That Fully Bronchodilate Airways. Front. Pharmacol. 2019, 10, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schreiber, R.; Uliyakina, I.; Kongsuphol, P.; Warth, R.; Mirza, M.; Martins, J.R.; Kunzelmann, K. Expression and Function of Epithelial Anoctamins. J. Biol. Chem. 2010, 285, 7838–7845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benarroch, E.E. Anoctamins (TMEM16 Proteins): Functions and Involvement in Neurologic Disease. Neurology 2017, 89, 722–729. [Google Scholar] [CrossRef] [PubMed]
- Berg, J.; Yang, H.; Jan, L.Y. Ca2+-Activated Cl− Channels at a Glance. J. Cell Sci. 2012, 125, 1367–1371. [Google Scholar] [CrossRef] [Green Version]
- Crottès, D.; Jan, L.Y. The Multifaceted Role of TMEM16A in Cancer. Cell Calcium. 2019, 82, 102050. [Google Scholar] [CrossRef]
- Duran, C.; Hartzell, H.C. Physiological Roles and Diseases of Tmem16/Anoctamin Proteins: Are They All Chloride Channels? Acta Pharmacol. Sin. 2011, 32, 685–692. [Google Scholar] [CrossRef]
- Duran, C.; Qu, Z.; Osunkoya, A.O.; Cui, Y.; Hartzell, H.C. ANOs 3–7 in the Anoctamin/Tmem16 Cl− Channel Family Are Intracellular Proteins. Am. J. Physiol. Cell Physiol. 2012, 302, C482–C493. [Google Scholar] [CrossRef] [Green Version]
- Kunzelmann, K.; Tian, Y.; Martins, J.R.; Faria, D.; Kongsuphol, P.; Ousingsawat, J.; Thevenod, F.; Roussa, E.; Rock, J.; Schreiber, R. Anoctamins. Pflüg. Arch. Eur. J. Physiol. 2011, 462, 195–208. [Google Scholar] [CrossRef]
- Oh, U.; Jung, J. Cellular Functions of TMEM16/Anoctamin. Pflüg. Arch. Eur. J. Physiol. 2016, 468, 443–453. [Google Scholar] [CrossRef] [Green Version]
- Pedemonte, N.; Galietta, L.J.V. Structure and Function of TMEM16 Proteins (Anoctamins). Physiol. Rev. 2014, 94, 419–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picollo, A.; Malvezzi, M.; Accardi, A. TMEM16 Proteins: Unknown Structure and Confusing Functions. J. Mol. Biol. 2015, 427, 94–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salzer, I.; Boehm, S. Calcium-Activated Chloride Channels: Potential Targets for Antinociceptive Therapy. Int. J. Biochem. Cell Biol. 2019, 111, 37–41. [Google Scholar] [CrossRef] [PubMed]
- Whitlock, J.M.; Hartzell, H.C. Anoctamins/TMEM16 Proteins: Chloride Channels Flirting with Lipids and Extracellular Vesicles. Annu. Rev. Physiol. 2017, 79, 119–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caputo, A.; Caci, E.; Ferrera, L.; Pedemonte, N.; Barsanti, C.; Sondo, E.; Pfeffer, U.; Ravazzolo, R.; Zegarra-Moran, O.; Galietta, L.J.V. TMEM16A, A Membrane Protein Associated with Calcium-Dependent Chloride Channel Activity. Science 2008, 322, 590–594. [Google Scholar] [CrossRef]
- Dutta, A.K.; Khimji, A.; Kresge, C.; Bugde, A.; Dougherty, M.; Esser, V.; Ueno, Y.; Glaser, S.S.; Alpini, G.; Rockey, D.C.; et al. Identification and Functional Characterization of TMEM16A, a Ca2+-Activated Cl− Channel Activated by Extracellular Nucleotides, in Biliary Epithelium. J. Biol. Chem. 2011, 286, 766–776. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.C.; Xiao, S.; Huang, F.; Harfe, B.D.; Jan, Y.N.; Jan, L.Y. Calcium-Activated Chloride Channels (CaCCs) Regulate Action Potential and Synaptic Response in Hippocampal Neurons. Neuron 2012, 74, 179–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, A.J.; Forrest, A.S.; Jepps, T.A.; Valencik, M.L.; Wiwchar, M.; Singer, C.A.; Sones, W.R.; Greenwood, I.A.; Leblanc, N. Expression Profile and Protein Translation of TMEM16A in Murine Smooth Muscle. Am. J. Physiol. Cell Physiol. 2010, 299, C948–C959. [Google Scholar] [CrossRef] [Green Version]
- Heinze, C.; Seniuk, A.; Sokolov, M.V.; Huebner, A.K.; Klementowicz, A.E.; Szijártó, I.A.; Schleifenbaum, J.; Vitzthum, H.; Gollasch, M.; Ehmke, H.; et al. Disruption of Vascular Ca2+-Activated Chloride Currents Lowers Blood Pressure. J. Clin. Investig. 2014, 124, 675–686. [Google Scholar] [CrossRef] [Green Version]
- Manoury, B.; Tamuleviciute, A.; Tammaro, P. TMEM16A/Anoctamin 1 Protein Mediates Calcium-Activated Chloride Currents in Pulmonary Arterial Smooth Muscle Cells. J. Physiol. 2010, 588, 2305–2314. [Google Scholar] [CrossRef]
- Thomas-Gatewood, C.; Neeb, Z.P.; Bulley, S.; Adebiyi, A.; Bannister, J.P.; Leo, M.D.; Jaggar, J.H. TMEM16A Channels Generate Ca2+-Activated Cl− Currents in Cerebral Artery Smooth Muscle Cells. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H1819–H1827. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Li, C.; Huai, R.; Qu, Z. Overexpression of ANO1/TMEM16A, an Arterial Ca2+-Activated Cl− Channel, Contributes to Spontaneous Hypertension. J. Mol. Cell. Cardiol. 2015, 82, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Dauner, K.; Möbus, C.; Frings, S.; Möhrlen, F. Targeted Expression of Anoctamin Calcium-Activated Chloride Channels in Rod Photoreceptor Terminals of the Rodent Retina. Investig. Ophthalmol. Vis. Sci. 2013, 54, 3126–3136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hengl, T.; Kaneko, H.; Dauner, K.; Vocke, K.; Frings, S.; Möhrlen, F. Molecular Components of Signal Amplification in Olfactory Sensory Cilia. Proc. Natl. Acad. Sci. USA 2010, 107, 6052–6057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pietra, G.; Dibattista, M.; Menini, A.; Reisert, J.; Boccaccio, A. The Ca2+-Activated Cl− Channel TMEM16B Regulates Action Potential Firing and Axonal Targeting in Olfactory Sensory Neurons. J. Gen. Physiol. 2016, 148, 293–311. [Google Scholar] [CrossRef] [Green Version]
- Stephan, A.B.; Shum, E.Y.; Hirsh, S.; Cygnar, K.D.; Reisert, J.; Zhao, H. ANO2 Is the Cilial Calcium-Activated Chloride Channel That May Mediate Olfactory Amplification. Proc. Natl. Acad. Sci. USA 2009, 106, 11776–11781. [Google Scholar] [CrossRef] [Green Version]
- Stöhr, H.; Heisig, J.B.; Benz, P.M.; Schöberl, S.; Milenkovic, V.M.; Strauss, O.; Aartsen, W.M.; Wijnholds, J.; Weber, B.H.F.; Schulz, H.L. TMEM16B, a Novel Protein with Calcium-Dependent Chloride Channel Activity, Associates with a Presynaptic Protein Complex in Photoreceptor Terminals. J. Neurosci. Off. J. Soc. Neurosci. 2009, 29, 6809–6818. [Google Scholar] [CrossRef]
- Ayoglu, B.; Mitsios, N.; Kockum, I.; Khademi, M.; Zandian, A.; Sjöberg, R.; Forsström, B.; Bredenberg, J.; Bomfim, I.L.; Holmgren, E.; et al. Anoctamin 2 Identified as an Autoimmune Target in Multiple Sclerosis. Proc. Natl. Acad. Sci. USA 2016, 113, 2188–2193. [Google Scholar] [CrossRef] [Green Version]
- Ha, G.E.; Lee, J.; Kwak, H.; Song, K.; Kwon, J.; Jung, S.-Y.; Hong, J.; Chang, G.-E.; Hwang, E.M.; Shin, H.-S.; et al. The Ca2+-Activated Chloride Channel Anoctamin-2 Mediates Spike-Frequency Adaptation and Regulates Sensory Transmission in Thalamocortical Neurons. Nat. Commun. 2016, 7, 13791. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, Z.; Xiao, S.; Tien, J.; Le, S.; Le, T.; Jan, L.Y.; Yang, H. Inferior Olivary TMEM16B Mediates Cerebellar Motor Learning. Neuron 2017, 95, 1103–1111.e4. [Google Scholar] [CrossRef]
- Suzuki, J.; Fujii, T.; Imao, T.; Ishihara, K.; Kuba, H.; Nagata, S. Calcium-Dependent Phospholipid Scramblase Activity of TMEM16 Protein Family Members. J. Biol. Chem. 2013, 288, 13305–13316. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, J.; Umeda, M.; Sims, P.J.; Nagata, S. Calcium-Dependent Phospholipid Scrambling by TMEM16F. Nature 2010, 468, 834–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reichhart, N.; Milenkovic, V.M.; Wetzel, C.H.; Strauß, O. Prediction of Functional Consequences of Missense Mutations in ANO4 Gene. Int. J. Mol. Sci. 2021, 22, 2732. [Google Scholar] [CrossRef] [PubMed]
- Maniero, C.; Scudieri, P.; Haris Shaikh, L.; Zhao, W.; Gurnell, M.; Galietta, L.J.V.; Brown, M.J. ANO4 (Anoctamin 4) Is a Novel Marker of Zona Glomerulosa That Regulates Stimulated Aldosterone Secretion. Hypertension 2019, 74, 1152–1159. [Google Scholar] [CrossRef] [PubMed]
- Terracciano, A.; Sanna, S.; Uda, M.; Deiana, B.; Usala, G.; Busonero, F.; Maschio, A.; Scally, M.; Patriciu, N.; Chen, W.-M.; et al. Genome-Wide Association Scan for Five Major Dimensions of Personality. Mol. Psychiatry 2010, 15, 647–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Athanasiu, L.; Mattingsdal, M.; Kähler, A.K.; Brown, A.; Gustafsson, O.; Agartz, I.; Giegling, I.; Muglia, P.; Cichon, S.; Rietschel, M.; et al. Gene Variants Associated with Schizophrenia in a Norwegian Genome-Wide Study Are Replicated in a Large European Cohort. J. Psychiatr. Res. 2010, 44, 748–753. [Google Scholar] [CrossRef] [Green Version]
- Sherva, R.; Tripodis, Y.; Bennett, D.A.; Chibnik, L.B.; Crane, P.K.; de Jager, P.L.; Farrer, L.A.; Saykin, A.J.; Shulman, J.M.; Naj, A.; et al. Genome-Wide Association Study of the Rate of Cognitive Decline in Alzheimer’s Disease. Alzheimers Dement. 2014, 10, 45–52. [Google Scholar] [CrossRef] [Green Version]
- Webb, B.T.; Guo, A.-Y.; Maher, B.S.; Zhao, Z.; van den Oord, E.J.; Kendler, K.S.; Riley, B.P.; Gillespie, N.A.; Prescott, C.A.; Middeldorp, C.M.; et al. Meta-Analyses of Genome-Wide Linkage Scans of Anxiety-Related Phenotypes. Eur. J. Hum. Genet. 2012, 20, 1078–1084. [Google Scholar] [CrossRef]
- Whitlock, J.M.; Yu, K.; Cui, Y.Y.; Hartzell, H.C. Anoctamin 5/TMEM16E Facilitates Muscle Precursor Cell Fusion. J. Gen. Physiol. 2018, 150, 1498–1509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Zanni, E.; Gradogna, A.; Picco, C.; Scholz-Starke, J.; Boccaccio, A. TMEM16E/ANO5 Mutations Related to Bone Dysplasia or Muscular Dystrophy Cause Opposite Effects on Lipid Scrambling. Hum. Mutat. 2020, 41, 1157–1170. [Google Scholar] [CrossRef] [Green Version]
- van Kruchten, R.; Mattheij, N.J.A.; Saunders, C.; Feijge, M.A.H.; Swieringa, F.; Wolfs, J.L.N.; Collins, P.W.; Heemskerk, J.W.M.; Bevers, E.M. Both TMEM16F-Dependent and TMEM16F-Independent Pathways Contribute to Phosphatidylserine Exposure in Platelet Apoptosis and Platelet Activation. Blood 2013, 121, 1850–1857. [Google Scholar] [CrossRef] [Green Version]
- Fujii, T.; Sakata, A.; Nishimura, S.; Eto, K.; Nagata, S. TMEM16F Is Required for Phosphatidylserine Exposure and Microparticle Release in Activated Mouse Platelets. Proc. Natl. Acad. Sci. USA 2015, 112, 12800–12805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foltz, S.J.; Cui, Y.Y.; Choo, H.J.; Hartzell, H.C. ANO5 Ensures Trafficking of Annexins in Wounded Myofibers. J. Cell Biol. 2021, 220, 7059. [Google Scholar] [CrossRef] [PubMed]
- Stamelou, M.; Charlesworth, G.; Cordivari, C.; Schneider, S.A.; Kägi, G.; Sheerin, U.-M.; Rubio-Agusti, I.; Batla, A.; Houlden, H.; Wood, N.W.; et al. The Phenotypic Spectrum of DYT24 Due to ANO3 Mutations. Mov. Disord. 2014, 29, 928–934. [Google Scholar] [CrossRef] [PubMed]
- Kaikkonen, E.; Rantapero, T.; Zhang, Q.; Taimen, P.; Laitinen, V.; Kallajoki, M.; Jambulingam, D.; Ettala, O.; Knaapila, J.; Boström, P.J.; et al. ANO7 Is Associated with Aggressive Prostate Cancer. Int. J. Cancer 2018, 143, 2479–2487. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Schreiber, R.; Kunzelmann, K. Anoctamins Are a Family of Ca2+-Activated Cl− Channels. J. Cell Sci. 2012, 125, 4991–4998. [Google Scholar] [CrossRef] [Green Version]
- Blanco, M.A.; Alečković, M.; Hua, Y.; Li, T.; Wei, Y.; Xu, Z.; Cristea, I.M.; Kang, Y. Identification of Staphylococcal Nuclease Domain-Containing 1 (SND1) as a Metadherin-Interacting Protein with Metastasis-Promoting Functions. J. Biol. Chem. 2011, 286, 19982–19992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaikkonen, E.; Takala, A.; Pursiheimo, J.-P.; Wahlström, G.; Schleutker, J. The Interactome of the Prostate-Specific Protein Anoctamin 7. Cancer Biomark. 2020, 28, 91–100. [Google Scholar] [CrossRef] [Green Version]
- Hammer, C.; Wanitchakool, P.; Sirianant, L.; Papiol, S.; Monnheimer, M.; Faria, D.; Ousingsawat, J.; Schramek, N.; Schmitt, C.; Margos, G.; et al. A Coding Variant of ANO10, Affecting Volume Regulation of Macrophages, Is Associated with Borrelia Seropositivity. Mol. Med. Camb. Mass 2015, 21, 26–37. [Google Scholar] [CrossRef]
- Petkovic, M.; Oses-Prieto, J.; Burlingame, A.; Jan, L.Y.; Jan, Y.N. TMEM16K Is an Interorganelle Regulator of Endosomal Sorting. Nat. Commun. 2020, 11, 3298. [Google Scholar] [CrossRef]
- Tsuji, T.; Cheng, J.; Tatematsu, T.; Ebata, A.; Kamikawa, H.; Fujita, A.; Gyobu, S.; Segawa, K.; Arai, H.; Taguchi, T.; et al. Predominant Localization of Phosphatidylserine at the Cytoplasmic Leaflet of the ER, and Its TMEM16K-Dependent Redistribution. Proc. Natl. Acad. Sci. USA 2019, 116, 13368–13373. [Google Scholar] [CrossRef] [Green Version]
- Schreiber, R.; Ousingsawat, J.; Kunzelmann, K. Targeting of Intracellular TMEM16 Proteins to the Plasma Membrane and Activation by Purinergic Signaling. Int. J. Mol. Sci. 2020, 21, 4065. [Google Scholar] [CrossRef]
- Katsurahara, K.; Shiozaki, A.; Kosuga, T.; Kudou, M.; Shoda, K.; Arita, T.; Konishi, H.; Komatsu, S.; Kubota, T.; Fujiwara, H.; et al. ANO9 Regulated Cell Cycle in Human Esophageal Squamous Cell Carcinoma. Ann. Surg. Oncol. 2020, 27, 3218–3230. [Google Scholar] [CrossRef] [PubMed]
- Kramer, J.; Hawley, R.S. The Spindle-Associated Transmembrane Protein Axs Identifies a Membranous Structure Ensheathing the Meiotic Spindle. Nat. Cell Biol. 2003, 5, 261–263. [Google Scholar] [CrossRef] [PubMed]
- Wanitchakool, P.; Ousingsawat, J.; Sirianant, L.; Cabrita, I.; Faria, D.; Schreiber, R.; Kunzelmann, K. Cellular Defects by Deletion of ANO10 Are Due to Deregulated Local Calcium Signaling. Cell. Signal. 2017, 30, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Balreira, A.; Boczonadi, V.; Barca, E.; Pyle, A.; Bansagi, B.; Appleton, M.; Graham, C.; Hargreaves, I.P.; Rasic, V.M.; Lochmüller, H.; et al. ANO10 Mutations Cause Ataxia and Coenzyme Q10 Deficiency. J. Neurol. 2014, 261, 2192–2198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vermeer, S.; Hoischen, A.; Meijer, R.P.P.; Gilissen, C.; Neveling, K.; Wieskamp, N.; de Brouwer, A.; Koenig, M.; Anheim, M.; Assoum, M.; et al. Targeted Next-Generation Sequencing of a 12.5 Mb Homozygous Region Reveals ANO10 Mutations in Patients with Autosomal-Recessive Cerebellar Ataxia. Am. J. Hum. Genet. 2010, 87, 813–819. [Google Scholar] [CrossRef] [Green Version]
- Jun, I.; Park, H.S.; Piao, H.; Han, J.W.; An, M.J.; Yun, B.G.; Zhang, X.; Cha, Y.H.; Shin, Y.K.; Yook, J.I.; et al. ANO9/TMEM16J Promotes Tumourigenesis via EGFR and Is a Novel Therapeutic Target for Pancreatic Cancer. Br. J. Cancer 2017, 117, 1798–1809. [Google Scholar] [CrossRef]
- Katsurahara, K.; Shiozaki, A.; Kosuga, T.; Shimizu, H.; Kudou, M.; Arita, T.; Konishi, H.; Komatsu, S.; Kubota, T.; Fujiwara, H.; et al. ANO9 Regulates PD-L2 Expression and Binding Ability to PD-1 in Gastric Cancer. Cancer Sci. 2021, 112, 1026–1037. [Google Scholar] [CrossRef]
- Ferrera, L.; Caputo, A.; Ubby, I.; Bussani, E.; Zegarra-Moran, O.; Ravazzolo, R.; Pagani, F.; Galietta, L.J.V. Regulation of TMEM16A Chloride Channel Properties by Alternative Splicing. J. Biol. Chem. 2009, 284, 33360–33368. [Google Scholar] [CrossRef] [Green Version]
- Han, J.; Kim, H.; Seo, D.-G.; Lee, G.; Jeung, E.-B.; Yu, F.H. Multiple Transcripts of Anoctamin Genes Expressed in the Mouse Submandibular Salivary Gland. J. Periodontal Implant Sci. 2015, 45, 69–75. [Google Scholar] [CrossRef] [Green Version]
- Huanosta-Gutiérrez, A.; Espino-Saldaña, A.E.; Reyes, J.P.; Pétriz, A.; Miledi, R.; Martínez-Torres, A. TMEM16A Alternative Splicing Isoforms in Xenopus Tropicalis: Distribution and Functional Properties. Biochem. Biophys. Res. Commun. 2014, 446, 1096–1101. [Google Scholar] [CrossRef]
- Ponissery Saidu, S.; Stephan, A.B.; Talaga, A.K.; Zhao, H.; Reisert, J. Channel Properties of the Splicing Isoforms of the Olfactory Calcium-Activated Chloride Channel Anoctamin 2. J. Gen. Physiol. 2013, 141, 691–703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segawa, K.; Suzuki, J.; Nagata, S. Constitutive Exposure of Phosphatidylserine on Viable Cells. Proc. Natl. Acad. Sci. USA 2011, 108, 19246–19251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strege, P.R.; Bernard, C.E.; Mazzone, A.; Linden, D.R.; Beyder, A.; Gibbons, S.J.; Farrugia, G. A Novel Exon in the Human Ca2+-Activated Cl− Channel Ano1 Imparts Greater Sensitivity to Intracellular Ca2+. Am. J. Physiol.-Gastrointest. Liver Physiol. 2015, 309, G743–G749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Xu, L.; Lau, Y.S.; Gao, Y.; Moore, S.A.; Han, R. A Novel ANO5 Splicing Variant in a LGMD2L Patient Leads to Production of a Truncated Aggregation-Prone Ano5 Peptide. J. Pathol. Clin. Res. 2018, 4, 135–145. [Google Scholar] [CrossRef] [Green Version]
- O’Driscoll, K.E.; Pipe, R.A.; Britton, F.C. Increased Complexity of Tmem16a/Anoctamin 1 Transcript Alternative Splicing. BMC Mol. Biol. 2011, 12, 35. [Google Scholar] [CrossRef] [Green Version]
- Mazzone, A.; Gibbons, S.J.; Bernard, C.E.; Nowsheen, S.; Middha, S.; Almada, L.L.; Ordog, T.; Kendrick, M.L.; Lombardo, K.R.; Shen, K.R.; et al. Identification and Characterization of a Novel Promoter for the Human ANO1 Gene Regulated by the Transcription Factor Signal Transducer and Activator of Transcription 6 (STAT6). FASEB J. 2015, 29, 152–163. [Google Scholar] [CrossRef] [Green Version]
- Mazzone, A.; Bernard, C.E.; Strege, P.R.; Beyder, A.; Galietta, L.J.V.; Pasricha, P.J.; Rae, J.L.; Parkman, H.P.; Linden, D.R.; Szurszewski, J.H.; et al. Altered Expression of Ano1 Variants in Human Diabetic Gastroparesis. J. Biol. Chem. 2011, 286, 13393–13403. [Google Scholar] [CrossRef] [Green Version]
- Sondo, E.; Scudieri, P.; Tomati, V.; Caci, E.; Mazzone, A.; Farrugia, G.; Ravazzolo, R.; Galietta, L.J.V. Non-Canonical Translation Start Sites in the TMEM16A Chloride Channel. Biochim. Biophys. Acta BBA Biomembr. 2014, 1838, 89–97. [Google Scholar] [CrossRef] [Green Version]
- Hawn, M.B.; Akin, E.; Hartzell, H.C.; Greenwood, I.A.; Leblanc, N. Molecular Mechanisms of Activation and Regulation of ANO1-Encoded Ca2+-Activated Cl− Channels. Channels 2021, 197, 5411. [Google Scholar] [CrossRef]
- Xiao, Q.; Yu, K.; Perez-Cornejo, P.; Cui, Y.; Arreola, J.; Hartzell, H.C. Voltage- and Calcium-Dependent Gating of TMEM16A/Ano1 Chloride Channels Are Physically Coupled by the First Intracellular Loop. Proc. Natl. Acad. Sci. USA 2011, 108, 8891–8896. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, C.M. Interaction of Tetraethylammonium Ion Derivatives with the Potassium Channels of Giant Axons. J. Gen. Physiol. 1971, 58, 413–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenwood, I.A.; Large, W.A. Modulation of the Decay of Ca2+-Activated Cl− Currents in Rabbit Portal Vein Smooth Muscle Cells by External Anions. J. Physiol. 1999, 516 Pt 2, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Yellen, G. Single Channel Seeks Permeant Ion for Brief but Intimate Relationship. J. Gen. Physiol. 1997, 110, 83–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, W.; Jung, S.-R.; Kim, K.-W.; Yeon, J.-H.; Park, C.-G.; Nam, J.H.; Hille, B.; Suh, B.-C. Allosteric Modulation of Alternatively Spliced Ca2+-Activated Cl− Channels TMEM16A by PI(4,5)P2 and CaMKII. Proc. Natl. Acad. Sci. USA 2020, 20, 117. [Google Scholar] [CrossRef]
- Dang, S.; Feng, S.; Tien, J.; Peters, C.J.; Bulkley, D.; Lolicato, M.; Zhao, J.; Zuberbühler, K.; Ye, W.; Qi, L.; et al. Cryo-EM Structures of the TMEM16A Calcium-Activated Chloride Channel. Nature 2017, 552, 426–429. [Google Scholar] [CrossRef] [PubMed]
- Lam, A.K.M.; Rheinberger, J.; Paulino, C.; Dutzler, R. Gating the Pore of the Calcium-Activated Chloride Channel TMEM16A. Nat. Commun. 2021, 12, 785. [Google Scholar] [CrossRef]
- Paulino, C.; Kalienkova, V.; Lam, A.K.M.; Neldner, Y.; Dutzler, R. Activation Mechanism of the Calcium-Activated Chloride Channel TMEM16A Revealed by Cryo-EM. Nature 2017, 552, 421–425. [Google Scholar] [CrossRef] [Green Version]
- Paulino, C.; Neldner, Y.; Lam, A.K.; Kalienkova, V.; Brunner, J.D.; Schenck, S.; Dutzler, R. Structural Basis for Anion Conduction in the Calcium-Activated Chloride Channel TMEM16A. eLife 2017, 6, e26232. [Google Scholar] [CrossRef] [Green Version]
- Brunner, J.D.; Lim, N.K.; Schenck, S.; Duerst, A.; Dutzler, R. X-Ray Structure of a Calcium-Activated TMEM16 Lipid Scramblase. Nature 2014, 516, 207–212. [Google Scholar] [CrossRef]
- Falzone, M.E.; Rheinberger, J.; Lee, B.-C.; Peyear, T.; Sasset, L.; Raczkowski, A.M.; Eng, E.T.; Di Lorenzo, A.; Andersen, O.S.; Nimigean, C.M.; et al. Structural Basis of Ca2+-Dependent Activation and Lipid Transport by a TMEM16 Scramblase. eLife 2019, 8, e43229. [Google Scholar] [CrossRef]
- Kalienkova, V.; Clerico Mosina, V.; Bryner, L.; Oostergetel, G.T.; Dutzler, R.; Paulino, C. Stepwise Activation Mechanism of the Scramblase NhTMEM16 Revealed by Cryo-EM. eLife 2019, 8, e44364. [Google Scholar] [CrossRef] [PubMed]
- Khelashvili, G.; Falzone, M.E.; Cheng, X.; Lee, B.-C.; Accardi, A.; Weinstein, H. Dynamic Modulation of the Lipid Translocation Groove Generates a Conductive Ion Channel in Ca2+-Bound nhTMEM16. Nat. Commun. 2019, 10, 4972. [Google Scholar] [CrossRef] [PubMed]
- Alvadia, C.; Lim, N.K.; Clerico Mosina, V.; Oostergetel, G.T.; Dutzler, R.; Paulino, C. Cryo-EM Structures and Functional Characterization of the Murine Lipid Scramblase TMEM16F. eLife 2019, 8, e44365. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Dang, S.; Han, T.W.; Ye, W.; Jin, P.; Cheng, T.; Li, J.; Jan, Y.N.; Jan, L.Y.; Cheng, Y. Cryo-EM Studies of TMEM16F Calcium-Activated Ion Channel Suggest Features Important for Lipid Scrambling. Cell Rep. 2019, 28, 567–579.e4. [Google Scholar] [CrossRef] [PubMed]
- Bethel, N.P.; Grabe, M. Atomistic Insight into Lipid Translocation by a TMEM16 Scramblase. Proc. Natl. Acad. Sci. USA 2016, 113, 14049–14054. [Google Scholar] [CrossRef] [Green Version]
- Stansfeld, P.J.; Goose, J.E.; Caffrey, M.; Carpenter, E.P.; Parker, J.L.; Newstead, S.; Sansom, M.S.P. MemProtMD: Automated Insertion of Membrane Protein Structures into Explicit Lipid Membranes. Structure 2015, 23, 1350–1361. [Google Scholar] [CrossRef] [Green Version]
- Jeng, G.; Aggarwal, M.; Yu, W.-P.; Chen, T.-Y. Independent Activation of Distinct Pores in Dimeric TMEM16A Channels. J. Gen. Physiol. 2016, 148, 393–404. [Google Scholar] [CrossRef] [Green Version]
- Lim, N.K.; Lam, A.K.M.; Dutzler, R. Independent Activation of Ion Conduction Pores in the Double-Barreled Calcium-Activated Chloride Channel TMEM16A. J. Gen. Physiol. 2016, 148, 375–392. [Google Scholar] [CrossRef] [Green Version]
- Gyobu, S.; Miyata, H.; Ikawa, M.; Yamazaki, D.; Takeshima, H.; Suzuki, J.; Nagata, S. A Role of TMEM16E Carrying a Scrambling Domain in Sperm Motility. Mol. Cell. Biol. 2016, 36, 645–659. [Google Scholar] [CrossRef] [Green Version]
- Gyobu, S.; Ishihara, K.; Suzuki, J.; Segawa, K.; Nagata, S. Characterization of the Scrambling Domain of the TMEM16 Family. Proc. Natl. Acad. Sci. USA 2017, 114, 6274–6279. [Google Scholar] [CrossRef] [Green Version]
- Peters, C.J.; Gilchrist, J.M.; Tien, J.; Bethel, N.P.; Qi, L.; Chen, T.; Wang, L.; Jan, Y.N.; Grabe, M.; Jan, L.Y. The Sixth Transmembrane Segment Is a Major Gating Component of the TMEM16A Calcium-Activated Chloride Channel. Neuron 2018, 97, 1063–1077.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinsdale, R.L.; Pipatpolkai, T.; Agostinelli, E.; Russell, A.J.; Stansfeld, P.J.; Tammaro, P. An Outer-Pore Gate Modulates the Pharmacology of the TMEM16A Channel. Proc. Natl. Acad. Sci. USA 2021, 118, 2118. [Google Scholar] [CrossRef] [PubMed]
- Lam, A.K.; Dutzler, R. Calcium-Dependent Electrostatic Control of Anion Access to the Pore of the Calcium-Activated Chloride Channel TMEM16A. eLife 2018, 7, e39122. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Zhang, S.; Ren, S.; Chen, Y.; Yuan, H.; Chai, R.; Yu, H.; Zhang, H.; Zhan, Y.; An, H. Two Ca2+-Binding Sites Cooperatively Couple Together in TMEM16A Channel. J. Membr. Biol. 2016, 249, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Jung, J.; Tak, M.H.; Wee, J.; Lee, B.; Jang, Y.; Chun, H.; Yang, D.-J.; Yang, Y.D.; Park, S.H.; et al. Two Helices in the Third Intracellular Loop Determine Anoctamin 1 (TMEM16A) Activation by Calcium. Pflugers Arch. 2015, 467, 1677–1687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pang, C.-L.; Yuan, H.-B.; Cao, T.-G.; Su, J.-G.; Chen, Y.-F.; Liu, H.; Yu, H.; Zhang, H.-L.; Zhan, Y.; An, H.-L.; et al. Molecular Simulation Assisted Identification of Ca2+ Binding Residues in TMEM16A. J. Comput. Aided Mol. Des. 2015, 29, 1035–1043. [Google Scholar] [CrossRef]
- Tien, J.; Peters, C.J.; Wong, X.M.; Cheng, T.; Jan, Y.N.; Jan, L.Y.; Yang, H. A Comprehensive Search for Calcium Binding Sites Critical for TMEM16A Calcium-Activated Chloride Channel Activity. eLife 2014, 3, e02772. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Kim, A.; David, T.; Palmer, D.; Jin, T.; Tien, J.; Huang, F.; Cheng, T.; Coughlin, S.R.; Jan, Y.N.; et al. TMEM16F Forms a Ca2+-Activated Cation Channel Required for Lipid Scrambling in Platelets during Blood Coagulation. Cell 2012, 151, 111–122. [Google Scholar] [CrossRef] [Green Version]
- Yu, K.; Duran, C.; Qu, Z.; Cui, Y.-Y.; Hartzell, H.C. Explaining Calcium-Dependent Gating of Anoctamin-1 Chloride Channels Requires a Revised Topology. Circ. Res. 2012, 110, 990–999. [Google Scholar] [CrossRef]
- Le, S.C.; Yang, H. An Additional Ca2+ Binding Site Allosterically Controls TMEM16A Activation. Cell Rep. 2020, 33, 108570. [Google Scholar] [CrossRef]
- Tak, M.H.; Jang, Y.; Son, W.S.; Yang, Y.D.; Oh, U. EF-Hand like Region in the N-Terminus of Anoctamin 1 Modulates Channel Activity by Ca2+ and Voltage. Exp. Neurobiol. 2019, 28, 658–669. [Google Scholar] [CrossRef] [PubMed]
- Roh, J.W.; Hwang, G.E.; Kim, W.K.; Nam, J.H. Ca2+ Sensitivity of Anoctamin 6/TMEM16F Is Regulated by the Putative Ca2+-Binding Reservoir at the N-Terminal Domain. Mol. Cells 2021, 44, 88–100. [Google Scholar] [CrossRef] [PubMed]
- Cenedese, V.; Betto, G.; Celsi, F.; Cherian, O.L.; Pifferi, S.; Menini, A. The Voltage Dependence of the TMEM16B/Anoctamin2 Calcium-Activated Chloride Channel Is Modified by Mutations in the First Putative Intracellular Loop. J. Gen. Physiol. 2012, 139, 285–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, Q.; Cui, Y. Acidic Amino Acids in the First Intracellular Loop Contribute to Voltage- and Calcium- Dependent Gating of Anoctamin1/TMEM16A. PLoS ONE 2014, 9, e99376. [Google Scholar] [CrossRef]
- Lee, B.-C.; Khelashvili, G.; Falzone, M.; Menon, A.K.; Weinstein, H.; Accardi, A. Gating Mechanism of the Extracellular Entry to the Lipid Pathway in a TMEM16 Scramblase. Nat. Commun. 2018, 9, 3251. [Google Scholar] [CrossRef]
- Lam, A.K.M.; Dutzler, R. Mechanism of Pore Opening in the Calcium-Activated Chloride Channel TMEM16A. Nat. Commun. 2021, 12, 786. [Google Scholar] [CrossRef]
- Jia, Z.; Chen, J. Specific PIP2 Binding Promotes Calcium Activation of TMEM16A Chloride Channels. Commun. Biol. 2021, 4, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Yu, K.; Hartzell, H.C.; Tajkhorshid, E. Lipids and Ions Traverse the Membrane by the Same Physical Pathway in the NhTMEM16 Scramblase. eLife 2017, 6, e28671. [Google Scholar] [CrossRef] [PubMed]
- Malvezzi, M.; Chalat, M.; Janjusevic, R.; Picollo, A.; Terashima, H.; Menon, A.K.; Accardi, A. Ca2+-Dependent Phospholipid Scrambling by a Reconstituted TMEM16 Ion Channel. Nat. Commun. 2013, 4, 2367. [Google Scholar] [CrossRef]
- Qu, Z.; Hartzell, H.C. Anion Permeation in Ca2+-Activated Cl− Channels. J. Gen. Physiol. 2000, 116, 825–844. [Google Scholar] [CrossRef] [Green Version]
- Ni, Y.-L.; Kuan, A.-S.; Chen, T.-Y. Activation and Inhibition of TMEM16A Calcium-Activated Chloride Channels. PLoS ONE 2014, 9, e86734. [Google Scholar] [CrossRef] [PubMed]
- Grubb, S.; Poulsen, K.A.; Juul, C.A.; Kyed, T.; Klausen, T.K.; Larsen, E.H.; Hoffmann, E.K. TMEM16F (Anoctamin 6), an Anion Channel of Delayed Ca2+ Activation. J. Gen. Physiol. 2013, 141, 585–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, D.M.; Chen, L.S.; Yu, W.-P.; Chen, T.-Y. Comparison of Ion Transport Determinants between a TMEM16 Chloride Channel and Phospholipid Scramblase. J. Gen. Physiol. 2019, 151, 518–531. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, T.; Iehara, T.; Sato, K.; Fujii, T.; Sakai, H.; Okada, Y. TMEM16F Is a Component of a Ca2+-Activated Cl− Channel but Not a Volume-Sensitive Outwardly Rectifying Cl− Channel. Am. J. Physiol. Cell Physiol. 2013, 304, C748–C759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stabilini, S.; Menini, A.; Pifferi, S. Anion and Cation Permeability of the Mouse TMEM16F Calcium-Activated Channel. Int. J. Mol. Sci. 2021, 22, 8578. [Google Scholar] [CrossRef] [PubMed]
- Peters, C.J.; Yu, H.; Tien, J.; Jan, Y.N.; Li, M.; Jan, L.Y. Four Basic Residues Critical for the Ion Selectivity and Pore Blocker Sensitivity of TMEM16A Calcium-Activated Chloride Channels. Proc. Natl. Acad. Sci. USA 2015, 112, 3547–3552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aoun, J.; Hayashi, M.; Sheikh, I.A.; Sarkar, P.; Saha, T.; Ghosh, P.; Bhowmick, R.; Ghosh, D.; Chatterjee, T.; Chakrabarti, P.; et al. Anoctamin 6 Contributes to Cl− Secretion in Accessory Cholera Enterotoxin (Ace)-Stimulated Diarrhea: An Essential Role for Phosphatidylinositol 4,5-Bisphosphate (PIP2) Signaling in Cholera. J. Biol. Chem. 2016, 291, 26816–26836. [Google Scholar] [CrossRef] [Green Version]
- Henkel, B.; Drose, D.R.; Ackels, T.; Oberland, S.; Spehr, M.; Neuhaus, E.M. Co-Expression of Anoctamins in Cilia of Olfactory Sensory Neurons. Chem. Senses 2015, 40, 73–87. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.J.; Jun, I.; Yoon, J.S.; Jung, J.; Kim, Y.K.; Kim, W.K.; Kim, B.J.; Song, J.; Kim, S.J.; Nam, J.H.; et al. Selective Serotonin Reuptake Inhibitors Facilitate ANO6 (TMEM16F) Current Activation and Phosphatidylserine Exposure. Pflugers Arch. 2015, 467, 2243–2256. [Google Scholar] [CrossRef]
- Muratori, C.; Pakhomov, A.G.; Gianulis, E.; Meads, J.; Casciola, M.; Mollica, P.A.; Pakhomova, O.N. Activation of the Phospholipid Scramblase TMEM16F by Nanosecond Pulsed Electric Fields (NsPEF) Facilitates Its Diverse Cytophysiological Effects. J. Biol. Chem. 2017, 292, 19381–19391. [Google Scholar] [CrossRef] [Green Version]
- Ousingsawat, J.; Wanitchakool, P.; Kmit, A.; Romao, A.M.; Jantarajit, W.; Schreiber, R.; Kunzelmann, K. Anoctamin 6 Mediates Effects Essential for Innate Immunity Downstream of P2X 7 Receptors in Macrophages. Nat. Commun. 2015, 6, 6245. [Google Scholar] [CrossRef] [PubMed]
- Simões, F.; Ousingsawat, J.; Wanitchakool, P.; Fonseca, A.; Cabrita, I.; Benedetto, R.; Schreiber, R.; Kunzelmann, K. CFTR Supports Cell Death through ROS-Dependent Activation of TMEM16F (Anoctamin 6). Pflüg. Arch. Eur. J. Physiol. 2018, 470, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Taylor, K.A.; Mahaut-Smith, M.P. A Major Interspecies Difference in the Ionic Selectivity of Megakaryocyte Ca2+-Activated Channels Sensitive to the TMEM16F Inhibitor CaCCinh-A01. Platelets 2019, 30, 962–966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bricogne, C.; Fine, M.; Pereira, P.M.; Sung, J.; Tijani, M.; Wang, Y.; Henriques, R.; Collins, M.K.; Hilgemann, D.W. TMEM16F Activation by Ca2+ Triggers Plasma Membrane Expansion and Directs PD-1 Trafficking. Sci. Rep. 2019, 9, 619. [Google Scholar] [CrossRef]
- Pomorski, T.; Menon, A.K. Lipid Flippases and Their Biological Functions. Cell. Mol. Life Sci. CMLS 2006, 63, 2908–2921. [Google Scholar] [CrossRef]
- Falzone, M.E.; Malvezzi, M.; Lee, B.-C.; Accardi, A. Known Structures and Unknown Mechanisms of TMEM16 Scramblases and Channels. J. Gen. Physiol. 2018, 150, 933–947. [Google Scholar] [CrossRef] [Green Version]
- Kalienkova, V.; Clerico Mosina, V.; Paulino, C. The Groovy TMEM16 Family: Molecular Mechanisms of Lipid Scrambling and Ion Conduction. J. Mol. Biol. 2021, 433, 166941. [Google Scholar] [CrossRef]
- Whitlock, J.M.; Hartzell, H.C. A Pore Idea: The Ion Conduction Pathway of TMEM16/ANO Proteins Is Composed Partly of Lipid. Pflüg. Arch. Eur. J. Physiol. 2016, 468, 455–473. [Google Scholar] [CrossRef] [Green Version]
- Kostritskii, A.Y.; Machtens, J.-P. Molecular Mechanisms of Ion Conduction and Ion Selectivity in TMEM16 Lipid Scramblases. Nat. Commun. 2021, 12, 2826. [Google Scholar] [CrossRef]
- Hartzell, C.; Putzier, I.; Arreola, J. Calcium-Activated Chloride Channels. Annu. Rev. Physiol. 2005, 67, 719–758. [Google Scholar] [CrossRef] [Green Version]
- Angermann, J.E.; Sanguinetti, A.R.; Kenyon, J.L.; Leblanc, N.; Greenwood, I.A. Mechanism of the Inhibition of Ca2+-Activated Cl− Currents by Phosphorylation in Pulmonary Arterial Smooth Muscle Cells. J. Gen. Physiol. 2006, 128, 73–87. [Google Scholar] [CrossRef] [Green Version]
- Arreola, J.; Melvin, J.E.; Begenisich, T. Activation of Calcium-Dependent Chloride Channels in Rat Parotid Acinar Cells. J. Gen. Physiol. 1996, 108, 35–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuruma, A.; Hartzell, H.C. Bimodal Control of a Ca2+-Activated Cl− Channel by Different Ca2+ Signals. J. Gen. Physiol. 2000, 115, 59–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, W.; Han, T.W.; Nassar, L.M.; Zubia, M.; Jan, Y.N.; Jan, L.Y. Phosphatidylinositol-(4,5)-Bisphosphate Regulates Calcium Gating of Small-Conductance Cation Channel TMEM16F. Proc. Natl. Acad. Sci. USA 2018, 115, E1667–E1674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Contreras-Vite, J.A.; Cruz-Rangel, S.; De Jesús-Pérez, J.J.; Figueroa, I.A.A.; Rodríguez-Menchaca, A.A.; Pérez-Cornejo, P.; Hartzell, H.C.; Arreola, J. Revealing the Activation Pathway for TMEM16A Chloride Channels from Macroscopic Currents and Kinetic Models. Pflüg. Arch. Eur. J. Physiol. 2016, 468, 1241–1257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Jesús-Pérez, J.J.; Cruz-Rangel, S.; Espino-Saldaña, Á.E.; Martínez-Torres, A.; Qu, Z.; Hartzell, H.C.; Corral-Fernandez, N.E.; Pérez-Cornejo, P.; Arreola, J. Phosphatidylinositol 4,5-Bisphosphate, Cholesterol, and Fatty Acids Modulate the Calcium-Activated Chloride Channel TMEM16A (ANO1). Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2018, 1863, 299–312. [Google Scholar] [CrossRef]
- Le, S.C.; Jia, Z.; Chen, J.; Yang, H. Molecular Basis of PIP2-Dependent Regulation of the Ca2+-Activated Chloride Channel TMEM16A. Nat. Commun. 2019, 10, 3769. [Google Scholar] [CrossRef]
- Tembo, M.; Carlson, A.E. PIP2 and Ca2+ Are Both Required to Open TMEM16a Channels in Xenopus Laevis Oocytes. Biophys. J. 2018, 114, 610a. [Google Scholar] [CrossRef]
- Lin, H.; Roh, J.; Woo, J.H.; Kim, S.J.; Nam, J.H. TMEM16F/ANO6, a Ca2+-Activated Anion Channel, Is Negatively Regulated by the Actin Cytoskeleton and Intracellular MgATP. Biochem. Biophys. Res. Commun. 2018, 503, 2348–2354. [Google Scholar] [CrossRef]
- Ye, W.; Han, T.W.; He, M.; Jan, Y.N.; Jan, L.Y. Dynamic Change of Electrostatic Field in TMEM16F Permeation Pathway Shifts Its Ion Selectivity. eLife 2019, 8, e45187. [Google Scholar] [CrossRef]
- Yuan, H.; Gao, C.; Chen, Y.; Jia, M.; Geng, J.; Zhang, H.; Zhan, Y.; Boland, L.M.; An, H. Divalent Cations Modulate TMEM16A Calcium-Activated Chloride Channels by a Common Mechanism. J. Membr. Biol. 2013, 246, 893–902. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.M.; Kwon, H.C.; Chen, T.-Y. Divalent Cation Modulation of Ion Permeation in TMEM16 Proteins. Int. J. Mol. Sci. 2021, 22, 2209. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.M.; Chen, L.S.; Jeng, G.; Yu, W.-P.; Chen, T.-Y. Cobalt Ion Interaction with TMEM16A Calcium-Activated Chloride Channel: Inhibition and Potentiation. PLoS ONE 2020, 15, e0231812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, K.; Zhu, J.; Qu, Z.; Cui, Y.-Y.; Hartzell, H.C. Activation of the Ano1 (TMEM16A) Chloride Channel by Calcium Is Not Mediated by Calmodulin. J. Gen. Physiol. 2014, 143, 253–267. [Google Scholar] [CrossRef] [Green Version]
- Vocke, K.; Dauner, K.; Hahn, A.; Ulbrich, A.; Broecker, J.; Keller, S.; Frings, S.; Möhrlen, F. Calmodulin-Dependent Activation and Inactivation of Anoctamin Calcium-Gated Chloride Channels. J. Gen. Physiol. 2013, 142, 381–404. [Google Scholar] [CrossRef] [Green Version]
- Perez-Cornejo, P.; Gokhale, A.; Duran, C.; Cui, Y.; Xiao, Q.; Hartzell, H.C.; Faundez, V. Anoctamin 1 (Tmem16A) Ca2+-Activated Chloride Channel Stoichiometrically Interacts with an Ezrin–Radixin–Moesin Network. Proc. Natl. Acad. Sci. USA 2012, 109, 10376–10381. [Google Scholar] [CrossRef] [Green Version]
- Terashima, H.; Picollo, A.; Accardi, A. Purified TMEM16A Is Sufficient to Form Ca2+-Activated Cl− Channels. Proc. Natl. Acad. Sci. USA 2013, 110, 19354–19359. [Google Scholar] [CrossRef] [Green Version]
- Chao, S.H.; Suzuki, Y.; Zysk, J.R.; Cheung, W.Y. Activation of Calmodulin by Various Metal Cations as a Function of Ionic Radius. Mol. Pharmacol. 1984, 26, 75–82. [Google Scholar]
- Tian, Y.; Kongsuphol, P.; Hug, M.; Ousingsawat, J.; Witzgall, R.; Schreiber, R.; Kunzelmann, K. Calmodulin-Dependent Activation of the Epithelial Calcium-Dependent Chloride Channel TMEM16A. FASEB J. 2011, 25, 1058–1068. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Yang, H.; Zheng, L.-Y.; Zhang, Z.; Tang, Y.-B.; Wang, G.-L.; Du, Y.-H.; Lv, X.-F.; Liu, J.; Zhou, J.-G.; et al. Downregulation of TMEM16A Calcium-Activated Chloride Channel Contributes to Cerebrovascular Remodeling during Hypertension by Promoting Basilar Smooth Muscle Cell Proliferation. Circulation 2012, 125, 697–707. [Google Scholar] [CrossRef] [Green Version]
- Jung, J.; Nam, J.H.; Park, H.W.; Oh, U.; Yoon, J.-H.; Lee, M.G. Dynamic Modulation of ANO1/TMEM16A HCO3− Permeability by Ca2+/Calmodulin. Proc. Natl. Acad. Sci. USA 2012, 110, 360–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenwood, I.A.; Ledoux, J.; Leblanc, N. Differential Regulation of Ca2+-Activated Cl− Currents in Rabbit Arterial and Portal Vein Smooth Muscle Cells by Ca2+-Calmodulin-Dependent Kinase. J. Physiol. 2001, 534, 395–408. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-X.; Lv, X.-F.; Yuan, F.; Li, X.-Y.; Ma, M.-M.; Liu, C.-Z.; Zhou, J.-G.; Wang, G.-L.; Guan, Y.-Y. Ca2+/Calmodulin-Dependent Protein Kinase II γ-Dependent Serine727 Phosphorylation Is Required for TMEM16A Ca2+-Activated Cl− Channel Regulation in Cerebrovascular Cells. Circ. J. Off. J. Jpn. Circ. Soc. 2018, 82, 903–913. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.X.; Kotlikoff, M.I. Inactivation of Calcium-Activated Chloride Channels in Smooth Muscle by Calcium/Calmodulin-Dependent Protein Kinase. Proc. Natl. Acad. Sci. USA 1997, 94, 14918–14923. [Google Scholar] [CrossRef] [Green Version]
- Ayon, R.J.; Hawn, M.B.; Aoun, J.; Wiwchar, M.; Forrest, A.S.; Cunningham, F.; Singer, C.A.; Valencik, M.L.; Greenwood, I.A.; Leblanc, N. Molecular Mechanism of TMEM16A Regulation: Role of CaMKII and PP1/PP2A. Am. J. Physiol.-Cell Physiol. 2019, 59, 418. [Google Scholar] [CrossRef]
- Wiwchar, M.; Ayon, R.; Greenwood, I.A.; Leblanc, N. Phosphorylation Alters the Pharmacology of Ca2+-Activated Cl− Channels in Rabbit Pulmonary Arterial Smooth Muscle Cells. Br. J. Pharmacol. 2009, 158, 1356–1365. [Google Scholar] [CrossRef] [Green Version]
- Sim, K.M.; Lee, Y.-S.; Kim, H.J.; Cho, C.-H.; Yi, G.-S.; Park, M.-J.; Hwang, E.M.; Park, J.-Y. Suppression of CaMKIIβ Inhibits ANO1-Mediated Glioblastoma Progression. Cells 2020, 9, 1079. [Google Scholar] [CrossRef]
- Chun, H.; Cho, H.; Choi, J.; Lee, J.; Kim, S.M.; Kim, H.; Oh, U. Protons Inhibit Anoctamin 1 by Competing with Calcium. Cell Calcium 2015, 58, 431–441. [Google Scholar] [CrossRef]
- Liang, P.; Yang, H. Molecular Underpinning of Intracellular PH Regulation on TMEM16F. J. Gen. Physiol. 2021, 153, 2704. [Google Scholar] [CrossRef]
- Han, Y.; Shewan, A.M.; Thorn, P. HCO3− Transport through Anoctamin/Transmembrane Protein ANO1/TMEM16A in Pancreatic Acinar Cells Regulates Luminal PH. J. Biol. Chem. 2016, 291, 20345–20352. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.G.; Ohana, E.; Park, H.W.; Yang, D.; Muallem, S. Molecular Mechanism of Pancreatic and Salivary Gland Fluid and HCO3− Secretion. Physiol. Rev. 2012, 92, 39–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segura-Covarrubias, G.; Aréchiga-Figueroa, I.A.; De Jesús-Pérez, J.J.; Sánchez-Solano, A.; Pérez-Cornejo, P.; Arreola, J. Voltage-Dependent Protonation of the Calcium Pocket Enable Activation of the Calcium-Activated Chloride Channel Anoctamin-1 (TMEM16A). Sci. Rep. 2020, 10, 6644. [Google Scholar] [CrossRef] [PubMed]
- Webb, B.A.; Chimenti, M.; Jacobson, M.P.; Barber, D.L. Dysregulated PH: A Perfect Storm for Cancer Progression. Nat. Rev. Cancer 2011, 11, 671–677. [Google Scholar] [CrossRef] [PubMed]
- Wanitchakool, P.; Wolf, L.; Koehl, G.E.; Sirianant, L.; Schreiber, R.; Kulkarni, S.; Duvvuri, U.; Kunzelmann, K. Role of Anoctamins in Cancer and Apoptosis. Philos. Trans. R. Soc. B Biol. Sci. 2014, 369, 20130096. [Google Scholar] [CrossRef]
- Cruz-Rangel, S.; Jesús-Pérez, J.J.D.; Aréchiga-Figueroa, I.A.; Rodríguez-Menchaca, A.A.; Pérez-Cornejo, P.; Hartzell, H.C.; Arreola, J. Extracellular Protons Enable Activation of the Calcium-Dependent Chloride Channel TMEM16A. J. Physiol. 2017, 595, 1515–1531. [Google Scholar] [CrossRef]
- Chiche, J.; Brahimi-Horn, M.C.; Pouysségur, J. Tumour Hypoxia Induces a Metabolic Shift Causing Acidosis: A Common Feature in Cancer. J. Cell. Mol. Med. 2010, 14, 771–794. [Google Scholar] [CrossRef] [Green Version]
- Chesler, M. Regulation and Modulation of PH in the Brain. Physiol. Rev. 2003, 83, 1183–1221. [Google Scholar] [CrossRef]
- Dmitriev, A.V.; Mangel, S.C. Circadian Clock Regulation of PH in the Rabbit Retina. J. Neurosci. Off. J. Soc. Neurosci. 2001, 21, 2897–2902. [Google Scholar] [CrossRef]
- Dmitriev, A.V.; Mangel, S.C. Retinal PH Reflects Retinal Energy Metabolism in the Day and Night. J. Neurophysiol. 2004, 91, 2404–2412. [Google Scholar] [CrossRef]
- Newman, E.A. Acid Efflux from Retinal Glial Cells Generated by Sodium Bicarbonate Cotransport. J. Neurosci. Off. J. Soc. Neurosci. 1996, 16, 159–168. [Google Scholar] [CrossRef]
- Chatton, J.Y.; Spring, K.R. Acidic PH of the Lateral Intercellular Spaces of MDCK Cells Cultured on Permeable Supports. J. Membr. Biol. 1994, 140, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Chu, S.; Montrose, M.H. Extracellular PH Regulation in Microdomains of Colonic Crypts: Effects of Short-Chain Fatty Acids. Proc. Natl. Acad. Sci. USA 1995, 92, 3303–3307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, S.; Tanaka, S.; Kaunitz, J.D.; Montrose, M.H. Dynamic Regulation of Gastric Surface PH by Luminal PH. J. Clin. Investig. 1999, 103, 605–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, P.J.; Chatton, J.Y.; Tran, P.H.; Bungay, P.M.; Spring, K.R. PH, Morphology, and Diffusion in Lateral Intercellular Spaces of Epithelial Cell Monolayers. Am. J. Physiol. 1994, 266, C73–C80. [Google Scholar] [CrossRef] [PubMed]
- Maouyo, D.; Chu, S.; Montrose, M.H. PH Heterogeneity at Intracellular and Extracellular Plasma Membrane Sites in HT29-C1 Cell Monolayers. Am. J. Physiol. Cell Physiol. 2000, 278, C973–C981. [Google Scholar] [CrossRef] [PubMed]
- Rechkemmer, G.; Wahl, M.; Kuschinsky, W.; von Engelhardt, W. PH-Microclimate at the Luminal Surface of the Intestinal Mucosa of Guinea Pig and Rat. Pflugers Arch. 1986, 407, 33–40. [Google Scholar] [CrossRef]
- Sandoval, M.; Burgos, J.; Sepúlveda, F.V.; Cid, L.P. Extracellular PH in Restricted Domains as a Gating Signal for Ion Channels Involved in Transepithelial Transport. Biol. Pharm. Bull. 2011, 34, 803–809. [Google Scholar] [CrossRef] [Green Version]
- Sondo, E.; Caci, E.; Galietta, L.J.V. The TMEM16A Chloride Channel as an Alternative Therapeutic Target in Cystic Fibrosis. Int. J. Biochem. Cell Biol. 2014, 52, 73–76. [Google Scholar] [CrossRef]
- Martins, J.R.; Faria, D.; Kongsuphol, P.; Reisch, B.; Schreiber, R.; Kunzelmann, K. Anoctamin 6 Is an Essential Component of the Outwardly Rectifying Chloride Channel. Proc. Natl. Acad. Sci. USA 2011, 108, 18168–18172. [Google Scholar] [CrossRef] [Green Version]
- Benedetto, R.; Ousingsawat, J.; Wanitchakool, P.; Zhang, Y.; Holtzman, M.J.; Amaral, M.; Rock, J.R.; Schreiber, R.; Kunzelmann, K. Epithelial Chloride Transport by CFTR Requires TMEM16A. Sci. Rep. 2017, 7, 12397. [Google Scholar] [CrossRef] [Green Version]
- Genovese, M.; Borrelli, A.; Venturini, A.; Guidone, D.; Caci, E.; Viscido, G.; Gambardella, G.; Bernardo, D.D.; Scudieri, P.; Galietta, L.J.V. TRPV4 and Purinergic Receptor Signalling Pathways Are Separately Linked in Airway Epithelia to CFTR and TMEM16A Chloride Channels. J. Physiol. 2019, 597, 5859–5878. [Google Scholar] [CrossRef] [PubMed]
- Hahn, A.; Faulhaber, J.; Srisawang, L.; Stortz, A.; Salomon, J.J.; Mall, M.A.; Frings, S.; Möhrlen, F. Cellular Distribution and Function of Ion Channels Involved in Transport Processes in Rat Tracheal Epithelium. Physiol. Rep. 2017, 5, e13290. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Zhang, H.; Wu, M.; Yang, H.; Kudo, M.; Peters, C.J.; Woodruff, P.G.; Solberg, O.D.; Donne, M.L.; Huang, X.; et al. Calcium-Activated Chloride Channel TMEM16A Modulates Mucin Secretion and Airway Smooth Muscle Contraction. Proc. Natl. Acad. Sci. USA 2012, 109, 16354–16359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondo, M.; Tsuji, M.; Hara, K.; Arimura, K.; Yagi, O.; Tagaya, E.; Takeyama, K.; Tamaoki, J. Chloride Ion Transport and Overexpression of TMEM16A in a Guinea-Pig Asthma Model. Clin. Exp. Allergy 2017, 47, 795–804. [Google Scholar] [CrossRef] [PubMed]
- Centeio, R.; Cabrita, I.; Benedetto, R.; Talbi, K.; Ousingsawat, J.; Schreiber, R.; Sullivan, J.K.; Kunzelmann, K. Pharmacological Inhibition and Activation of the Ca2+ Activated Cl− Channel TMEM16A. Int. J. Mol. Sci. 2020, 21, 2557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guarascio, D.M.; Gonzalez-Velandia, K.Y.; Hernandez-Clavijo, A.; Menini, A.; Pifferi, S. Functional Expression of TMEM16A in Taste Bud Cells. J. Physiol. 2021, 599, 3697–3714. [Google Scholar] [CrossRef] [PubMed]
- Henriques, T.; Agostinelli, E.; Hernandez-Clavijo, A.; Maurya, D.K.; Rock, J.R.; Harfe, B.D.; Menini, A.; Pifferi, S. TMEM16A Calcium-Activated Chloride Currents in Supporting Cells of the Mouse Olfactory Epithelium. J. Gen. Physiol. 2019, 151, 954–966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, W.; Suh, B.-C. Differential Regulation of Ca2+-Activated Cl− Channel TMEM16A Splice Variants by Membrane PI(4,5)P2. Int. J. Mol. Sci. 2021, 22, 4088. [Google Scholar] [CrossRef]
- Tembo, M.; Wozniak, K.L.; Bainbridge, R.E.; Carlson, A.E. Phosphatidylinositol 4,5-Bisphosphate (PIP2) and Ca2+ Are Both Required to Open the Cl− Channel TMEM16A. J. Biol. Chem. 2019, 294, 12556–12564. [Google Scholar] [CrossRef]
- Kloner, R.A.; King, K.S.; Harrington, M.G. No-Reflow Phenomenon in the Heart and Brain. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H550–H562. [Google Scholar] [CrossRef]
- Bulley, S.; Jaggar, J.H. Cl− Channels in Smooth Muscle Cells. Pflugers Arch. 2014, 466, 861–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, M.-M.; Lou, J.; Song, B.-L.; Gong, Y.-F.; Li, Y.-C.; Yu, C.-J.; Wang, Q.-S.; Ma, T.-X.; Ma, K.; Hartzell, H.C.; et al. Hypoxia Augments the Calcium-Activated Chloride Current Carried by Anoctamin-1 in Cardiac Vascular Endothelial Cells of Neonatal Mice. Br. J. Pharmacol. 2014, 171, 3680–3692. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Xia, Y.; Paudel, O.; Yang, X.-R.; Sham, J.S.K. Chronic Hypoxia-Induced Upregulation of Ca2+-Activated Cl− Channel in Pulmonary Arterial Myocytes: A Mechanism Contributing to Enhanced Vasoreactivity. J. Physiol. 2012, 590, 3507–3521. [Google Scholar] [CrossRef] [PubMed]
- Papp, R.; Nagaraj, C.; Zabini, D.; Nagy, B.M.; Lengyel, M.; Skofic Maurer, D.; Sharma, N.; Egemnazarov, B.; Kovacs, G.; Kwapiszewska, G.; et al. Targeting TMEM16A to Reverse Vasoconstriction and Remodelling in Idiopathic Pulmonary Arterial Hypertension. Eur. Respir. J. 2019, 53, 1800965. [Google Scholar] [CrossRef] [PubMed]
- Blount, A.; Zhang, S.; Chestnut, M.; Hixon, B.; Skinner, D.; Sorscher, E.J.; Woodworth, B.A. Transepithelial Ion Transport Is Suppressed in Hypoxic Sinonasal Epithelium. The Laryngoscope 2011, 121, 1929–1934. [Google Scholar] [CrossRef] [Green Version]
- Ousingsawat, J.; Wanitchakool, P.; Schreiber, R.; Kunzelmann, K. Contribution of TMEM16F to Pyroptotic Cell Death. Cell Death Dis. 2018, 9, 1–11. [Google Scholar] [CrossRef]
- Schreiber, R.; Buchholz, B.; Kraus, A.; Schley, G.; Scholz, J.; Ousingsawat, J.; Kunzelmann, K. Lipid Peroxidation Drives Renal Cyst Growth In Vitro through Activation of TMEM16A. J. Am. Soc. Nephrol. 2019, 30, 228–242. [Google Scholar] [CrossRef] [Green Version]
- Schreiber, R.; Ousingsawat, J.; Wanitchakool, P.; Sirianant, L.; Benedetto, R.; Reiss, K.; Kunzelmann, K. Regulation of TMEM16A/ANO1 and TMEM16F/ANO6 Ion Currents and Phospholipid Scrambling by Ca2+ and Plasma Membrane Lipid. J. Physiol. 2018, 596, 217–229. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.; Oh, U. Anoctamin 1 Mediates Thermal Pain as a Heat Sensor. Curr. Neuropharmacol. 2013, 11, 641–651. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.; Yang, Y.D.; Lee, J.; Lee, B.; Kim, T.; Jang, Y.; Back, S.K.; Na, H.S.; Harfe, B.D.; Wang, F.; et al. The Calcium-Activated Chloride Channel Anoctamin 1 Acts as a Heat Sensor in Nociceptive Neurons. Nat. Neurosci. 2012, 15, 1015–1021. [Google Scholar] [CrossRef]
- Lin, H.; Jun, I.; Woo, J.H.; Lee, M.G.; Kim, S.J.; Nam, J.H. Temperature-Dependent Increase in the Calcium Sensitivity and Acceleration of Activation of ANO6 Chloride Channel Variants. Sci. Rep. 2019, 9, 6706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brauchi, S.; Orio, P.; Latorre, R. Clues to Understanding Cold Sensation: Thermodynamics and Electrophysiological Analysis of the Cold Receptor TRPM8. Proc. Natl. Acad. Sci. USA 2004, 101, 15494–15499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latorre, R.; Brauchi, S.; Orta, G.; Zaelzer, C.; Vargas, G. ThermoTRP Channels as Modular Proteins with Allosteric Gating. Cell Calcium 2007, 42, 427–438. [Google Scholar] [CrossRef] [PubMed]
- Voets, T. Quantifying and modeling the temperature-dependent gating of TRP channels. In Reviews of Physiology, Biochemistry and Pharmacology; Nilius, B., Amara, S.G., Gudermann, T., Jahn, R., Lill, R., Offermanns, S., Petersen, O.H., Eds.; Springer: Berlin/Heidelberg, Germany, 2012; Volume 162, pp. 91–119. ISBN 978-3-642-29256-9. [Google Scholar]
- Clapham, D.E.; Miller, C. A Thermodynamic Framework for Understanding Temperature Sensing by Transient Receptor Potential (TRP) Channels. Proc. Natl. Acad. Sci. USA 2011, 108, 19492–19497. [Google Scholar] [CrossRef] [Green Version]
- Lee, B.; Cho, H.; Jung, J.; Yang, Y.D.; Yang, D.-J.; Oh, U. Anoctamin 1 Contributes to Inflammatory and Nerve-Injury Induced Hypersensitivity. Mol. Pain 2014, 10, 5. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Linley, J.E.; Du, X.; Zhang, X.; Ooi, L.; Zhang, H.; Gamper, N. The Acute Nociceptive Signals Induced by Bradykinin in Rat Sensory Neurons Are Mediated by Inhibition of M-Type K+ Channels and Activation of Ca2+-Activated Cl– Channels. J. Clin. Investig. 2010, 120, 1240–1252. [Google Scholar] [CrossRef] [Green Version]
- Jang, W.; Kim, J.Y.; Cui, S.; Jo, J.; Lee, B.-C.; Lee, Y.; Kwon, K.-S.; Park, C.-S.; Kim, C. The Anoctamin Family Channel Subdued Mediates Thermal Nociception in Drosophila. J. Biol. Chem. 2015, 290, 2521–2528. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, R.; Sakuragi, T.; Noji, H.; Nagata, S. Single-Molecule Analysis of Phospholipid Scrambling by TMEM16F. Proc. Natl. Acad. Sci. USA 2018, 115, 3066–3071. [Google Scholar] [CrossRef] [Green Version]
- Pritchard, H.A.T.; Leblanc, N.; Albert, A.P.; Greenwood, I.A. Inhibitory Role of Phosphatidylinositol 4,5-Bisphosphate on TMEM16A-Encoded Calcium-Activated Chloride Channels in Rat Pulmonary Artery. Br. J. Pharmacol. 2014, 171, 4311–4321. [Google Scholar] [CrossRef] [Green Version]
- Ta, C.M.; Acheson, K.E.; Rorsman, N.J.G.; Jongkind, R.C.; Tammaro, P. Contrasting Effects of Phosphatidylinositol 4,5-Bisphosphate on Cloned TMEM16A and TMEM16B Channels. Br. J. Pharmacol. 2017, 174, 2984–2999. [Google Scholar] [CrossRef] [Green Version]
- Yu, K.; Jiang, T.; Cui, Y.; Tajkhorshid, E.; Hartzell, H.C. A Network of Phosphatidylinositol 4,5-Bisphosphate Binding Sites Regulates Gating of the Ca2+-Activated Cl− Channel ANO1 (TMEM16A). Proc. Natl. Acad. Sci. USA 2019, 116, 19952–19962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arreola, J.; Hartzell, H.C. Wasted TMEM16A Channels Are Rescued by Phosphatidylinositol 4,5-Bisphosphate. Cell Calcium 2019, 84, 102103. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.-C.; Menon, A.K.; Accardi, A. The nhTMEM16 Scramblase Is Also a Nonselective Ion Channel. Biophys. J. 2016, 111, 1919–1924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sones, W.R.; Davis, A.J.; Leblanc, N.; Greenwood, I.A. Cholesterol Depletion Alters Amplitude and Pharmacology of Vascular Calcium-Activated Chloride Channels. Cardiovasc. Res. 2010, 87, 476–484. [Google Scholar] [CrossRef] [Green Version]
- Elinder, F.; Liin, S. Actions and Mechanisms of Polyunsaturated Fatty Acids on Voltage-Gated Ion Channels. Front. Physiol. 2017, 8, 43. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Yan, X.; Li, R.; Zhang, A.; Niu, Z.; Cai, Z.; Duan, W.; Li, X.; Zhang, H. Increased TMEM16A Involved in Alveolar Fluid Clearance After Lipopolysaccharide Stimulation. Inflammation 2016, 39, 881–890. [Google Scholar] [CrossRef]
- Zhang, A.; Yan, X.; Li, H.; Gu, Z.; Zhang, P.; Zhang, H.; Li, Y.; Yu, H. TMEM16A Protein Attenuates Lipopolysaccharide-Mediated Inflammatory Response of Human Lung Epithelial Cell Line A549. Exp. Lung Res. 2014, 40, 237–250. [Google Scholar] [CrossRef]
- Sui, J.; Zhang, C.; Fang, X.; Wang, J.; Li, Y.; Wang, J.; Wang, L.; Dong, J.; Zhou, Z.; Li, C.; et al. Dual Role of Ca2+-Activated Cl− Channel Transmembrane Member 16A in Lipopolysaccharide-Induced Intestinal Epithelial Barrier Dysfunction in Vitro. Cell Death Dis. 2020, 11, 1–12. [Google Scholar] [CrossRef]
- Opal, S.M.; Scannon, P.J.; Vincent, J.-L.; White, M.; Carroll, S.F.; Palardy, J.E.; Parejo, N.A.; Pribble, J.P.; Lemke, J.H. Relationship between Plasma Levels of Lipopolysaccharide (LPS) and LPS-Binding Protein in Patients with Severe Sepsis and Septic Shock. J. Infect. Dis. 1999, 180, 1584–1589. [Google Scholar] [CrossRef] [Green Version]
- Sirianant, L.; Ousingsawat, J.; Wanitchakool, P.; Schreiber, R.; Kunzelmann, K. Cellular Volume Regulation by Anoctamin 6: Ca2+, Phospholipase A2 and Osmosensing. Pflüg. Arch. Eur. J. Physiol. 2016, 468, 335–349. [Google Scholar] [CrossRef]
- Yoo, J.; Cui, Q. Curvature Generation and Pressure Profile Modulation in Membrane by Lysolipids: Insights from Coarse-Grained Simulations. Biophys. J. 2009, 97, 2267–2276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, D.L.; Desiderio, D.M.; Miller, D.D.; Tolley, B.; Tigyi, G.J. Direct Quantitative Analysis of Lysophosphatidic Acid Molecular Species by Stable Isotope Dilution Electrospray Ionization Liquid Chromatography–Mass Spectrometry. Anal. Biochem. 2001, 292, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Sano, T.; Baker, D.; Virag, T.; Wada, A.; Yatomi, Y.; Kobayashi, T.; Igarashi, Y.; Tigyi, G. Multiple Mechanisms Linked to Platelet Activation Result in Lysophosphatidic Acid and Sphingosine 1-Phosphate Generation in Blood. J. Biol. Chem. 2002, 277, 21197–21206. [Google Scholar] [CrossRef] [Green Version]
- Bolen, A.L.; Naren, A.P.; Yarlagadda, S.; Beranova-Giorgianni, S.; Chen, L.; Norman, D.; Baker, D.L.; Rowland, M.M.; Best, M.D.; Sano, T.; et al. The Phospholipase A1 Activity of Lysophospholipase A-I Links Platelet Activation to LPA Production during Blood Coagulation. J. Lipid Res. 2011, 52, 958–970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eichholtz, T.; Jalink, K.; Fahrenfort, I.; Moolenaar, W.H. The Bioactive Phospholipid Lysophosphatidic Acid Is Released from Activated Platelets. Biochem. J. 1993, 291, 677–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Andrews, D.A.; Low, P.S. Lysophosphatidic Acid Opens a Ca2+ Channel in Human Erythrocytes. Blood 2000, 95, 2420–2425. [Google Scholar] [CrossRef] [PubMed]
- Baig, A.A.; Haining, E.J.; Geuss, E.; Beck, S.; Swieringa, F.; Wanitchakool, P.; Schuhmann, M.K.; Stegner, D.; Kunzelmann, K.; Kleinschnitz, C.; et al. TMEM16F-Mediated Platelet Membrane Phospholipid Scrambling Is Critical for Hemostasis and Thrombosis but Not Thromboinflammation in Mice—Brief Report. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 2152–2157. [Google Scholar] [CrossRef] [Green Version]
- Chung, S.-M.; Bae, O.-N.; Lim, K.-M.; Noh, J.-Y.; Lee, M.-Y.; Jung, Y.-S.; Chung, J.-H. Lysophosphatidic Acid Induces Thrombogenic Activity Through Phosphatidylserine Exposure and Procoagulant Microvesicle Generation in Human Erythrocytes. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 414–421. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Liu, G.; Chen, H.; Borst, O.; Gawaz, M.; Vortkamp, A.; Schreiber, R.; Kunzelmann, K.; Lang, F. Involvement of Ca2+ Activated Cl− Channel Ano6 in Platelet Activation and Apoptosis. Cell. Physiol. Biochem. 2015, 37, 1934–1944. [Google Scholar] [CrossRef]
- Öhlinger, T.; Müllner, E.W.; Fritz, M.; Sauer, T.; Werning, M.; Baron, D.M.; Salzer, U. Lysophosphatidic Acid-Induced pro-Thrombotic Phosphatidylserine Exposure and Ionophore-Induced Microvesiculation Is Mediated by the Scramblase TMEM16F in Erythrocytes. Blood Cells. Mol. Dis. 2020, 83, 102426. [Google Scholar] [CrossRef]
- Suzuki, T.; Suzuki, J.; Nagata, S. Functional Swapping between Transmembrane Proteins TMEM16A and TMEM16F. J. Biol. Chem. 2014, 289, 7438–7447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, T.; Le, S.C.; Zhang, Y.; Liang, P.; Yang, H. Evidence That Polyphenols Do Not Inhibit the Phospholipid Scramblase TMEM16F. J. Biol. Chem. 2020, 295, 12537–12544. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Dutta, A.; Kresge, C.; Bugde, A.; Feranchak, A.P. Bile Acids Stimulate Cholangiocyte Fluid Secretion by Activation of Transmembrane Member 16A Cl− Channels. Hepatology 2018, 68, 187–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunningham, S.A.; Awayda, M.S.; Bubien, J.K.; Ismailov, I.I.; Arrate, M.P.; Berdiev, B.K.; Benos, D.J.; Fuller, C.M. Cloning of an Epithelial Chloride Channel from Bovine Trachea. J. Biol. Chem. 1996, 271, 7244. [Google Scholar] [CrossRef]
- Yurtsever, Z.; Sala-Rabanal, M.; Randolph, D.T.; Scheaffer, S.M.; Roswit, W.T.; Alevy, Y.G.; Patel, A.C.; Heier, R.F.; Romero, A.G.; Nichols, C.G.; et al. Self-Cleavage of Human CLCA1 Protein by a Novel Internal Metalloprotease Domain Controls Calcium-Activated Chloride Channel Activation. J. Biol. Chem. 2012, 287, 42138–42149. [Google Scholar] [CrossRef] [Green Version]
- Patel, A.C.; Brett, T.J.; Holtzman, M.J. The Role of CLCA Proteins in Inflammatory Airway Disease. Annu. Rev. Physiol. 2009, 71, 425–449. [Google Scholar] [CrossRef] [Green Version]
- Alevy, Y.G.; Patel, A.C.; Romero, A.G.; Patel, D.A.; Tucker, J.; Roswit, W.T.; Miller, C.A.; Heier, R.F.; Byers, D.E.; Brett, T.J.; et al. IL-13–Induced Airway Mucus Production Is Attenuated by MAPK13 Inhibition. J. Clin. Investig. 2012, 122, 4555–4568. [Google Scholar] [CrossRef] [Green Version]
- Britton, F.C.; Ohya, S.; Horowitz, B.; Greenwood, I.A. Comparison of the Properties of CLCA1 Generated Currents and ICl(Ca) in Murine Portal Vein Smooth Muscle Cells. J. Physiol. 2002, 539, 107–117. [Google Scholar] [CrossRef]
- Elble, R.C.; Ji, G.; Nehrke, K.; DeBiasio, J.; Kingsley, P.D.; Kotlikoff, M.I.; Pauli, B.U. Molecular and Functional Characterization of a Murine Calcium-Activated Chloride Channel Expressed in Smooth Muscle. J. Biol. Chem. 2002, 277, 18586–18591. [Google Scholar] [CrossRef] [Green Version]
- Gandhi, R.; Elble, R.C.; Gruber, A.D.; Schreur, K.D.; Ji, H.-L.; Fuller, C.M.; Pauli, B.U. Molecular and Functional Characterization of a Calcium-Sensitive Chloride Channel from Mouse Lung. J. Biol. Chem. 1998, 273, 32096–32101. [Google Scholar] [CrossRef] [Green Version]
- Greenwood, I.A.; Miller, L.J.; Ohya, S.; Horowitz, B. The Large Conductance Potassium Channel β-Subunit Can Interact with and Modulate the Functional Properties of a Calcium-Activated Chloride Channel, CLCA1. J. Biol. Chem. 2002, 277, 22119–22122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sala-Rabanal, M.; Yurtsever, Z.; Nichols, C.G.; Brett, T.J. Secreted CLCA1 Modulates TMEM16A to Activate Ca2+-Dependent Chloride Currents in Human Cells. eLife 2015, 4, e05875. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Ramena, G.; Yin, Y.; Premkumar, L.; Elble, R.C. CLCA2 Is a Positive Regulator of Store-Operated Calcium Entry and TMEM16A. PLoS ONE 2018, 13, e0196512. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, Y.; Koyama, R.; Maruyama, R.; Hirano, T.; Tamura, M.; Sugisaka, J.; Suzuki, H.; Idogawa, M.; Shinomura, Y.; Tokino, T. CLCA2, a Target of the P53 Family, Negatively Regulates Cancer Cell Migration and Invasion. Cancer Biol. Ther. 2012, 13, 1512–1521. [Google Scholar] [CrossRef] [Green Version]
- Shinmura, K.; Igarashi, H.; Kato, H.; Kawanishi, Y.; Inoue, Y.; Nakamura, S.; Ogawa, H.; Yamashita, T.; Kawase, A.; Funai, K.; et al. CLCA2 as a Novel Immunohistochemical Marker for Differential Diagnosis of Squamous Cell Carcinoma from Adenocarcinoma of the Lung. Dis. Markers 2014, 2014, 619273. [Google Scholar] [CrossRef]
- Shiwarski, D.J.; Shao, C.; Bill, A.; Kim, J.; Xiao, D.; Bertrand, C.A.; Seethala, R.S.; Sano, D.; Myers, J.N.; Ha, P.; et al. To “Grow” or “Go”: TMEM16A Expression as a Switch between Tumor Growth and Metastasis in SCCHN. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 4673–4688. [Google Scholar] [CrossRef] [Green Version]
- Sala-Rabanal, M.; Yurtsever, Z.; Berry, K.N.; Nichols, C.G.; Brett, T.J. Modulation of TMEM16A Channel Activity by the von Willebrand Factor Type A (VWA) Domain of the Calcium-Activated Chloride Channel Regulator 1 (CLCA1). J. Biol. Chem. 2017, 292, 9164–9174. [Google Scholar] [CrossRef] [Green Version]
- Centeio, R.; Ousingsawat, J.; Schreiber, R.; Kunzelmann, K. CLCA1 Regulates Airway Mucus Production and Ion Secretion Through TMEM16A. Int. J. Mol. Sci. 2021, 22, 5133. [Google Scholar] [CrossRef]
- Barhanin, J.; Lesage, F.; Guillemare, E.; Fink, M.; Lazdunski, M.; Romey, G. K(V)LQT1 and LsK (MinK) Proteins Associate to Form the I(Ks) Cardiac Potassium Current. Nature 1996, 384, 78–80. [Google Scholar] [CrossRef]
- Sanguinetti, M.C.; Curran, M.E.; Zou, A.; Shen, J.; Spector, P.S.; Atkinson, D.L.; Keating, M.T. Coassembly of K(V)LQT1 and MinK (IsK) Proteins to Form Cardiac I(Ks) Potassium Channel. Nature 1996, 384, 80–83. [Google Scholar] [CrossRef] [PubMed]
- Ávalos Prado, P.; Häfner, S.; Comoglio, Y.; Wdziekonski, B.; Duranton, C.; Attali, B.; Barhanin, J.; Sandoz, G. KCNE1 Is an Auxiliary Subunit of Two Distinct Ion Channel Superfamilies. Cell 2021, 184, 534–544.e11. [Google Scholar] [CrossRef] [PubMed]
- McIntyre, J.C.; Joiner, A.M.; Zhang, L.; Iñiguez-Lluhí, J.; Martens, J.R. SUMOylation Regulates Ciliary Localization of Olfactory Signaling Proteins. J. Cell Sci. 2015, 128, 1934–1945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hood, J.L.; Emeson, R.B. Editing of Neurotransmitter Receptor and Ion Channel RNAs in the Nervous System. Curr. Top. Microbiol. Immunol. 2012, 353, 61–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, M.; Kleine-Weber, H.; Pöhlmann, S. A Multibasic Cleavage Site in the Spike Protein of SARS-CoV-2 Is Essential for Infection of Human Lung Cells. Mol. Cell 2020, 78, 779–784.e5. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- van den Eijnde, S.M.; van den Hoff, M.J.; Reutelingsperger, C.P.; van Heerde, W.L.; Henfling, M.E.; Vermeij-Keers, C.; Schutte, B.; Borgers, M.; Ramaekers, F.C. Transient Expression of Phosphatidylserine at Cell-Cell Contact Areas Is Required for Myotube Formation. J. Cell Sci. 2001, 114, 3631–3642. [Google Scholar] [CrossRef]
- Helming, L.; Gordon, S. Molecular Mediators of Macrophage Fusion. Trends Cell Biol. 2009, 19, 514–522. [Google Scholar] [CrossRef]
- Lyden, T.W.; Ng, A.K.; Rote, N.S. Modulation of Phosphatidylserine Epitope Expression by BeWo Cells during Forskolin Treatment. Placenta 1993, 14, 177–186. [Google Scholar] [CrossRef]
- Rival, C.M.; Xu, W.; Shankman, L.S.; Morioka, S.; Arandjelovic, S.; Lee, C.S.; Wheeler, K.M.; Smith, R.P.; Haney, L.B.; Isakson, B.E.; et al. Phosphatidylserine on Viable Sperm and Phagocytic Machinery in Oocytes Regulate Mammalian Fertilization. Nat. Commun. 2019, 10, 4456. [Google Scholar] [CrossRef] [Green Version]
- Verma, S.K.; Leikina, E.; Melikov, K.; Gebert, C.; Kram, V.; Young, M.F.; Uygur, B.; Chernomordik, L.V. Cell-Surface Phosphatidylserine Regulates Osteoclast Precursor Fusion. J. Biol. Chem. 2018, 293, 254–270. [Google Scholar] [CrossRef] [Green Version]
- Tao, K.; Tzou, P.L.; Nouhin, J.; Bonilla, H.; Jagannathan, P.; Shafer, R.W. SARS-CoV-2 Antiviral Therapy. Clin. Microbiol. Rev. 2021, 34, e00109-21. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Topological diagram of TMEM16x proteins, showing the position of the alternative spliced exons and a range of functional domains. The boxes (broken lines) highlight the TMs involved in the formation of the ion/lipid pathways in TMEM16x. The approximate position of each splicing variant is labelled using the nomenclature described in the main text. The diagrams also show the approximate position of a range of functional domains, including (1) the principal Ca2+-binding pocket, which encompasses conserved acidic residues located in TM6-8; (2) additional regulatory Ca2+-binding sites; (3) putative calmodulin (CaM)-binding sites; (4) putative phosphatidylinositol 4,5-bisphosphate (PIP2)-binding sites; and (5) the pH-sensitive domain on the extracellular loop between TM5 and TM6 of TMEM16A.
Figure 1.
Topological diagram of TMEM16x proteins, showing the position of the alternative spliced exons and a range of functional domains. The boxes (broken lines) highlight the TMs involved in the formation of the ion/lipid pathways in TMEM16x. The approximate position of each splicing variant is labelled using the nomenclature described in the main text. The diagrams also show the approximate position of a range of functional domains, including (1) the principal Ca2+-binding pocket, which encompasses conserved acidic residues located in TM6-8; (2) additional regulatory Ca2+-binding sites; (3) putative calmodulin (CaM)-binding sites; (4) putative phosphatidylinositol 4,5-bisphosphate (PIP2)-binding sites; and (5) the pH-sensitive domain on the extracellular loop between TM5 and TM6 of TMEM16A.

Figure 2.
Structural alignment of the experimentally determined Ca2+-bound structure of TMEM16A channel, and a model of the TMEM16A channel in the open state. (A) Left: Lateral view of the dimeric Ca2+-bound TMEM16A cryo-EM structure (PDB ID: 5OYB, blue). Right: TMEM16A open-state model (from [122], grey). The Ca2+ ions are shown in green. (B) Left: Single monomer of the Ca2+-bound TMEM16A cryo-EM structure (5OYB, blue), aligned with the open-state model (grey) [122]. The Ca2+ ions are shown in green. Right: Magnification of the gate-forming pore residues (V539 and I636, from [122]), in the Ca2+-bound TMEM16A cryo-EM structure (5OYB, blue), and of the TMEM16A open-state model (grey) [122], as indicated.
Figure 2.
Structural alignment of the experimentally determined Ca2+-bound structure of TMEM16A channel, and a model of the TMEM16A channel in the open state. (A) Left: Lateral view of the dimeric Ca2+-bound TMEM16A cryo-EM structure (PDB ID: 5OYB, blue). Right: TMEM16A open-state model (from [122], grey). The Ca2+ ions are shown in green. (B) Left: Single monomer of the Ca2+-bound TMEM16A cryo-EM structure (5OYB, blue), aligned with the open-state model (grey) [122]. The Ca2+ ions are shown in green. Right: Magnification of the gate-forming pore residues (V539 and I636, from [122]), in the Ca2+-bound TMEM16A cryo-EM structure (5OYB, blue), and of the TMEM16A open-state model (grey) [122], as indicated.

Figure 3.
TMEM16A close and open conformation diagram. Diagrammatic representation of a single TMEM16A channel pore and related gating mechanisms. Ca2+ binding induces opening of the steric gate in TM6 and attenuation of the electrostatic gate. The movement of the TM6 helix during gating is represented as a tilt of the inner portion of the pore and opening of the outer pore. The green and red backgrounds depict positive and negative electrostatic potentials in the pore, respectively.
Figure 3.
TMEM16A close and open conformation diagram. Diagrammatic representation of a single TMEM16A channel pore and related gating mechanisms. Ca2+ binding induces opening of the steric gate in TM6 and attenuation of the electrostatic gate. The movement of the TM6 helix during gating is represented as a tilt of the inner portion of the pore and opening of the outer pore. The green and red backgrounds depict positive and negative electrostatic potentials in the pore, respectively.

Figure 4.
Summary of the principal factors that control TMEM16x function. Schematic representation of various physiological modulators of TMEM16x activity. A TMEM16x protein in the plasma membrane of a generic cell is shown in blue. Factors that control TMEM16x activity are connected to the protein with red or blue lines to denote inhibitory or stimulatory influences, respectively. The black dashed arrows indicate the modulations for which an underlying mechanism has not yet been fully defined and/or conflicting evidence has been proposed.
Figure 4.
Summary of the principal factors that control TMEM16x function. Schematic representation of various physiological modulators of TMEM16x activity. A TMEM16x protein in the plasma membrane of a generic cell is shown in blue. Factors that control TMEM16x activity are connected to the protein with red or blue lines to denote inhibitory or stimulatory influences, respectively. The black dashed arrows indicate the modulations for which an underlying mechanism has not yet been fully defined and/or conflicting evidence has been proposed.


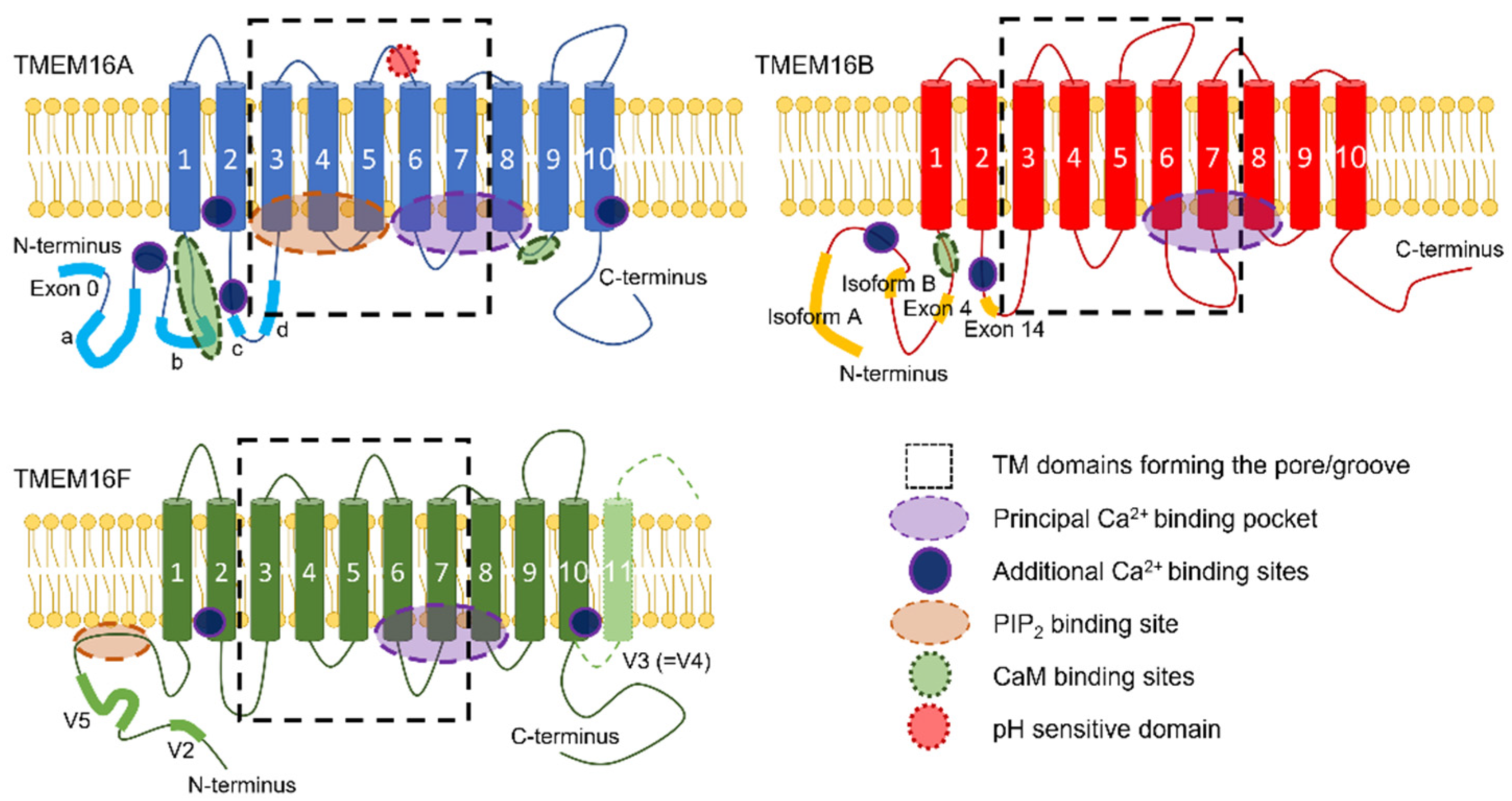{kind=link}
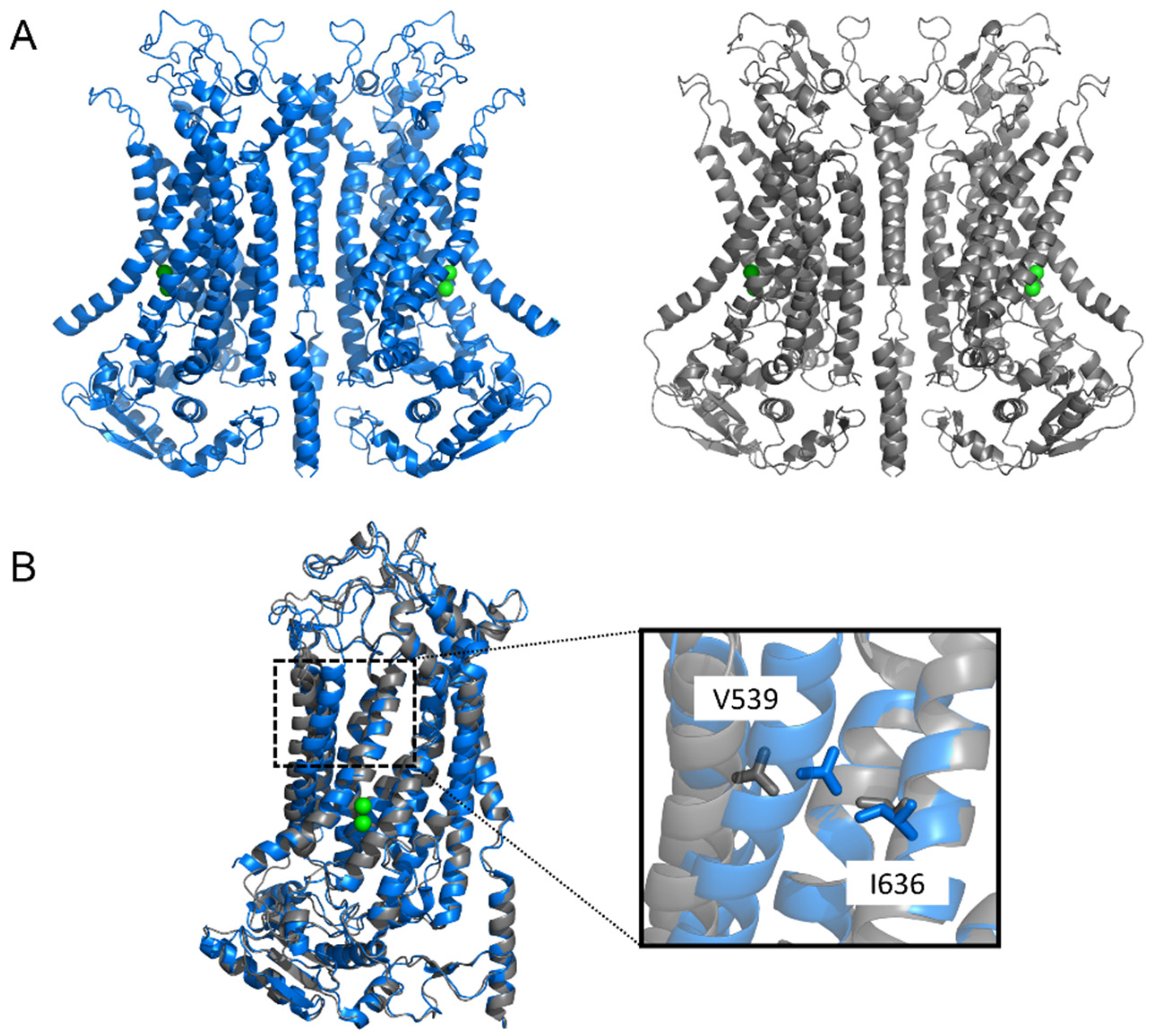{kind=link}
{kind=link}
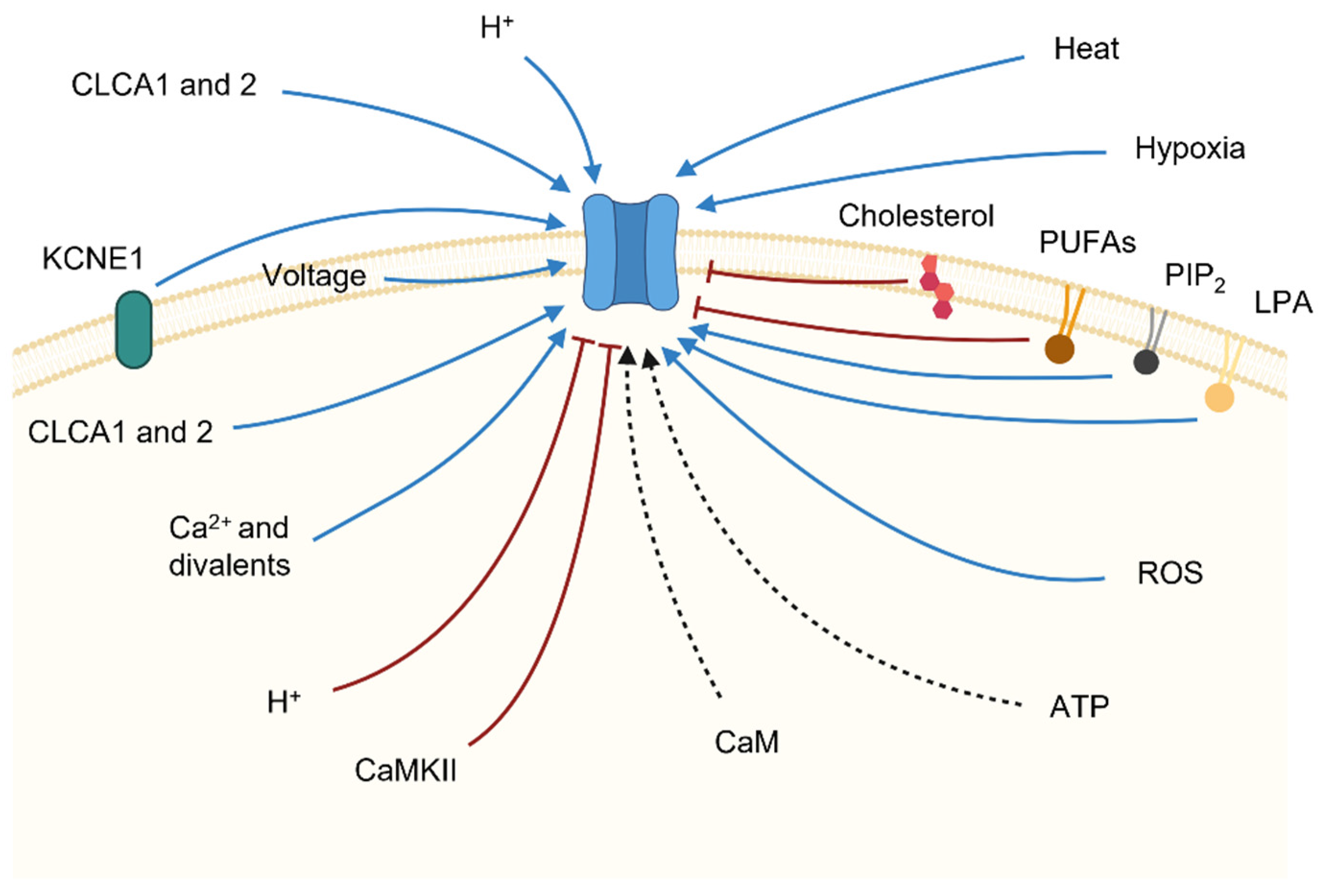{kind=link}
Table 1.
List of the key factors controlling TMEM16x activities.
| Modulator | TMEM16x | Effect |
|---|---|---|
| Ca2+, Sr2+, Ni2+, Ba2+ | CaCCs, A, B, F | Agonist |
| Co2+, Zn2+, Mg2+ | A | Antagonist |
| Mg2+, Gd3+ | A | Charge screening |
| Calmodulin | A, B | Modulation of ion channel properties (contrasting results) |
| CaMKII | CaCCs, A | Current inhibition |
| pHi | F | Inhibition of channel and scramblase activities |
| pHi | A | Current modulation |
| pHo | A | Current activation |
| ATP | A, F | Current activation/reduction of current rundown (contrasting effects) |
| Hypoxia | A | Current activation |
| ROS | A, F | Activation of scramblase and channel activities |
| Heat | A, F | Current activation; Modulation of scramblase activity |
| PIP2 | A, F | Activation of channel and scramblase activities |
| PIP2 | CaCCs, B | Current inhibition |
| Plasmalemmal lipids | afTMEM16, nhTMEM16 | Contrasting effects |
| Cholesterol | CaCCs, A | Current inhibition |
| Fatty acids | A | Current inhibition |
| LPS | A | Increase in current density |
| LPA | A, F | Activation of channel and scramblase activities |
| AA | F | Current inhibition |
| Bile acids (UDCA and TUDCA) | A | Current activation |
| CLACs | CaCCs, A | Increase in current density |
| KCNE1 | A | Increase in current density |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Agostinelli, E.; Tammaro, P. Polymodal Control of TMEM16x Channels and Scramblases. Int. J. Mol. Sci. 2022, 23, 1580. https://doi.org/10.3390/ijms23031580
AMA Style
Agostinelli E, Tammaro P. Polymodal Control of TMEM16x Channels and Scramblases. International Journal of Molecular Sciences. 2022; 23(3):1580. https://doi.org/10.3390/ijms23031580
Chicago/Turabian StyleAgostinelli, Emilio, and Paolo Tammaro. 2022. "Polymodal Control of TMEM16x Channels and Scramblases" International Journal of Molecular Sciences 23, no. 3: 1580. https://doi.org/10.3390/ijms23031580
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.