
Molecular Mechanisms of Kidney Injury and Repair

 , , , , , ,
, , , , , ,  and
and 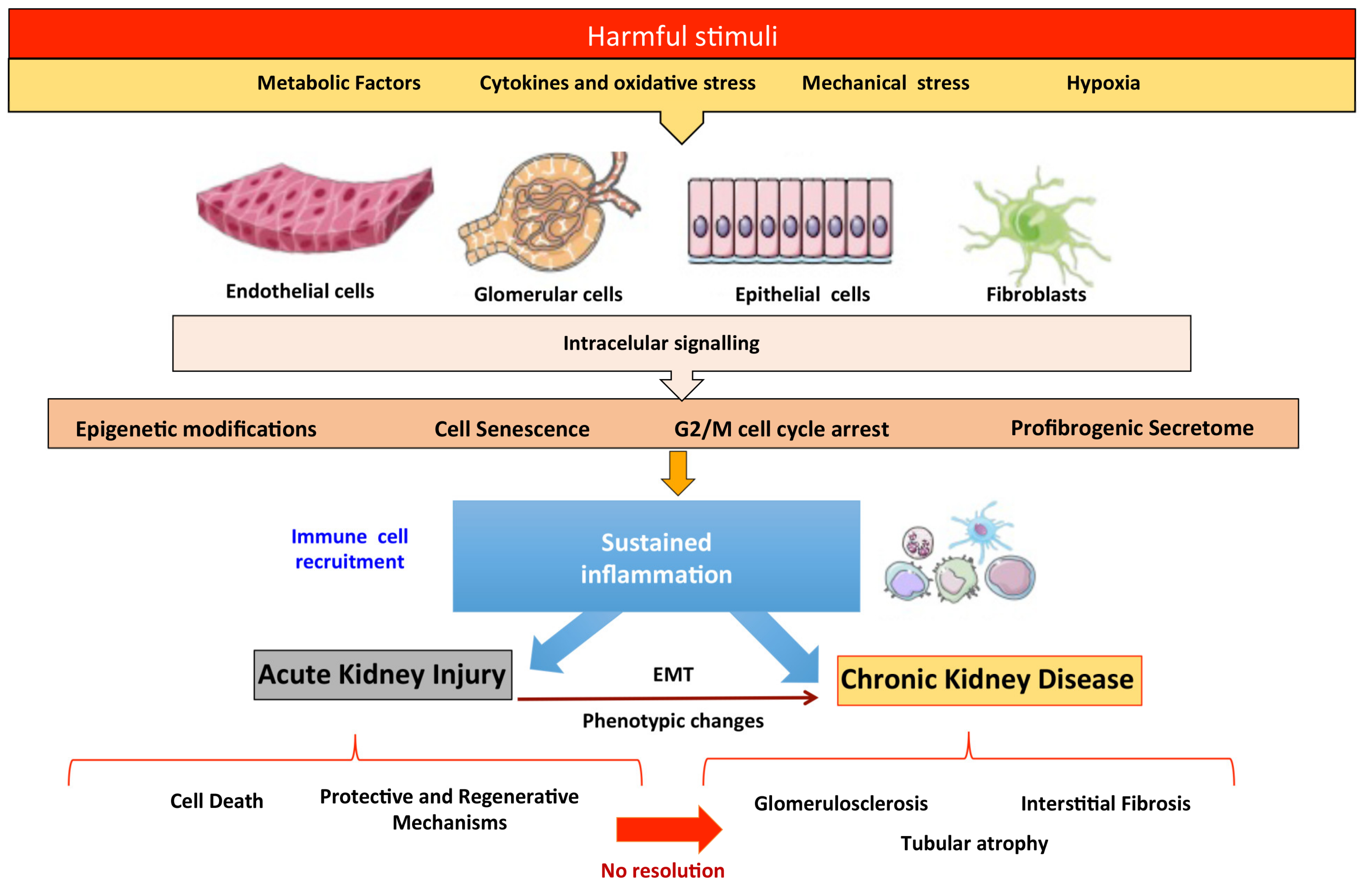{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Molecular and Cellular Events Contributing to the AKI-to-CKD Transition
2.1. Macrophages in Kidney Injury
2.2. Regulated Cell Death
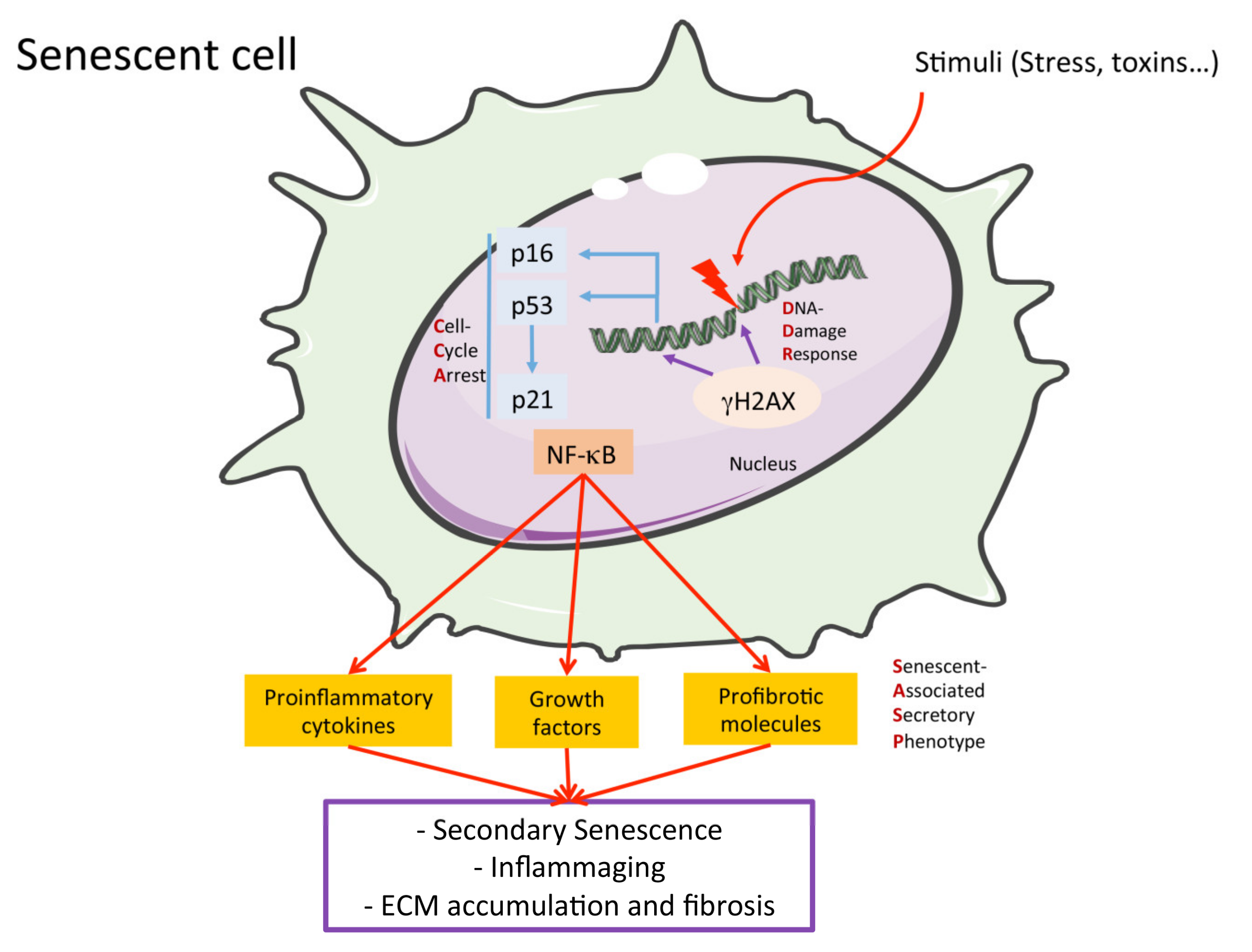
2.3. Cellular Senescence
2.4. The Senescence-Associated Secretory Phenotype (SAPS)
2.5. Polyploidization: A Question of Size; Samson or Delilah
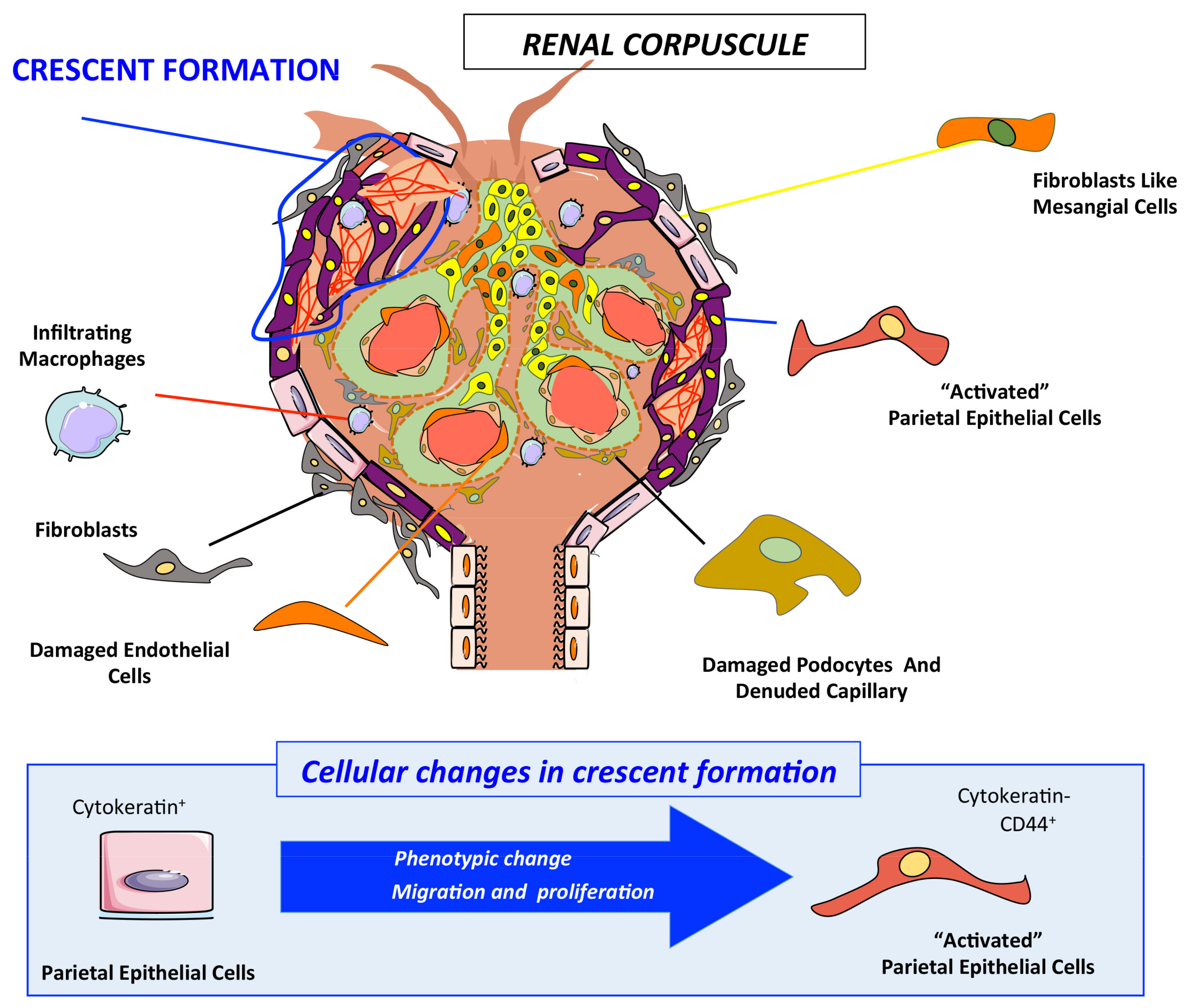
2.6. Podocyte Damage and Activation of Parietal Epithelial Cells
3. Recovery and Repair Strategies
3.1. Resident Renal Progenitor Cells
3.2. Stem Cells and the Reparative Secretome
3.3. Macrophages in Repair
3.4. Senescent Cell Clearance: Senotherapies
4. AKI/CKD-to-Cancer Transition
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ortiz, A.; Roger, M.; Jiménez, V.M.; Perez, J.C.R.; Furlano, M.; Atxer, L.S.; Zurro, D.G.; Casabona, C.M.R.; Zurro, D.G.; Gómez, C.G.; et al. RICORS2040: The need for collaborative research in chronic kidney disease. Clin. Kidney J. 2021. [Google Scholar] [CrossRef]
- Chawla, L.S.; Eggers, P.W.; Star, R.A.; Kimmel, P.L. Acute Kidney Injury and Chronic Kidney Disease as Interconnected Syndromes. N. Engl. J. Med. 2014, 371, 58–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz-Ortega, M.; Rayego-Mateos, S.; Lamas, S.; Ortiz, A.; Rodrigues-Diez, R.R. Targeting the progression of chronic kidney disease. Nat. Rev. Nephrol. 2020, 16, 269–288. [Google Scholar] [CrossRef] [PubMed]
- Ucero, A.C.; Benito-Martin, A.; Izquierdo, M.C.; Sanchez-Niño, M.D.; Sanz, A.B.; Ramos, A.M.; Berzal, S.; Ruiz-Ortega, M.; Egido, J.; Ortiz, A. Unilateral ureteral obstruction: Beyond obstruction. Int. Urol. Nephrol. 2014, 46, 765–776. [Google Scholar] [CrossRef] [PubMed]
- Linkermann, A.; Stockwell, B.R.; Krautwald, S.; Anders, H.-J. Regulated cell death and inflammation: An auto-amplification loop causes organ failure. Nat. Rev. Immunol. 2014, 14, 759–767. [Google Scholar] [CrossRef]
- Rayego-Mateos, S.; Campillo, S.; Rodrigues-Diez, R.R.; Tejera-Muñoz, A.; Marquez-Exposito, L.; Goldschmeding, R.; Rodríguez-Puyol, D.; Calleros, L.; Ruiz-Ortega, M. Interplay between extracellular matrix components and cellular and molecular mechanisms in kidney fibrosis. Clin. Sci. 2021, 135, 1999–2029. [Google Scholar] [CrossRef]
- Patel, A.A.; Ginhoux, F.; Yona, S. Monocytes, macrophages, dendritic cells and neutrophils: An update on lifespan kinetics in health and disease. Immunology 2021, 163, 250–261. [Google Scholar] [CrossRef]
- Lech, M.; Gröbmayr, R.; Ryu, M.; Lorenz, G.; Hartter, I.; Mulay, S.R.; Susanti, H.E.; Kobayashi, K.S.; Flavell, R.A.; Anders, H.-J. Macrophage Phenotype Controls Long-Term AKI Outcomes—Kidney Regeneration versus Atrophy. J. Am. Soc. Nephrol. 2014, 25, 292–304. [Google Scholar] [CrossRef]
- Wynn, T.A.; Vannella, K.M. Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity 2016, 44, 450–462. [Google Scholar] [CrossRef] [Green Version]
- Kinsey, G.R. Macrophage Dynamics in AKI to CKD Progression. J. Am. Soc. Nephrol. 2014, 25, 209–211. [Google Scholar] [CrossRef]
- Huen, S.C.; Cantley, L.G. Macrophages in Renal Injury and Repair. Annu. Rev. Physiol. 2017, 79, 449–469. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Huen, S.; Nishio, H.; Nishio, S.; Lee, H.K.; Choi, B.-S.; Ruhrberg, C.; Cantley, L.G. Distinct Macrophage Phenotypes Contribute to Kidney Injury and Repair. J. Am. Soc. Nephrol. 2011, 22, 317–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantero-Navarro, E.; Rayego-Mateos, S.; Orejudo, M.; Tejedor-Santamaria, L.; Tejera-Muñoz, A.; Sanz, A.B.; Marquez-Exposito, L.; Marchant, V.; Santos-Sanchez, L.; Egido, J.; et al. Role of Macrophages and Related Cytokines in Kidney Disease. Front. Med. 2021, 8, 688060. [Google Scholar] [CrossRef] [PubMed]
- Tian, S.; Zhang, L.; Tang, J.; Guo, X.; Dong, K.; Chen, S.-Y. HMGB1 exacerbates renal tubulointerstitial fibrosis through facilitating M1 macrophage phenotype at the early stage of obstructive injury. Am. J. Physiol. Renal Physiol. 2015, 308, F69–F75. [Google Scholar] [CrossRef]
- Zhao, Y.; Ding, Z.; Ge, W.; Liu, J.; Xu, X.; Cheng, R.; Zhang, J. Riclinoctaose Attenuates Renal Ischemia-Reperfusion Injury by the Regulation of Macrophage Polarization. Front. Pharmacol. 2021, 12, 745425. [Google Scholar] [CrossRef]
- Li, X.; Mu, G.; Song, C.; Zhou, L.; He, L.; Jin, Q.; Lu, Z. Role of M2 Macrophages in Sepsis-Induced Acute Kidney Injury. Shock 2018, 50, 233–239. [Google Scholar] [CrossRef]
- Lv, L.L.; Tang, P.M.-K.; Li, C.J.; You, Y.K.; Li, J.; Huang, X.-R.; Ni, J.; Feng, M.; Liu, B.C.; Lan, H.-Y. The pattern recognition receptor, Mincle, is essential for maintaining the M1 macrophage phenotype in acute renal inflammation. Kidney Int. 2017, 91, 587–602. [Google Scholar] [CrossRef]
- Wen, Y.; Lu, X.; Ren, J.; Privratsky, J.R.; Yang, B.; Rudemiller, N.P.; Zhang, J.; Griffiths, R.; Jain, M.K.; Nedospasov, S.A.; et al. KLF4 in Macrophages Attenuates TNF α-Mediated Kidney Injury and Fibrosis. J. Am. Soc. Nephrol. 2019, 30, 1925–1938. [Google Scholar] [CrossRef]
- Wu, H.; Ma, J.; Wang, P.; Corpuz, T.M.; Panchapakesan, U.; Wyburn, K.R.; Chadban, S.J. HMGB1 Contributes to Kidney Ischemia Reperfusion Injury. J. Am. Soc. Nephrol. 2010, 21, 1878–1890. [Google Scholar] [CrossRef] [Green Version]
- Privratsky, J.R.; Zhang, J.; Lu, X.; Rudemiller, N.; Wei, Q.; Yu, Y.-R.; Gunn, M.D.; Crowley, S.D. Interleukin 1 receptor (IL-1R1) activation exacerbates toxin-induced acute kidney injury. Am. J. Physiol. Renal Physiol. 2018, 315, F682–F691. [Google Scholar] [CrossRef]
- Awad, A.S.; You, H.; Gao, T.; Cooper, T.K.; Nedospasov, S.A.; Vacher, J.; Wilkinson, P.F.; Farrell, F.X.; Brian Reeves, W. Macrophage-derived tumor necrosis factor-α mediates diabetic renal injury. Kidney Int. 2015, 88, 722–733. [Google Scholar] [CrossRef] [Green Version]
- Xing, L.; Zhongqian, L.; Chunmei, S.; Pingfa, C.; Lei, H.; Qin, J.; Genhua, M.; Yijun, D. Activation of M1 macrophages in sepsis-induced acute kidney injury in response to heparin-binding protein. PLoS ONE 2018, 13, e0196423. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.-L.; Lv, L.-L.; Tang, T.-T.; Wang, B.; Feng, Y.; Zhou, L.-T.; Cao, J.-Y.; Tang, R.-N.; Wu, M.; Liu, H.; et al. HIF-1α inducing exosomal microRNA-23a expression mediates the cross-talk between tubular epithelial cells and macrophages in tubulointerstitial inflammation. Kidney Int. 2019, 95, 388–404. [Google Scholar] [CrossRef] [PubMed]
- Komada, T.; Chung, H.; Lau, A.; Platnich, J.M.; Beck, P.L.; Benediktsson, H.; Duff, H.J.; Jenne, C.N.; Muruve, D.A. Macrophage Uptake of Necrotic Cell DNA Activates the AIM2 Inflammasome to Regulate a Proinflammatory Phenotype in CKD. J. Am. Soc. Nephrol. 2018, 29, 1165–1181. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Ren, J.; Gui, Y.; Wei, W.; Shu, B.; Lu, Q.; Xue, X.; Sun, X.; He, W.; Yang, J.; et al. Wnt/β -Catenin–Promoted Macrophage Alternative Activation Contributes to Kidney Fibrosis. J. Am. Soc. Nephrol. 2018, 29, 182–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klessens, C.Q.F.; Zandbergen, M.; Wolterbeek, R.; Bruijn, J.A.; Rabelink, T.J.; Bajema, I.M.; IJpelaar, D.H.T. Macrophages in diabetic nephropathy in patients with type 2 diabetes. Nephrol. Dial. Transpl. 2017, 32, 1322–1329. [Google Scholar] [CrossRef]
- Nelson, P.J.; Rees, A.J.; Griffin, M.D.; Hughes, J.; Kurts, C.; Duffield, J. The Renal Mononuclear Phagocytic System. J. Am. Soc. Nephrol. 2012, 23, 194–203. [Google Scholar] [CrossRef]
- Wang, X.; Chen, J.; Xu, J.; Xie, J.; Harris, D.C.H.; Zheng, G. The Role of Macrophages in Kidney Fibrosis. Front. Physiol. 2021, 12, 705838. [Google Scholar] [CrossRef]
- Kim, M.-G.; Yang, J.; Ko, Y.S.; Lee, H.Y.; Oh, S.W.; Cho, W.Y.; Jo, S.-K. Impact of aging on transition of acute kidney injury to chronic kidney disease. Sci. Rep. 2019, 9, 18445. [Google Scholar] [CrossRef] [Green Version]
- Solez, K.; Morel-Maroger, L.; Sraer, J.-D. The Morphology of “Acute Tubular Necrosis” in Man. Medicine 1979, 58, 362–376. [Google Scholar] [CrossRef]
- Steen Olsen, T.; Steen Olsen, H.; Hansen, H.E. Tubular ultrastructure in acute renal failure in man: Epithelial necrosis and regeneration. Virchows Arch. A Pathol. Anat. Histopathol. 1985, 406, 75–89. [Google Scholar] [CrossRef] [PubMed]
- Bonventre, J.V.; Yang, L. Cellular pathophysiology of ischemic acute kidney injury. J. Clin. Investig. 2011, 121, 4210–4221. [Google Scholar] [CrossRef] [PubMed]
- Sanz, A.B.; Santamaría, B.; Ruiz-Ortega, M.; Egido, J.; Ortiz, A. Mechanisms of Renal Apoptosis in Health and Disease. J. Am. Soc. Nephrol. 2008, 19, 1634–1642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belavgeni, A.; Meyer, C.; Stumpf, J.; Hugo, C.; Linkermann, A. Ferroptosis and Necroptosis in the Kidney. Cell Chem. Biol. 2020, 27, 448–462. [Google Scholar] [CrossRef]
- Martin-Sanchez, D.; Poveda, J.; Fontecha-Barriuso, M.; Ruiz-Andres, O.; Sanchez-Niño, M.D.; Ruiz-Ortega, M.; Ortiz, A.; Sanz, A.B. Targeting of regulated necrosis in kidney disease. Nefrología 2018, 38, 125–135. [Google Scholar] [CrossRef]
- Sancho-Martínez, S.M.; Prieto-García, L.; Prieto, M.; Fuentes-Calvo, I.; López-Novoa, J.M.; Morales, A.I.; Martínez-Salgado, C.; López-Hernández, F.J. N -acetylcysteine transforms necrosis into apoptosis and affords tailored protection from cisplatin cytotoxicity. Toxicol. Appl. Pharmacol. 2018, 349, 83–93. [Google Scholar] [CrossRef]
- Martin-Sanchez, D.; Fontecha-Barriuso, M.; Carrasco, S.; Sanchez-Niño, M.D.; von Mässenhausen, A.; Linkermann, A.; Cannata-Ortiz, P.; Ruiz-Ortega, M.; Egido, J.; Ortiz, A.; et al. TWEAK and RIPK1 mediate a second wave of cell death during AKI. Proc. Natl. Acad. Sci. USA 2018, 115, 4182–4187. [Google Scholar] [CrossRef] [Green Version]
- Linkermann, A.; Skouta, R.; Himmerkus, N.; Mulay, S.R.; Dewitz, C.; De Zen, F.; Prokai, A.; Zuchtriegel, G.; Krombach, F.; Welz, P.-S.; et al. Synchronized renal tubular cell death involves ferroptosis. Proc. Natl. Acad. Sci. USA 2014, 111, 16836–16841. [Google Scholar] [CrossRef] [Green Version]
- Linkermann, A.; Brasen, J.H.; Darding, M.; Jin, M.K.; Sanz, A.B.; Heller, J.-O.; De Zen, F.; Weinlich, R.; Ortiz, A.; Walczak, H.; et al. Two independent pathways of regulated necrosis mediate ischemia-reperfusion injury. Proc. Natl. Acad. Sci. USA 2013, 110, 12024–12029. [Google Scholar] [CrossRef] [Green Version]
- Martin-Sanchez, D.; Ruiz-Andres, O.; Poveda, J.; Carrasco, S.; Cannata-Ortiz, P.; Sanchez-Niño, M.D.; Ruiz Ortega, M.; Egido, J.; Linkermann, A.; Ortiz, A.; et al. Ferroptosis, but Not Necroptosis, Is Important in Nephrotoxic Folic Acid–Induced AKI. J. Am. Soc. Nephrol. 2017, 28, 218–229. [Google Scholar] [CrossRef]
- He, Z.; Liao, W.; Song, Q.; Li, B.; Liu, J.; Xiong, Y.; Song, C.; Yang, S. Role of ferroptosis induced by a high concentration of calcium oxalate in the formation and development of urolithiasis. Int. J. Mol. Med. 2020, 47, 289–301. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Ma, H.; Shao, J.; Wu, J.; Zhou, L.; Zhang, Z.; Wang, Y.; Huang, Z.; Ren, J.; Liu, S.; et al. A Role for Tubular Necroptosis in Cisplatin-Induced AKI. J. Am. Soc. Nephrol. 2015, 26, 2647–2658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulay, S.R.; Honarpisheh, M.M.; Foresto-Neto, O.; Shi, C.; Desai, J.; Zhao, Z.B.; Marschner, J.A.; Popper, B.; Buhl, E.M.; Boor, P.; et al. Mitochondria Permeability Transition versus Necroptosis in Oxalate-Induced AKI. J. Am. Soc. Nephrol. 2019, 30, 1857–1869. [Google Scholar] [CrossRef]
- Mulay, S.R.; Linkermann, A.; Anders, H.-J. Necroinflammation in Kidney Disease. J. Am. Soc. Nephrol. 2016, 27, 27–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, H.; Xu, J.; Xu, Z.; Yang, B.; Luo, P.; He, Q. Defining therapeutic targets for renal fibrosis: Exploiting the biology of pathogenesis. Biomed. Pharmacother. 2021, 143, 112115. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.-C.; Tang, T.-T.; Lv, L.-L.; Lan, H.-Y. Renal tubule injury: A driving force toward chronic kidney disease. Kidney Int. 2018, 93, 568–579. [Google Scholar] [CrossRef] [PubMed]
- Schelling, J.R. Tubular atrophy in the pathogenesis of chronic kidney disease progression. Pediatr. Nephrol. 2016, 31, 693–706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitamoto, K.; Machida, Y.; Uchida, J.; Izumi, Y.; Shiota, M.; Nakao, T.; Iwao, H.; Yukimura, T.; Nakatani, T.; Miura, K. Effects of Liposome Clodronate on Renal Leukocyte Populations and Renal Fibrosis in Murine Obstructive Nephropathy. J. Pharmacol. Sci. 2009, 111, 285–292. [Google Scholar] [CrossRef] [Green Version]
- Tan, T.K.; Zheng, G.; Hsu, T.-T.; Ra Lee, S.; Zhang, J.; Zhao, Y.; Tian, X.; Wang, Y.; Min Wang, Y.; Cao, Q.; et al. Matrix metalloproteinase-9 of tubular and macrophage origin contributes to the pathogenesis of renal fibrosis via macrophage recruitment through osteopontin cleavage. Lab. Investig. 2013, 93, 434–449. [Google Scholar] [CrossRef] [Green Version]
- Martin-Sanchez, D.; Fontecha-Barriuso, M.; Martinez-Moreno, J.M.; Ramos, A.M.; Sanchez-Niño, M.D.; Guerrero-Hue, M.; Moreno, J.A.; Ortiz, A.; Sanz, A.B. Ferroptosis and kidney disease. Nefrología 2020, 40, 384–394. [Google Scholar] [CrossRef]
- Chen, H.; Fang, Y.; Wu, J.; Chen, H.; Zou, Z.; Zhang, X.; Shao, J.; Xu, Y. RIPK3-MLKL-mediated necroinflammation contributes to AKI progression to CKD. Cell Death Dis. 2018, 9, 878. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Huang, C.; Yi, H.; Cao, Q.; Zhao, Y.; Chen, J.; Chen, X.; Pollock, C. RIPK3 blockade attenuates kidney fibrosis in a folic acid model of renal injury. FASEB J. 2020, 34, 10286–10298. [Google Scholar] [CrossRef] [PubMed]
- Pastrana, D.V. mSphere of Influence: It’s Not Me, It’s you-how Donor Factors Influence Kidney Transplant Outcomes. mSphere 2020, 5, e00964-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, B.; Wan, Y.; Chen, R.; Zhang, C.; Li, X.; Meng, F.; Glaser, S.; Wu, N.; Zhou, T.; Li, S.; et al. The emerging role of cellular senescence in renal diseases. J. Cell. Mol. Med. 2020, 24, 2087–2097. [Google Scholar] [CrossRef]
- Valentijn, F.A.; Falke, L.L.; Nguyen, T.Q.; Goldschmeding, R. Cellular senescence in the aging and diseased kidney. J. Cell Commun. Signal. 2018, 12, 69–82. [Google Scholar] [CrossRef] [Green Version]
- Marquez-Exposito, L.; Tejedor-Santamaria, L.; Santos-Sanchez, L.; Valentijn, F.A.; Cantero-Navarro, E.; Rayego-Mateos, S.; Rodrigues-Diez, R.R.; Tejera-Muñoz, A.; Marchant, V.; Sanz, A.B.; et al. Acute Kidney Injury is aggravated in Aged Mice by the Exacerbation of Proinflammatory Processes. Front. Pharmacol. 2021, 12, 662020. [Google Scholar] [CrossRef]
- Knoppert, S.N.; Valentijn, F.A.; Nguyen, T.Q.; Goldschmeding, R.; Falke, L.L. Cellular Senescence and the Kidney: Potential Therapeutic Targets and Tools. Front. Pharmacol. 2019, 10, 770. [Google Scholar] [CrossRef]
- Muñoz-Espín, D.; Serrano, M. Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef]
- Melk, A.; Schmidt, B.M.W.; Takeuchi, O.; Sawitzki, B.; Rayner, D.C.; Halloran, P.F. Expression of p16INK4a and other cell cycle regulator and senescence associated genes in aging human kidney. Kidney Int. 2004, 65, 510–520. [Google Scholar] [CrossRef] [Green Version]
- Branzei, D.; Foiani, M. Regulation of DNA repair throughout the cell cycle. Nat. Rev. Mol. Cell Biol. 2008, 9, 297–308. [Google Scholar] [CrossRef]
- Gire, V.; Dulić, V. Senescence from G2 arrest, revisited. Cell Cycle 2015, 14, 297–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Sullivan, E.D.; Hughes, J.; Ferenbach, D.A. Renal Aging: Causes and Consequences. J. Am. Soc. Nephrol. 2017, 28, 407–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonventre, J.V. Maladaptive Proximal Tubule Repair: Cell Cycle Arrest. Nephron Clin. Pract. 2014, 127, 61–64. [Google Scholar] [CrossRef] [PubMed]
- Schroth, J.; Thiemermann, C.; Henson, S.M. Senescence and the Aging Immune System as Major Drivers of Chronic Kidney Disease. Front. Cell Dev. Biol. 2020, 8, 564461. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Yanagita, M. Immunology of the ageing kidney. Nat. Rev. Nephrol. 2019, 15, 625–640. [Google Scholar] [CrossRef]
- Acosta, J.C.; Banito, A.; Wuestefeld, T.; Georgilis, A.; Janich, P.; Morton, J.P.; Athineos, D.; Kang, T.-W.; Lasitschka, F.; Andrulis, M.; et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell Biol. 2013, 15, 978–990. [Google Scholar] [CrossRef]
- Cao, Q.; Harris, D.C.H.; Wang, Y. Macrophages in Kidney Injury, Inflammation, and Fibrosis. Physiology 2015, 30, 183–194. [Google Scholar] [CrossRef]
- Sato, Y.; Yanagita, M. Immune cells and inflammation in AKI to CKD progression. Am. J. Physiol. Renal Physiol. 2018, 315, F1501–F1512. [Google Scholar] [CrossRef]
- Rabb, H.; Griffin, M.D.; McKay, D.B.; Swaminathan, S.; Pickkers, P.; Rosner, M.H.; Kellum, J.A.; Ronco, C. Inflammation in AKI: Current Understanding, Key Questions, and Knowledge Gaps. J. Am. Soc. Nephrol. 2016, 27, 371–379. [Google Scholar] [CrossRef] [Green Version]
- Deshmane, S.L.; Kremlev, S.; Amini, S.; Sawaya, B.E. Monocyte Chemoattractant Protein-1 (MCP-1): An Overview. J. Interf. Cytokine Res. 2009, 29, 313–326. [Google Scholar] [CrossRef]
- Mansour, S.G.; Puthumana, J.; Coca, S.G.; Gentry, M.; Parikh, C.R. Biomarkers for the detection of renal fibrosis and prediction of renal outcomes: A systematic review. BMC Nephrol. 2017, 18, 72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satirapoj, B. Tubulointerstitial Biomarkers for Diabetic Nephropathy. J. Diabetes Res. 2018, 2018, 2852398. [Google Scholar] [CrossRef] [PubMed]
- Sanz, A.B.; Sanchez-Niño, M.D.; Ramos, A.M.; Moreno, J.A.; Santamaria, B.; Ruiz-Ortega, M.; Egido, J.; Ortiz, A. NF-κB in Renal Inflammation. J. Am. Soc. Nephrol. 2010, 21, 1254–1262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poveda, J.; Sanz, A.B.; Carrasco, S.; Ruiz-Ortega, M.; Cannata-Ortiz, P.; Sanchez-Niño, M.D.; Ortiz, A. Bcl3: A regulator of NF-κB inducible by TWEAK in acute kidney injury with anti-inflammatory and antiapoptotic properties in tubular cells. Exp. Mol. Med. 2017, 49, e352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno, J.A.; Izquierdo, M.C.; Sanchez-Niño, M.D.; Suárez-Alvarez, B.; Lopez-Larrea, C.; Jakubowski, A.; Blanco, J.; Ramirez, R.; Selgas, R.; Ruiz-Ortega, M.; et al. The Inflammatory Cytokines TWEAK and TNFα Reduce Renal Klotho Expression through NFκB. J. Am. Soc. Nephrol. 2011, 22, 1315–1325. [Google Scholar] [CrossRef] [Green Version]
- Ferrucci, L.; Fabbri, E. Inflammageing: Chronic inflammation in ageing, cardiovascular disease, and frailty. Nat. Rev. Cardiol. 2018, 15, 505–522. [Google Scholar] [CrossRef]
- Wiggins, K.A.; Parry, A.J.; Cassidy, L.D.; Humphry, M.; Webster, S.J.; Goodall, J.C.; Narita, M.; Clarke, M.C.H. IL-1α cleavage by inflammatory caspases of the noncanonical inflammasome controls the senescence-associated secretory phenotype. Aging Cell 2019, 18, e12946. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Duran, I.; Tarrats, N.; Hari, P.; Acosta, J.C. Measuring the Inflammasome in Oncogene-Induced Senescence. Methods Mol Biol. 2019, 1896, 57–70. [Google Scholar] [CrossRef]
- Zhang, P.; Lu, B.; Zhu, R.; Yang, D.; Liu, W.; Wang, Q.; Ji, N.; Chen, Q.; Ding, Y.; Liang, X.; et al. Hyperglycemia accelerates inflammaging in the gingival epithelium through inflammasomes activation. J. Periodontal Res. 2021, 56, 667–678. [Google Scholar] [CrossRef]
- Sari, F.T.; Sari, F.T.; Sari, F.T.; Arfian, N.; Sari, D.C.R. Effect of kidney ischemia/reperfusion injury on proliferation, apoptosis, and cellular senescence in acute kidney injury in mice. Med. J. Malays. 2020, 75, 20–23. [Google Scholar]
- Li, C.; Xie, N.; Li, Y.; Liu, C.; Hou, F.F.; Wang, J. N-acetylcysteine ameliorates cisplatin-induced renal senescence and renal interstitial fibrosis through sirtuin1 activation and p53 deacetylation. Free Radic. Biol. Med. 2019, 130, 512–527. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.-M.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-β: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Meng, X.-M.; Ng, Y.-Y.; Ma, F.Y.; Zhou, S.; Zhang, Y.; Yang, C.; Huang, X.-R.; Xiao, J.; Wang, Y.-Y.; et al. TGF-β/Smad3 signalling regulates the transition of bone marrow-derived macrophages into myofibroblasts during tissue fibrosis. Oncotarget 2016, 7, 8809–8822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rayego-Mateos, S.; Morgado-Pascual, J.L.; Rodrigues-Diez, R.R.; Rodrigues-Diez, R.; Falke, L.L.; Mezzano, S.; Ortiz, A.; Egido, J.; Goldschmeding, R.; Ruiz-Ortega, M. Connective tissue growth factor induces renal fibrosis via epidermal growth factor receptor activation. J. Pathol. 2018, 244, 227–241. [Google Scholar] [CrossRef]
- Meunier, F. Antifungal therapy in the immunocompromised host. Mycoses 1988, 31 (Suppl. S2), 54–58. [Google Scholar]
- Ruiz-Ortega, M.; Rodríguez-Vita, J.; Sanchez-Lopez, E.; Carvajal, G.; Egido, J. TGF-beta signaling in vascular fibrosis. Cardiovasc. Res. 2007, 74, 196–206. [Google Scholar] [CrossRef] [Green Version]
- Rayego-Mateos, S.; Rodrigues-Diez, R.; Morgado-Pascual, J.L.; Valentijn, F.; Valdivielso, J.M.; Goldschmeding, R.; Ruiz-Ortega, M. Role of Epidermal Growth Factor Receptor (EGFR) and Its Ligands in Kidney Inflammation and Damage. Mediat. Inflamm. 2018, 2018, 8739473. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-Ortega, M.; Lamas, S.; Ortiz, A. Antifibrotic Agents for the Management of CKD: A Review. Am. J. Kidney Dis. 2022, S0272-6386(21)01051-9, (published online ahead of print, 6 January 2022). [Google Scholar] [CrossRef]
- Van Deursen, J.M. The role of senescent cells in ageing. Nature 2014, 509, 439–446. [Google Scholar] [CrossRef] [Green Version]
- Ovadya, Y.; Krizhanovsky, V. Senescent cells: SASPected drivers of age-related pathologies. Biogerontology 2014, 15, 627–642. [Google Scholar] [CrossRef]
- Losick, V.P. Wound-Induced Polyploidy Is Required for Tissue Repair. Adv. Wound Care 2016, 5, 271–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gjelsvik, K.J.; Besen-McNally, R.; Losick, V.P. Solving the Polyploid Mystery in Health and Disease. Trends Genet. 2019, 35, 6–14. [Google Scholar] [CrossRef] [PubMed]
- Minamishima, Y.A.; Nakayama, K.; Nakayama, K.-I. Recovery of liver mass without proliferation of hepatocytes after partial hepatectomy in Skp2-deficient mice. Cancer Res. 2002, 62, 995–999. [Google Scholar] [PubMed]
- Miyaoka, Y.; Ebato, K.; Kato, H.; Arakawa, S.; Shimizu, S.; Miyajima, A. Hypertrophy and unconventional cell division of hepatocytes underlie liver regeneration. Curr. Biol. 2012, 22, 1166–1175. [Google Scholar] [CrossRef] [Green Version]
- Wilkinson, P.D.; Alencastro, F.; Delgado, E.R.; Leek, M.P.; Weirich, M.P.; Otero, P.A.; Roy, N.; Brown, W.K.; Oertel, M.; Duncan, A.W. Polyploid Hepatocytes Facilitate Adaptation and Regeneration to Chronic Liver Injury. Am. J. Pathol. 2019, 189, 1241–1255. [Google Scholar] [CrossRef]
- Lazzeri, E.; Angelotti, M.L.; Peired, A.; Conte, C.; Marschner, J.A.; Maggi, L.; Mazzinghi, B.; Lombardi, D.; Melica, M.E.; Nardi, S.; et al. Endocycle-related tubular cell hypertrophy and progenitor proliferation recover renal function after acute kidney injury. Nat. Commun. 2018, 9, 1344. [Google Scholar] [CrossRef]
- Megyesi, J.; Safirstein, R.L.; Price, P.M. Induction of p21WAF1/CIP1/SDI1 in kidney tubule cells affects the course of cisplatin-induced acute renal failure. J. Clin. Investig. 1998, 101, 777–782. [Google Scholar] [CrossRef] [Green Version]
- Nagata, M.; Yamaguchi, Y.; Komatsu, Y.; Ito, K. Mitosis and the presence of binucleate cells among glomerular podocytes in diseased human kidneys. Nephron 1995, 70, 68–71. [Google Scholar] [CrossRef]
- Petermann, A.T.; Pippin, J.; Hiromura, K.; Monkawa, T.; Durvasula, R.; Couser, W.G.; Kopp, J.; Shankland, S.J. Mitotic cell cycle proteins increase in podocytes despite lack of proliferation. Kidney Int. 2003, 63, 113–122. [Google Scholar] [CrossRef] [Green Version]
- Griffin, S.V.; Petermann, A.T.; Durvasula, R.V.; Shankland, S.J. Podocyte proliferation and differentiation in glomerular disease: Role of cell-cycle regulatory proteins. Nephrol. Dial. Transpl. 2003, 18 (Suppl. 6), vi8–vi13. [Google Scholar] [CrossRef] [Green Version]
- Yu, D.; Petermann, A.; Kunter, U.; Rong, S.; Shankland, S.J.; Floege, J. Urinary podocyte loss is a more specific marker of ongoing glomerular damage than proteinuria. J. Am. Soc. Nephrol. 2005, 16, 1733–1741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Najafian, B.; Tøndel, C.; Svarstad, E.; Gubler, M.-C.; Oliveira, J.-P.; Mauer, M. Accumulation of Globotriaosylceramide in Podocytes in Fabry Nephropathy Is Associated with Progressive Podocyte Loss. J. Am. Soc. Nephrol. 2020, 31, 865–875. [Google Scholar] [CrossRef] [PubMed]
- Floege, J.; Johnson, R.J.; Alpers, C.E.; Fatemi-Nainie, S.; Richardson, C.A.; Gordon, K.; Couser, W.G. Visceral glomerular epithelial cells can proliferate in vivo and synthesize platelet-derived growth factor B-chain. Am. J. Pathol. 1993, 142, 637–650. [Google Scholar] [PubMed]
- Kriz, W.; Hähnel, B.; Rösener, S.; Elger, M. Long-term treatment of rats with FGF-2 results in focal segmental glomerulosclerosis. Kidney Int. 1995, 48, 1435–1450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagata, M. Podocyte injury and its consequences. Kidney Int. 2016, 89, 1221–1230. [Google Scholar] [CrossRef]
- Nagata, M.; Kriz, W. Glomerular damage after uninephrectomy in young rats. II. Mechanical stress on podocytes as a pathway to sclerosis. Kidney Int. 1992, 42, 148–160. [Google Scholar] [CrossRef] [Green Version]
- Chagnac, A.; Zingerman, B.; Rozen-Zvi, B.; Herman-Edelstein, M. Consequences of Glomerular Hyperfiltration: The Role of Physical Forces in the Pathogenesis of Chronic Kidney Disease in Diabetes and Obesity. Nephron 2019, 143, 38–42. [Google Scholar] [CrossRef]
- Lee, E.Y.; Chung, C.H.; Khoury, C.C.; Yeo, T.K.; Pyagay, P.E.; Wang, A.; Chen, S. The monocyte chemoattractant protein-1/CCR2 loop, inducible by TGF-beta, increases podocyte motility and albumin permeability. Am. J. Physiol. Renal Physiol. 2009, 297, F85–F94. [Google Scholar] [CrossRef] [Green Version]
- Tarabra, E.; Giunti, S.; Barutta, F.; Salvidio, G.; Burt, D.; Deferrari, G.; Gambino, R.; Vergola, D.; Pinach, S.; Perin, P.C.; et al. Effect of the monocyte chemoattractant protein-1/CC chemokine receptor 2 system on nephrin expression in streptozotocin-treated mice and human cultured podocytes. Diabetes 2009, 58, 2109–2118. [Google Scholar] [CrossRef] [Green Version]
- Angeletti, A.; Cantarelli, C.; Petrosyan, A.; Andrighetto, S.; Budge, K.; D’Agati, V.D.; Hartzell, S.; Malvi, D.; Donadei, C.; Thurman, J.M.; et al. Loss of decay-accelerating factor triggers podocyte injury and glomerulosclerosis. J. Exp. Med. 2020, 217, e20191699. [Google Scholar] [CrossRef]
- Trimarchi, H.; Ortiz, A.; Sánchez-Niño, M.D. Lyso-Gb3 Increases αvβ3 Integrin Gene Expression in Cultured Human Podocytes in Fabry Nephropathy. J. Clin. Med. 2020, 9, 3659. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Niño, M.D.; Carpio, D.; Sanz, A.B.; Ruiz-Ortega, M.; Mezzano, S.; Ortiz, A. Lyso-Gb3 activates Notch1 in human podocytes. Hum. Mol. Genet. 2015, 24, 5720–5732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Niño, M.D.; Poveda, J.; Sanz, A.B.; Carrasco, S.; Ruiz-Ortega, M.; Selgas, R.; Egido, J.; Ortiz, A. 3,4-DGE is cytotoxic and decreases HSP27/HSPB1 in podocytes. Arch. Toxicol. 2014, 88, 597–608. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Niño, M.D.; Sanz, A.B.; Sanchez-Lopez, E.; Ruiz-Ortega, M.; Benito-Martin, A.; Saleem, M.A.; Mathieson, P.W.; Mezzano, S.; Egido, J.; Ortiz, A. HSP27/HSPB1 as an adaptive podocyte antiapoptotic protein activated by high glucose and angiotensin II. Lab. Investig. 2012, 92, 32–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Niño, M.D.; Sanz, A.B.; Carrasco, S.; Saleem, M.A.; Mathieson, P.W.; Valdivielso, J.M.; Ruiz-Ortega, M.; Egido, J.; Ortiz, A. Globotriaosylsphingosine actions on human glomerular podocytes: Implications for Fabry nephropathy. Nephrol. Dial. Transpl. 2011, 26, 1797–1802. [Google Scholar] [CrossRef] [Green Version]
- Rubio-Navarro, A.; Sanchez-Niño, M.D.; Guerrero-Hue, M.; García-Caballero, C.; Gutiérrez, E.; Yuste, C.; Sevillano, Á.; Praga, M.; Egea, J.; Román, E.; et al. Podocytes are new cellular targets of haemoglobin-mediated renal damage. J. Pathol. 2018, 244, 296–310. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Lin, L.; Tao, X.; Song, Y.; Cui, J.; Wan, J. The role of podocyte damage in the etiology of ischemia-reperfusion acute kidney injury and post-injury fibrosis. BMC Nephrol. 2019, 20, 106. [Google Scholar] [CrossRef] [Green Version]
- Inagi, R.; Ishimoto, Y.; Nangaku, M. Proteostasis in endoplasmic reticulum--new mechanisms in kidney disease. Nat. Rev. Nephrol. 2014, 10, 369–378. [Google Scholar] [CrossRef]
- Chen, S.; Meng, X.-F.; Zhang, C. Role of NADPH oxidase-mediated reactive oxygen species in podocyte injury. BioMed Res. Int. 2013, 2013, 839761. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Hitomi, H.; Diah, S.; Deguchi, K.; Mori, H.; Masaki, T.; Nakano, D.; Kobori, H.; Nishiyama, A. Roles of Na+/H+ exchanger type 1 and intracellular pH in angiotensin II-induced reactive oxygen species generation and podocyte apoptosis. J. Pharmacol. Sci. 2013, 122, 176–183. [Google Scholar] [CrossRef] [Green Version]
- Nagase, M.; Fujita, T. Role of Rac1-mineralocorticoid-receptor signalling in renal and cardiac disease. Nat. Rev. Nephrol. 2013, 9, 86–98. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.Y.; Dryer, S.E. RAGE and αVβ3-integrin are essential for suPAR signaling in podocytes. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 166186. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Sheng, X.-X.; Zhang, H.-J. DUSP26 regulates podocyte oxidative stress and fibrosis in a mouse model with diabetic nephropathy through the mediation of ROS. Biochem. Biophys. Res. Commun. 2019, 515, 410–416. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Liu, W.; Xiao, J.; Zhang, Y.; Chen, Y.; Luo, C.; Huang, Q.; Peng, F.; Gong, W.; Li, S.; et al. FOXO3a accumulation and activation accelerate oxidative stress-induced podocyte injury. FASEB J. 2020, 34, 13300–13316. [Google Scholar] [CrossRef]
- Lan, X.; Kumar, V.; Jha, A.; Aslam, R.; Wang, H.; Chen, K.; Yu, Y.; He, W.; Chen, F.; Luo, H.; et al. EDA2R mediates podocyte injury in high glucose milieu. Biochimie 2020, 174, 74–83. [Google Scholar] [CrossRef]
- Chen, Z.; Wan, X.; Hou, Q.; Shi, S.; Wang, L.; Chen, P.; Zhu, X.; Zeng, C.; Qin, W.; Zhou, W.; et al. GADD45B mediates podocyte injury in zebrafish by activating the ROS-GADD45B-p38 pathway. Cell Death Dis. 2016, 7, e2068. [Google Scholar] [CrossRef] [Green Version]
- Jennette, J.C.; Nachman, P.H. ANCA Glomerulonephritis and Vasculitis. Clin. J. Am. Soc. Nephrol. 2017, 12, 1680–1691. [Google Scholar] [CrossRef]
- Cerullo, D.; Rottoli, D.; Corna, D.; Rizzo, P.; Abbate, M.; Macconi, D.; Benigni, A.; Remuzzi, G.; Zoja, C. Characterization of a Rat Model of Myeloperoxidase-Anti-Neutrophil Cytoplasmic Antibody-Associated Crescentic Glomerulonephritis. Nephron 2021, 145, 428–444. [Google Scholar] [CrossRef]
- Shankland, S.J.; Smeets, B.; Pippin, J.W.; Moeller, M.J. The emergence of the glomerular parietal epithelial cell. Nat. Rev. Nephrol. 2014, 10, 158–173. [Google Scholar] [CrossRef]
- Guettier, C.; Nochy, D.; Jacquot, C.; Mandet, C.; Camilleri, J.P.; Bariety, J. Immunohistochemical demonstration of parietal epithelial cells and macrophages in human proliferative extra-capillary lesions. Virchows Arch. A Pathol. Anat. Histopathol. 1986, 409, 739–748. [Google Scholar] [CrossRef]
- Fogo, A.B.; Lusco, M.A.; Najafian, B.; Alpers, C.E. AJKD Atlas of Renal Pathology: Pauci-immune Necrotizing Crescentic Glomerulonephritis. Am. J. Kidney Dis. 2016, 68, e31–e32. [Google Scholar] [CrossRef] [PubMed]
- Puelles, V.G.; Fleck, D.; Ortz, L.; Papadouri, S.; Strieder, T.; Böhner, A.M.C.; van der Wolde, J.W.; Vogt, M.; Saritas, T.; Kuppe, C.; et al. Novel 3D analysis using optical tissue clearing documents the evolution of murine rapidly progressive glomerulonephritis. Kidney Int. 2019, 96, 505–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anguiano, L.; Kain, R.; Anders, H.-J. The glomerular crescent: Triggers, evolution, resolution, and implications for therapy. Curr. Opin. Nephrol. Hypertens. 2020, 29, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Eymael, J.; Sharma, S.; Loeven, M.A.; Wetzels, J.F.; Mooren, F.; Florquin, S.; Deegens, J.K.; Willemsen, B.K.; Sharma, V.; van Kuppevelt, T.H.; et al. CD44 is required for the pathogenesis of experimental crescentic glomerulonephritis and collapsing focal segmental glomerulosclerosis. Kidney Int. 2018, 93, 626–642. [Google Scholar] [CrossRef] [PubMed]
- Lazareth, H.; Henique, C.; Lenoir, O.; Puelles, V.G.; Flamant, M.; Bollée, G.; Fligny, C.; Camus, M.; Guyonnet, L.; Millien, C.; et al. The tetraspanin CD9 controls migration and proliferation of parietal epithelial cells and glomerular disease progression. Nat. Commun. 2019, 10, 3303. [Google Scholar] [CrossRef]
- Kuppe, C.; van Roeyen, C.; Leuchtle, K.; Kabgani, N.; Vogt, M.; Van Zandvoort, M.; Smeets, B.; Floege, J.; Gröne, H.-J.; Moeller, M.J. Investigations of Glucocorticoid Action in GN. J. Am. Soc. Nephrol. 2017, 28, 1408–1420. [Google Scholar] [CrossRef] [Green Version]
- Bollée, G.; Flamant, M.; Schordan, S.; Fligny, C.; Rumpel, E.; Milon, M.; Schordan, E.; Sabaa, N.; Vandermeersch, S.; Galaup, A.; et al. Epidermal growth factor receptor promotes glomerular injury and renal failure in rapidly progressive crescentic glomerulonephritis. Nat. Med. 2011, 17, 1242–1250. [Google Scholar] [CrossRef] [Green Version]
- D’Agati, V.D.; Kaskel, F.J.; Falk, R.J. Focal segmental glomerulosclerosis. N. Engl. J. Med. 2011, 365, 2398–2411. [Google Scholar] [CrossRef] [Green Version]
- Kriz, W.; Hosser, H.; Hähnel, B.; Simons, J.L.; Provoost, A.P. Development of vascular pole-associated glomerulosclerosis in the Fawn-hooded rat. J. Am. Soc. Nephrol. 1998, 9, 381–396. [Google Scholar] [CrossRef]
- Nagata, M.; Kobayashi, N.; Hara, S. Focal segmental glomerulosclerosis; why does it occur segmentally? Pflug. Arch. 2017, 469, 983–988. [Google Scholar] [CrossRef]
- Donzella, G.A.; Schols, D.; Lin, S.W.; Esté, J.A.; Nagashima, K.A.; Maddon, P.J.; Allaway, G.P.; Sakmar, T.P.; Henson, G.; De Clercq, E.; et al. AMD3100, a small molecule inhibitor of HIV-1 entry via the CXCR4 co-receptor. Nat. Med. 1998, 4, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.; Cordes, K.; Li, L.; Hood, L.; Couser, W.G.; Shankland, S.J.; Igarashi, P. Hematopoietic stem cells contribute to the regeneration of renal tubules after renal ischemia-reperfusion injury in mice. J. Am. Soc. Nephrol. 2003, 14, 1188–1199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Cohen, A.; Hudson, T.E.; Motlagh, D.; Amrani, D.L.; Duffield, J.S. Mobilized human hematopoietic stem/progenitor cells promote kidney repair after ischemia/reperfusion injury. Circulation 2010, 121, 2211–2220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Kong, Y.; Xie, C.; Zhou, L. Stem/progenitor cell in kidney: Characteristics, homing, coordination, and maintenance. Stem Cell Res. Ther. 2021, 12, 197. [Google Scholar] [CrossRef]
- Gupta, S.; Verfaillie, C.; Chmielewski, D.; Kren, S.; Eidman, K.; Connaire, J.; Heremans, Y.; Lund, T.; Blackstad, M.; Jiang, Y.; et al. Isolation and characterization of kidney-derived stem cells. J. Am. Soc. Nephrol. 2006, 17, 3028–3040. [Google Scholar] [CrossRef] [Green Version]
- Sagrinati, C.; Netti, G.S.; Mazzinghi, B.; Lazzeri, E.; Liotta, F.; Frosali, F.; Ronconi, E.; Meini, C.; Gacci, M.; Squecco, R.; et al. Isolation and characterization of multipotent progenitor cells from the Bowman’s capsule of adult human kidneys. J. Am. Soc. Nephrol. 2006, 17, 2443–2456. [Google Scholar] [CrossRef] [Green Version]
- Benigni, A.; Morigi, M.; Rizzo, P.; Gagliardini, E.; Rota, C.; Abbate, M.; Ghezzi, S.; Remuzzi, A.; Remuzzi, G. Inhibiting angiotensin-converting enzyme promotes renal repair by limiting progenitor cell proliferation and restoring the glomerular architecture. Am. J. Pathol. 2011, 179, 628–638. [Google Scholar] [CrossRef]
- Zhang, J.; Pippin, J.W.; Vaughan, M.R.; Krofft, R.D.; Taniguchi, Y.; Romagnani, P.; Nelson, P.J.; Liu, Z.-H.; Shankland, S.J. Retinoids augment the expression of podocyte proteins by glomerular parietal epithelial cells in experimental glomerular disease. Nephron. Exp. Nephrol. 2012, 121, e23–e37. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Pippin, J.W.; Krofft, R.D.; Naito, S.; Liu, Z.-H.; Shankland, S.J. Podocyte repopulation by renal progenitor cells following glucocorticoids treatment in experimental FSGS. Am. J. Physiol. Renal Physiol. 2013, 304, F1375–F1389. [Google Scholar] [CrossRef]
- Shankland, S.J.; Pippin, J.W.; Duffield, J.S. Progenitor cells and podocyte regeneration. Semin. Nephrol. 2014, 34, 418–428. [Google Scholar] [CrossRef] [Green Version]
- Romagnani, P.; Lasagni, L.; Remuzzi, G. Renal progenitors: An evolutionary conserved strategy for kidney regeneration. Nat. Rev. Nephrol. 2013, 9, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Bariety, J.; Mandet, C.; Hill, G.S.; Bruneval, P. Parietal podocytes in normal human glomeruli. J. Am. Soc. Nephrol. 2006, 17, 2770–2780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peired, A.; Angelotti, M.L.; Ronconi, E.; la Marca, G.; Mazzinghi, B.; Sisti, A.; Lombardi, D.; Giocaliere, E.; Della Bona, M.; Villanelli, F.; et al. Proteinuria impairs podocyte regeneration by sequestering retinoic acid. J. Am. Soc. Nephrol. 2013, 24, 1756–1768. [Google Scholar] [CrossRef] [Green Version]
- Angelotti, M.L.; Ronconi, E.; Ballerini, L.; Peired, A.; Mazzinghi, B.; Sagrinati, C.; Parente, E.; Gacci, M.; Carini, M.; Rotondi, M.; et al. Characterization of renal progenitors committed toward tubular lineage and their regenerative potential in renal tubular injury. Stem Cells 2012, 30, 1714–1725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ronconi, E.; Sagrinati, C.; Angelotti, M.L.; Lazzeri, E.; Mazzinghi, B.; Ballerini, L.; Parente, E.; Becherucci, F.; Gacci, M.; Carini, M.; et al. Regeneration of glomerular podocytes by human renal progenitors. J. Am. Soc. Nephrol. 2009, 20, 322–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romagnani, P. Toward the identification of a “renopoietic system”? Stem Cells 2009, 27, 2247–2253. [Google Scholar] [CrossRef] [Green Version]
- Lasagni, L.; Romagnani, P. Glomerular epithelial stem cells: The good, the bad, and the ugly. J. Am. Soc. Nephrol. 2010, 21, 1612–1619. [Google Scholar] [CrossRef] [Green Version]
- Andeen, N.K.; Nguyen, T.Q.; Steegh, F.; Hudkins, K.L.; Najafian, B.; Alpers, C.E. The phenotypes of podocytes and parietal epithelial cells may overlap in diabetic nephropathy. Kidney Int. 2015, 88, 1099–1107. [Google Scholar] [CrossRef] [Green Version]
- Eng, D.G.; Sunseri, M.W.; Kaverina, N.V.; Roeder, S.S.; Pippin, J.W.; Shankland, S.J. Glomerular parietal epithelial cells contribute to adult podocyte regeneration in experimental focal segmental glomerulosclerosis. Kidney Int. 2015, 88, 999–1012. [Google Scholar] [CrossRef] [Green Version]
- Berger, K.; Schulte, K.; Boor, P.; Kuppe, C.; van Kuppevelt, T.H.; Floege, J.; Smeets, B.; Moeller, M.J. The regenerative potential of parietal epithelial cells in adult mice. J. Am. Soc. Nephrol. 2014, 25, 693–705. [Google Scholar] [CrossRef] [Green Version]
- Poulsom, R.; Little, M.H. Parietal epithelial cells regenerate podocytes. J. Am. Soc. Nephrol. 2009, 20, 231–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shankland, S.J.; Anders, H.-J.; Romagnani, P. Glomerular parietal epithelial cells in kidney physiology, pathology, and repair. Curr. Opin. Nephrol. Hypertens. 2013, 22, 302–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lasagni, L.; Ballerini, L.; Angelotti, M.L.; Parente, E.; Sagrinati, C.; Mazzinghi, B.; Peired, A.; Ronconi, E.; Becherucci, F.; Bani, D.; et al. Notch activation differentially regulates renal progenitors proliferation and differentiation toward the podocyte lineage in glomerular disorders. Stem Cells 2010, 28, 1674–1685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darisipudi, M.N.; Kulkarni, O.P.; Sayyed, S.G.; Ryu, M.; Migliorini, A.; Sagrinati, C.; Parente, E.; Vater, A.; Eulberg, D.; Klussmann, S.; et al. Dual blockade of the homeostatic chemokine CXCL12 and the proinflammatory chemokine CCL2 has additive protective effects on diabetic kidney disease. Am. J. Pathol. 2011, 179, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Smeets, B.; Uhlig, S.; Fuss, A.; Mooren, F.; Wetzels, J.F.M.; Floege, J.; Moeller, M.J. Tracing the origin of glomerular extracapillary lesions from parietal epithelial cells. J. Am. Soc. Nephrol. 2009, 20, 2604–2615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Appel, D.; Kershaw, D.B.; Smeets, B.; Yuan, G.; Fuss, A.; Frye, B.; Elger, M.; Kriz, W.; Floege, J.; Moeller, M.J. Recruitment of podocytes from glomerular parietal epithelial cells. J. Am. Soc. Nephrol. 2009, 20, 333–343. [Google Scholar] [CrossRef] [Green Version]
- Smeets, B.; Angelotti, M.L.; Rizzo, P.; Dijkman, H.; Lazzeri, E.; Mooren, F.; Ballerini, L.; Parente, E.; Sagrinati, C.; Mazzinghi, B.; et al. Renal progenitor cells contribute to hyperplastic lesions of podocytopathies and crescentic glomerulonephritis. J. Am. Soc. Nephrol. 2009, 20, 2593–2603. [Google Scholar] [CrossRef] [Green Version]
- Lindgren, D.; Boström, A.-K.; Nilsson, K.; Hansson, J.; Sjölund, J.; Möller, C.; Jirström, K.; Nilsson, E.; Landberg, G.; Axelson, H.; et al. Isolation and characterization of progenitor-like cells from human renal proximal tubules. Am. J. Pathol. 2011, 178, 828–837. [Google Scholar] [CrossRef] [Green Version]
- Smeets, B.; Boor, P.; Dijkman, H.; Sharma, S.V.; Jirak, P.; Mooren, F.; Berger, K.; Bornemann, J.; Gelman, I.H.; Floege, J.; et al. Proximal tubular cells contain a phenotypically distinct, scattered cell population involved in tubular regeneration. J. Pathol. 2013, 229, 645–659. [Google Scholar] [CrossRef]
- Kang, H.M.; Huang, S.; Reidy, K.; Han, S.H.; Chinga, F.; Susztak, K. Sox9-Positive Progenitor Cells Play a Key Role in Renal Tubule Epithelial Regeneration in Mice. Cell Rep. 2016, 14, 861–871. [Google Scholar] [CrossRef] [Green Version]
- Maeshima, A.; Yamashita, S.; Nojima, Y. Identification of Renal Progenitor-Like Tubular Cells that Participate in the Regeneration Processes of the Kidney. J. Am. Soc. Nephrol. 2003, 14, 3138–3146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osafune, K. iPSC technology-based regenerative medicine for kidney diseases. Clin. Exp. Nephrol. 2021, 25, 574–584. [Google Scholar] [CrossRef] [PubMed]
- Rota, C.; Morigi, M.; Imberti, B. Stem Cell Therapies in Kidney Diseases: Progress and Challenges. Int. J. Mol. Sci. 2019, 20, 2790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Storti, G.; Favi, E.; Albanesi, F.; Kim, B.-S.; Cervelli, V. Adipose-Derived Stem/Stromal Cells in Kidney Transplantation: Status Quo and Future Perspectives. Int. J. Mol. Sci. 2021, 22, 11188. [Google Scholar] [CrossRef] [PubMed]
- Toyohara, T.; Mae, S.-I.; Sueta, S.-I.; Inoue, T.; Yamagishi, Y.; Kawamoto, T.; Kasahara, T.; Hoshina, A.; Toyoda, T.; Tanaka, H.; et al. Cell Therapy Using Human Induced Pluripotent Stem Cell-Derived Renal Progenitors Ameliorates Acute Kidney Injury in Mice. Stem Cells Transl. Med. 2015, 4, 980–992. [Google Scholar] [CrossRef]
- Imberti, B.; Tomasoni, S.; Ciampi, O.; Pezzotta, A.; Derosas, M.; Xinaris, C.; Rizzo, P.; Papadimou, E.; Novelli, R.; Benigni, A.; et al. Renal progenitors derived from human iPSCs engraft and restore function in a mouse model of acute kidney injury. Sci. Rep. 2015, 5, 8826. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Cheng, F.; Pan, S.; Liu, Z. Stem cells: A potential treatment option for kidney diseases. Stem Cell Res. Ther. 2020, 11, 249. [Google Scholar] [CrossRef]
- Ranghino, A.; Bruno, S.; Bussolati, B.; Moggio, A.; Dimuccio, V.; Tapparo, M.; Biancone, L.; Gontero, P.; Frea, B.; Camussi, G. The effects of glomerular and tubular renal progenitors and derived extracellular vesicles on recovery from acute kidney injury. Stem Cell Res. Ther. 2017, 8, 24. [Google Scholar] [CrossRef] [Green Version]
- Tkach, M.; Théry, C. Communication by Extracellular Vesicles: Where We Are and Where We Need to Go. Cell 2016, 164, 1226–1232. [Google Scholar] [CrossRef] [Green Version]
- Yáñez-Mó, M.; Siljander, P.R.-M.; Andreu, Z.; Bedina Zavec, A.; Borràs, F.E.; Buzas, E.I.; Buzas, K.; Casal, E.; Cappello, F.; Carvalho, J.; et al. Biological properties of extracellular vesicles and their physiological functions. J. Extracell. Vesicles 2015, 4, 27066. [Google Scholar] [CrossRef] [Green Version]
- Tögel, F.E.; Westenfelder, C. Kidney protection and regeneration following acute injury: Progress through stem cell therapy. Am. J. Kidney Dis. 2012, 60, 1012–1022. [Google Scholar] [CrossRef] [PubMed]
- Swaminathan, M.; Stafford-Smith, M.; Chertow, G.M.; Warnock, D.G.; Paragamian, V.; Brenner, R.M.; Lellouche, F.; Fox-Robichaud, A.; Atta, M.G.; Melby, S.; et al. Allogeneic Mesenchymal Stem Cells for Treatment of AKI after Cardiac Surgery. J. Am. Soc. Nephrol. 2018, 29, 260–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Packham, D.K.; Fraser, I.R.; Kerr, P.G.; Segal, K.R. Allogeneic Mesenchymal Precursor Cells (MPC) in Diabetic Nephropathy: A Randomized, Placebo-controlled, Dose Escalation Study. EBioMedicine 2016, 12, 263–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saad, A.; Dietz, A.B.; Herrmann, S.M.S.; Hickson, L.J.; Glockner, J.F.; McKusick, M.A.; Misra, S.; Bjarnason, H.; Armstrong, A.S.; Gastineau, D.A.; et al. Autologous Mesenchymal Stem Cells Increase Cortical Perfusion in Renovascular Disease. J. Am. Soc. Nephrol. 2017, 28, 2777–2785. [Google Scholar] [CrossRef] [Green Version]
- Deng, D.; Zhang, P.; Guo, Y.; Lim, T.O. A randomised double-blind, placebo-controlled trial of allogeneic umbilical cord-derived mesenchymal stem cell for lupus nephritis. Ann. Rheum. Dis. 2017, 76, 1436–1439. [Google Scholar] [CrossRef]
- Reinders, M.E.J.; de Fijter, J.W.; Roelofs, H.; Bajema, I.M.; de Vries, D.K.; Schaapherder, A.F.; Claas, F.H.J.; van Miert, P.P.M.C.; Roelen, D.L.; van Kooten, C.; et al. Autologous bone marrow-derived mesenchymal stromal cells for the treatment of allograft rejection after renal transplantation: Results of a phase I study. Stem Cells Transl. Med. 2013, 2, 107–111. [Google Scholar] [CrossRef]
- Pan, G.-H.; Chen, Z.; Xu, L.; Zhu, J.-H.; Xiang, P.; Ma, J.-J.; Peng, Y.-W.; Li, G.-H.; Chen, X.-Y.; Fang, J.-L.; et al. Low-dose tacrolimus combined with donor-derived mesenchymal stem cells after renal transplantation: A prospective, non-randomized study. Oncotarget 2016, 7, 12089–12101. [Google Scholar] [CrossRef] [Green Version]
- Erpicum, P.; Weekers, L.; Detry, O.; Bonvoisin, C.; Delbouille, M.-H.; Grégoire, C.; Baudoux, E.; Briquet, A.; Lechanteur, C.; Maggipinto, G.; et al. Infusion of third-party mesenchymal stromal cells after kidney transplantation: A phase I-II, open-label, clinical study. Kidney Int. 2019, 95, 693–707. [Google Scholar] [CrossRef]
- Birtwistle, L.; Chen, X.-M.; Pollock, C. Mesenchymal Stem Cell-Derived Extracellular Vesicles to the Rescue of Renal Injury. Int. J. Mol. Sci. 2021, 22, 6596. [Google Scholar] [CrossRef]
- Vizoso, F.J.; Eiro, N.; Cid, S.; Schneider, J.; Perez-Fernandez, R. Mesenchymal Stem Cell Secretome: Toward Cell-Free Therapeutic Strategies in Regenerative Medicine. Int. J. Mol. Sci. 2017, 18, 1852. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.; Liu, S.; Wang, C.; Zhang, C.; Wen, Y.; Zhang, K.; Chen, S.; Huang, H.; Liu, Y.; Wu, L.; et al. Embryonic stem cell-derived extracellular vesicles promote the recovery of kidney injury. Stem Cell Res. Ther. 2021, 12, 379. [Google Scholar] [CrossRef] [PubMed]
- Grange, C.; Tapparo, M.; Bruno, S.; Chatterjee, D.; Quesenberry, P.J.; Tetta, C.; Camussi, G. Biodistribution of mesenchymal stem cell-derived extracellular vesicles in a model of acute kidney injury monitored by optical imaging. Int. J. Mol. Med. 2014, 33, 1055–1063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nassar, W.; El-Ansary, M.; Sabry, D.; Mostafa, M.A.; Fayad, T.; Kotb, E.; Temraz, M.; Saad, A.-N.; Essa, W.; Adel, H. Umbilical cord mesenchymal stem cells derived extracellular vesicles can safely ameliorate the progression of chronic kidney diseases. Biomater. Res. 2016, 20, 21. [Google Scholar] [CrossRef] [Green Version]
- Cao, Q.; Wang, Y.; Zheng, D.; Sun, Y.; Wang, C.; Wang, X.M.; Lee, V.W.S.; Wang, Y.; Zheng, G.; Tan, T.K.; et al. Failed renoprotection by alternatively activated bone marrow macrophages is due to a proliferation-dependent phenotype switch in vivo. Kidney Int. 2014, 85, 794–806. [Google Scholar] [CrossRef] [PubMed]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Harris, D.C.H. Macrophages in Renal Disease. J. Am. Soc. Nephrol. 2011, 22, 21–27. [Google Scholar] [CrossRef] [Green Version]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef]
- Baek, J.-H.; Zeng, R.; Weinmann-Menke, J.; Valerius, M.T.; Wada, Y.; Ajay, A.K.; Colonna, M.; Kelley, V.R. IL-34 mediates acute kidney injury and worsens subsequent chronic kidney disease. J. Clin. Investig. 2015, 125, 3198–3214. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.-L.; Li, B.; Rao, S.; Yeo, E.-J.; Hudson, T.E.; Nowlin, B.T.; Pei, H.; Chen, L.; Zheng, J.J.; Carroll, T.J.; et al. Macrophage Wnt7b is critical for kidney repair and regeneration. Proc. Natl. Acad. Sci. USA 2010, 107, 4194–4199. [Google Scholar] [CrossRef] [Green Version]
- Arai, S.; Kitada, K.; Yamazaki, T.; Takai, R.; Zhang, X.; Tsugawa, Y.; Sugisawa, R.; Matsumoto, A.; Mori, M.; Yoshihara, Y.; et al. Apoptosis inhibitor of macrophage protein enhances intraluminal debris clearance and ameliorates acute kidney injury in mice. Nat. Med. 2016, 22, 183–193. [Google Scholar] [CrossRef]
- Sola, A.; Weigert, A.; Jung, M.; Vinuesa, E.; Brecht, K.; Weis, N.; Brüne, B.; Borregaard, N.; Hotter, G. Sphingosine-1-phosphate signalling induces the production of Lcn-2 by macrophages to promote kidney regeneration. J. Pathol. 2011, 225, 597–608. [Google Scholar] [CrossRef] [PubMed]
- Lv, J.; Chen, J.; Wang, M.; Yan, F. Klotho alleviates indoxyl sulfate-induced heart failure and kidney damage by promoting M2 macrophage polarization. Aging 2020, 12, 9139–9150. [Google Scholar] [CrossRef] [PubMed]
- Barrera-Chimal, J.; Estrela, G.R.; Lechner, S.M.; Giraud, S.; El Moghrabi, S.; Kaaki, S.; Kolkhof, P.; Hauet, T.; Jaisser, F. The myeloid mineralocorticoid receptor controls inflammatory and fibrotic responses after renal injury via macrophage interleukin-4 receptor signaling. Kidney Int. 2018, 93, 1344–1355. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Fernández, B.; Valiño-Rivas, L.; Sánchez-Niño, M.D.; Ortiz, A. Albuminuria Downregulation of the Anti-Aging Factor Klotho: The Missing Link Potentially Explaining the Association of Pathological Albuminuria with Premature Death. Adv. Ther. 2020, 37, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Fernandez, B.; Izquierdo, M.C.; Valiño-Rivas, L.; Nastou, D.; Sanz, A.B.; Ortiz, A.; Sanchez-Niño, M.D. Albumin downregulates Klotho in tubular cells. Nephrol. Dial. Transpl. 2018, 33, 1712–1722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mertens, C.; Kuchler, L.; Sola, A.; Guiteras, R.; Grein, S.; Brüne, B.; von Knethen, A.; Jung, M. Macrophage-Derived Iron-Bound Lipocalin-2 Correlates with Renal Recovery Markers Following Sepsis-Induced Kidney Damage. Int. J. Mol. Sci. 2020, 21, 7527. [Google Scholar] [CrossRef]
- Jung, M.; Brüne, B.; Hotter, G.; Sola, A. Macrophage-derived Lipocalin-2 contributes to ischemic resistance mechanisms by protecting from renal injury. Sci. Rep. 2016, 6, 21950. [Google Scholar] [CrossRef]
- Flaquer, M.; Franquesa, M.; Vidal, A.; Bolaños, N.; Torras, J.; Lloberas, N.; Herrero-Fresneda, I.; Grinyó, J.M.; Cruzado, J.M. Hepatocyte growth factor gene therapy enhances infiltration of macrophages and may induce kidney repair in db/db mice as a model of diabetes. Diabetologia 2012, 55, 2059–2068. [Google Scholar] [CrossRef] [Green Version]
- Guiteras, R.; Sola, A.; Flaquer, M.; Hotter, G.; Torras, J.; Grinyó, J.M.; Cruzado, J.M. Macrophage Overexpressing NGAL Ameliorated Kidney Fibrosis in the UUO Mice Model. Cell. Physiol. Biochem. 2017, 42, 1945–1960. [Google Scholar] [CrossRef]
- Guiteras, R.; Sola, A.; Flaquer, M.; Manonelles, A.; Hotter, G.; Cruzado, J.M. Exploring macrophage cell therapy on Diabetic Kidney Disease. J. Cell. Mol. Med. 2019, 23, 841–851. [Google Scholar] [CrossRef]
- Jung, M.; Brüne, B.; von Knethen, A.; Guiteras, R.; Cruzado, J.M.; Hotter, G.; Sola, A. Lipocalin-2 abrogates epithelial cell cycle arrest by PPARγ inhibition. Lab. Investig. 2018, 98, 1408–1422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.; Fessler, M.B.; Qu, P.; Heymann, J.; Kopp, J.B. Macrophage polarization in innate immune responses contributing to pathogenesis of chronic kidney disease. BMC Nephrol. 2020, 21, 270. [Google Scholar] [CrossRef]
- Meng, X.-M.; Tang, P.M.-K.; Li, J.; Lan, H.Y. Macrophage Phenotype in Kidney Injury and Repair. Kidney Dis. 2015, 1, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhou, L.; Liu, Y. Cellular Senescence in Kidney Fibrosis: Pathologic Significance and Therapeutic Strategies. Front. Pharmacol. 2020, 11, 601325. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, A.; Ziyadeh, F.N.; Neilson, E.G. Expression of apoptosis-regulatory genes in renal proximal tubular epithelial cells exposed to high ambient glucose and in diabetic kidneys. J. Investig. Med. 1997, 45, 50–56. [Google Scholar] [PubMed]
- Ortiz, A.; Lorz, C.; Catalán, M.P.; Danoff, T.M.; Yamasaki, Y.; Egido, J.; Neilson, E.G. Expression of apoptosis regulatory proteins in tubular epithelium stressed in culture or following acute renal failure. Kidney Int. 2000, 57, 969–981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nayman, A.H.; Siginc, H.; Zemheri, E.; Yencilek, F.; Yildirim, A.; Telci, D. Dual-Inhibition of mTOR and Bcl-2 Enhances the Anti-tumor Effect of Everolimus against Renal Cell Carcinoma In Vitro and In Vivo. J. Cancer 2019, 10, 1466–1478. [Google Scholar] [CrossRef] [Green Version]
- Chang, J.; Wang, Y.; Shao, L.; Laberge, R.-M.; Demaria, M.; Campisi, J.; Janakiraman, K.; Sharpless, N.E.; Ding, S.; Feng, W.; et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat. Med. 2016, 22, 78–83. [Google Scholar] [CrossRef] [Green Version]
- Mylonas, K.J.; O’Sullivan, E.D.; Humphries, D.; Baird, D.P.; Docherty, M.-H.; Neely, S.A.; Krimpenfort, P.J.; Melk, A.; Schmitt, R.; Ferreira-Gonzalez, S.; et al. Cellular senescence inhibits renal regeneration after injury in mice, with senolytic treatment promoting repair. Sci. Transl. Med. 2021, 13, eabb0203. [Google Scholar] [CrossRef]
- Kirkland, J.L.; Tchkonia, T. Senolytic drugs: From discovery to translation. J. Intern. Med. 2020, 288, 518–536. [Google Scholar] [CrossRef]
- Li, C.; Shen, Y.; Huang, L.; Liu, C.; Wang, J. Senolytic therapy ameliorates renal fibrosis postacute kidney injury by alleviating renal senescence. FASEB J. 2021, 35, e21229. [Google Scholar] [CrossRef] [PubMed]
- Hickson, L.J.; Langhi Prata, L.G.P.; Bobart, S.A.; Evans, T.K.; Giorgadze, N.; Hashmi, S.K.; Herrmann, S.M.; Jensen, M.D.; Jia, Q.; Jordan, K.L.; et al. Senolytics decrease senescent cells in humans: Preliminary report from a clinical trial of Dasatinib plus Quercetin in individuals with diabetic kidney disease. EBioMedicine 2019, 47, 446–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peired, A.J.; Lazzeri, E.; Guzzi, F.; Anders, H.-J.; Romagnani, P. From kidney injury to kidney cancer. Kidney Int. 2021, 100, 55–66. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rayego-Mateos, S.; Marquez-Expósito, L.; Rodrigues-Diez, R.; Sanz, A.B.; Guiteras, R.; Doladé, N.; Rubio-Soto, I.; Manonelles, A.; Codina, S.; Ortiz, A.; et al. Molecular Mechanisms of Kidney Injury and Repair. Int. J. Mol. Sci. 2022, 23, 1542. https://doi.org/10.3390/ijms23031542
Rayego-Mateos S, Marquez-Expósito L, Rodrigues-Diez R, Sanz AB, Guiteras R, Doladé N, Rubio-Soto I, Manonelles A, Codina S, Ortiz A, et al. Molecular Mechanisms of Kidney Injury and Repair. International Journal of Molecular Sciences. 2022; 23(3):1542. https://doi.org/10.3390/ijms23031542
Chicago/Turabian StyleRayego-Mateos, Sandra, Laura Marquez-Expósito, Raquel Rodrigues-Diez, Ana B. Sanz, Roser Guiteras, Nuria Doladé, Irene Rubio-Soto, Anna Manonelles, Sergi Codina, Alberto Ortiz, and et al. 2022. "Molecular Mechanisms of Kidney Injury and Repair" International Journal of Molecular Sciences 23, no. 3: 1542. https://doi.org/10.3390/ijms23031542