
ER Unfolded Protein Response in Liver In Vivo Is Characterized by Reduced, Not Increased, De Novo Lipogenesis and Cholesterol Synthesis Rates with Uptake of Fatty Acids from Adipose Tissue: Integrated Gene Expression, Translation Rates and Metabolic Fluxes

 , , ,
, , ,
Abstract
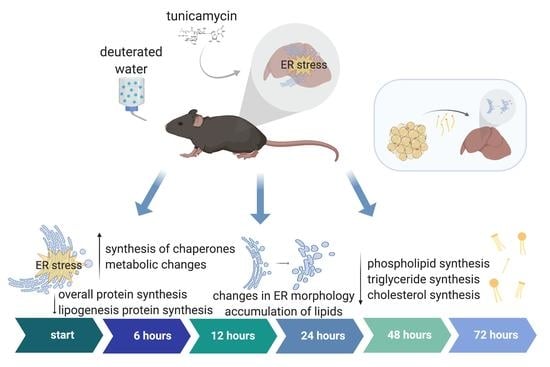
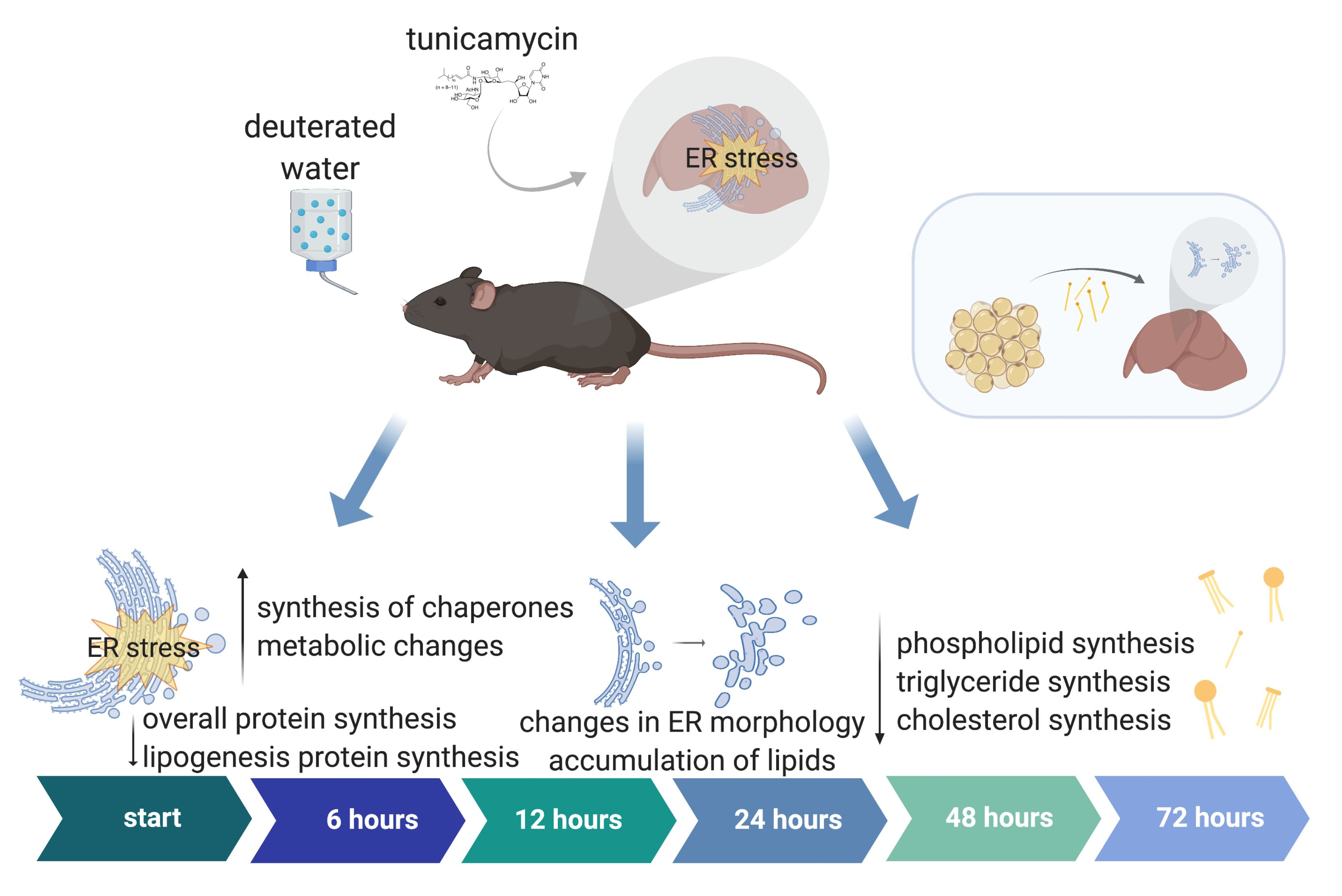
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Background
2. Results
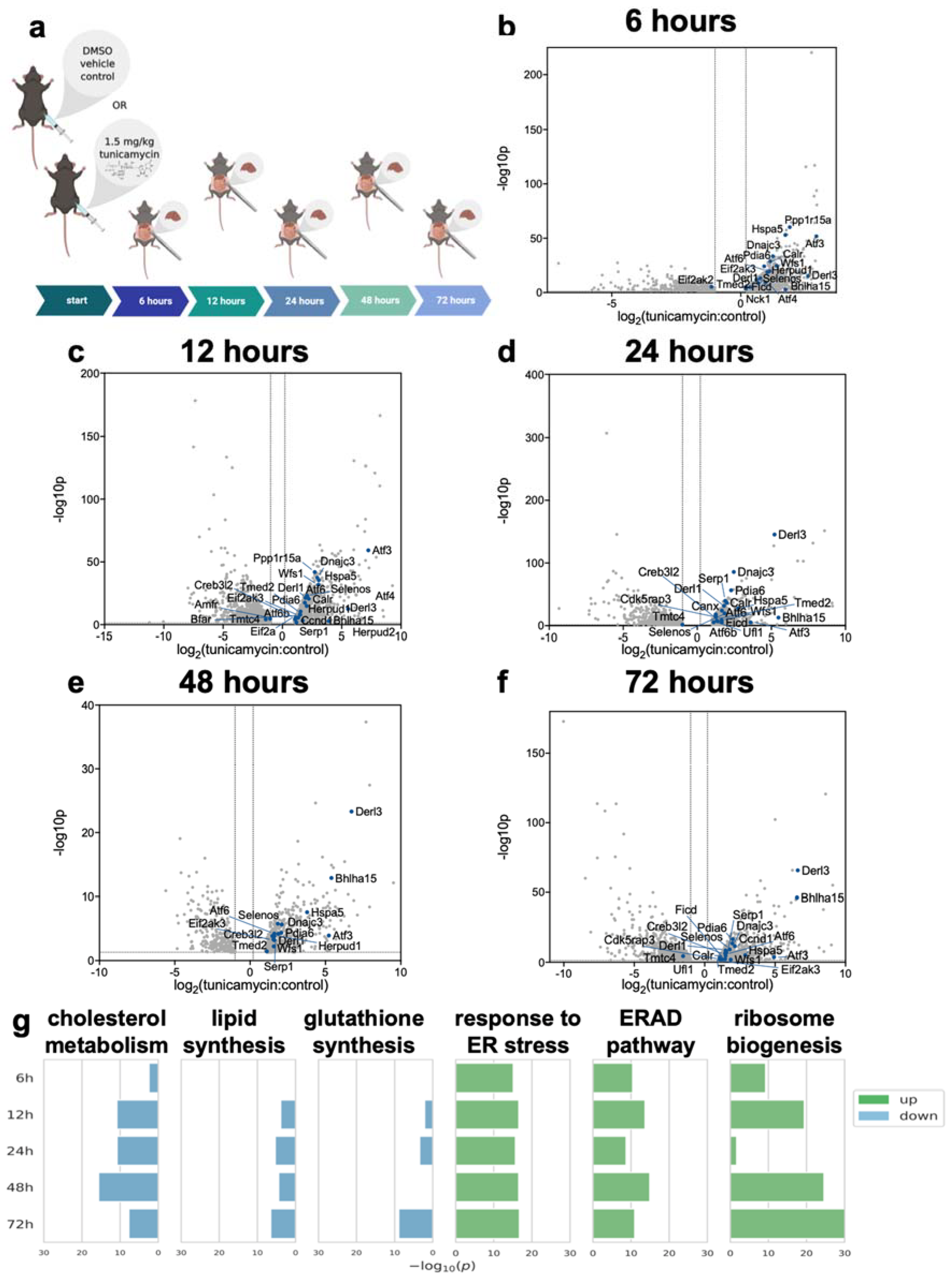
2.1. RNA-Seq of Mouse Liver under Induced UPRER Reveals Decreased Expression of Genes Involved in Lipid and Cholesterol Synthesis as Well as Many Downregulated and Some Upregulated Gene Ontologies
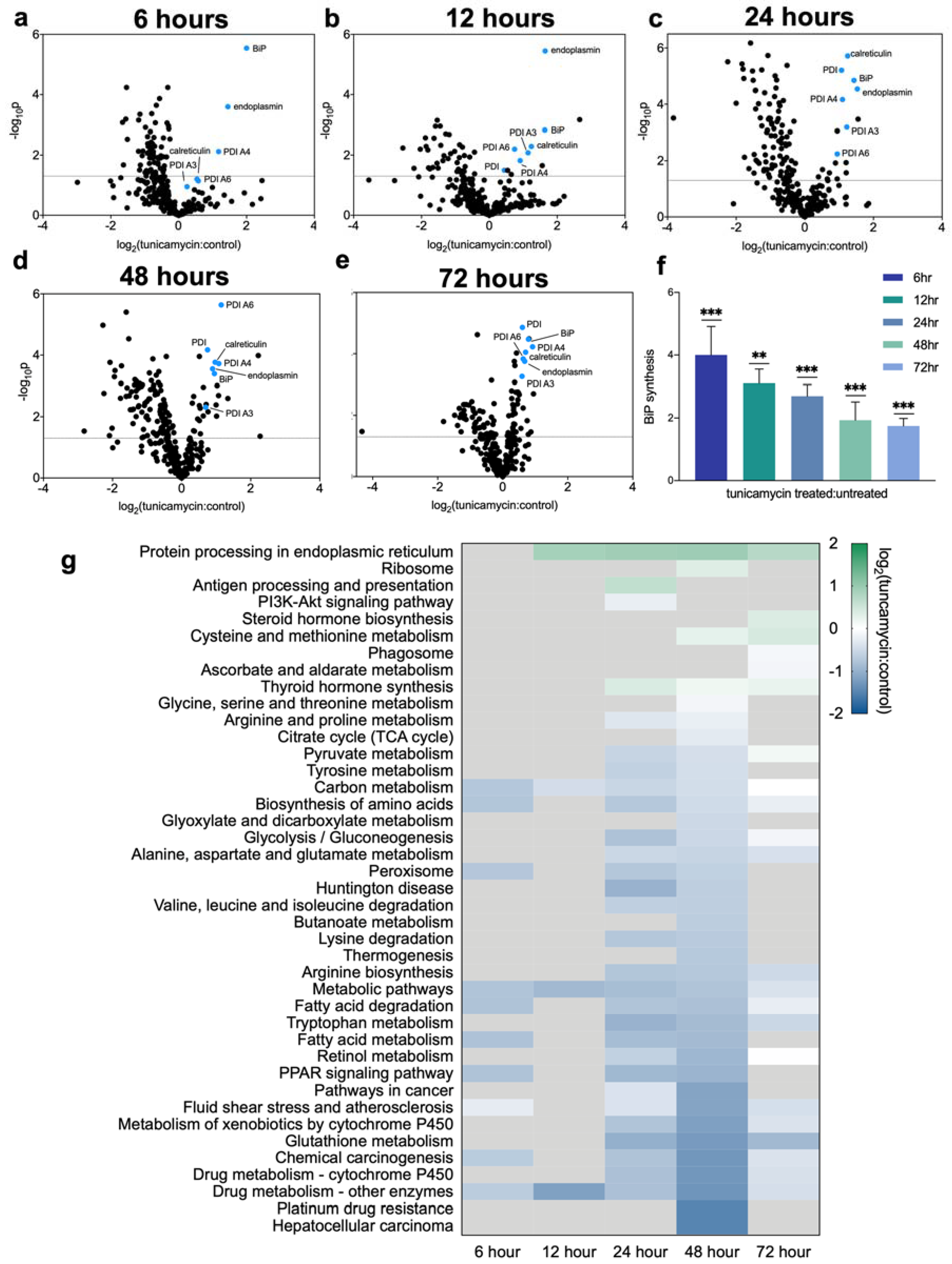
2.2. Dynamic Proteomic Measurements Reveal Decreased Global Protein Synthesis Rates, including Those Involved in Lipid Synthesis, but Not for Key UPRER Proteins
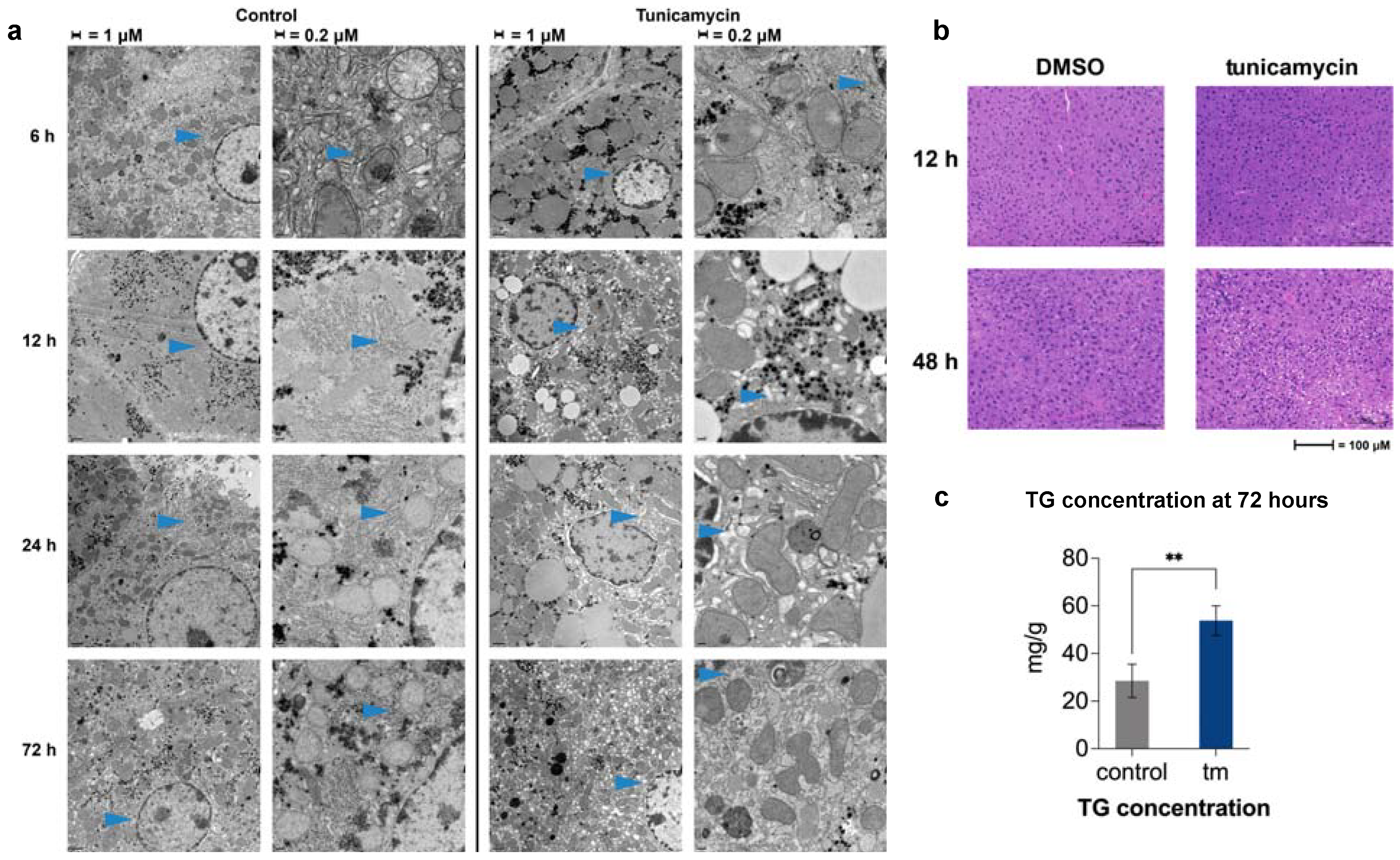
2.3. UPRER Induction Causes Lipid Accumulation by Histology
2.4. Changes in ER Morphology by Electron Microscopy
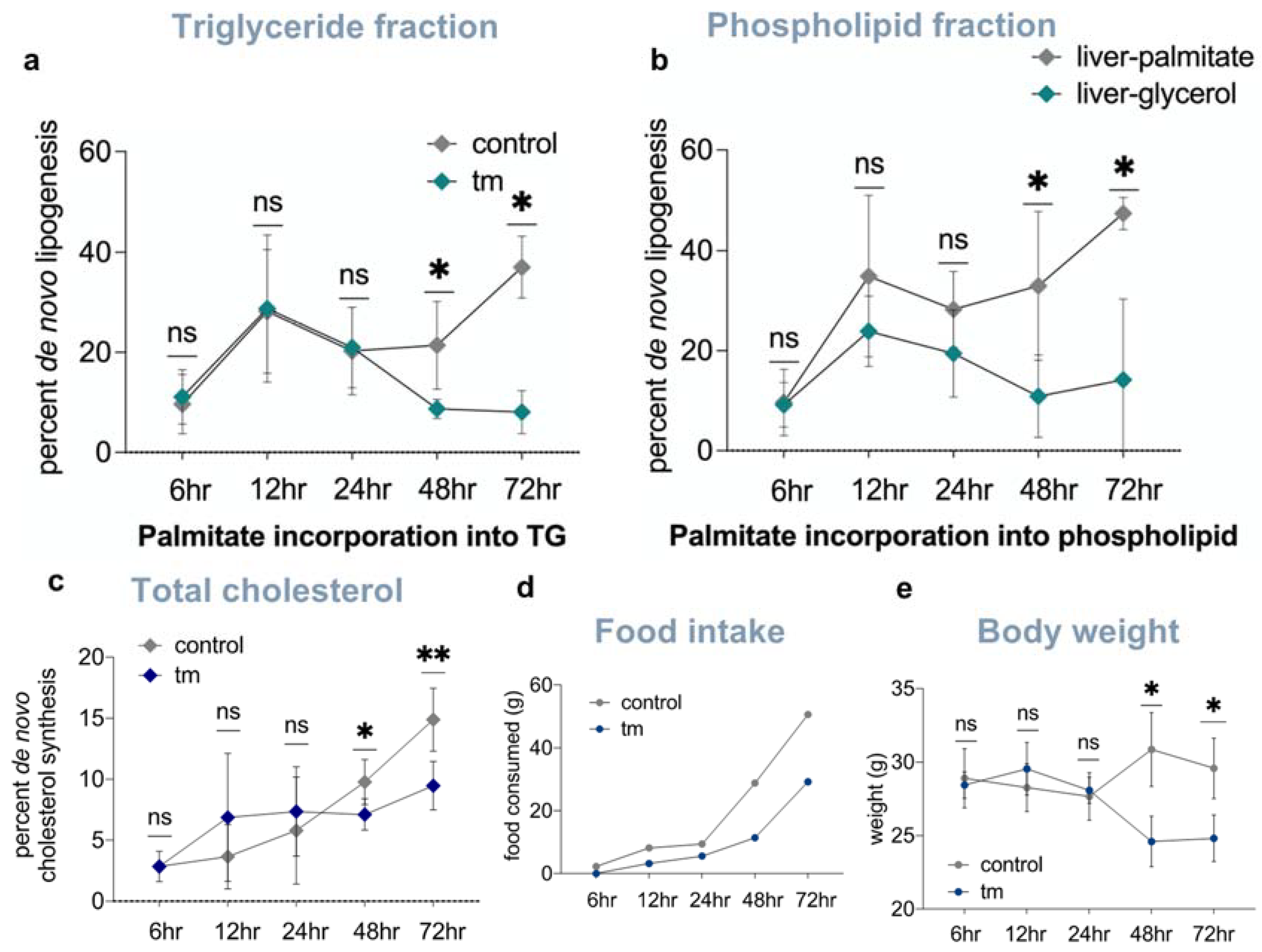
2.5. Lipid and Cholesterol Synthesis Rates Are Decreased Post-UPRER Induction
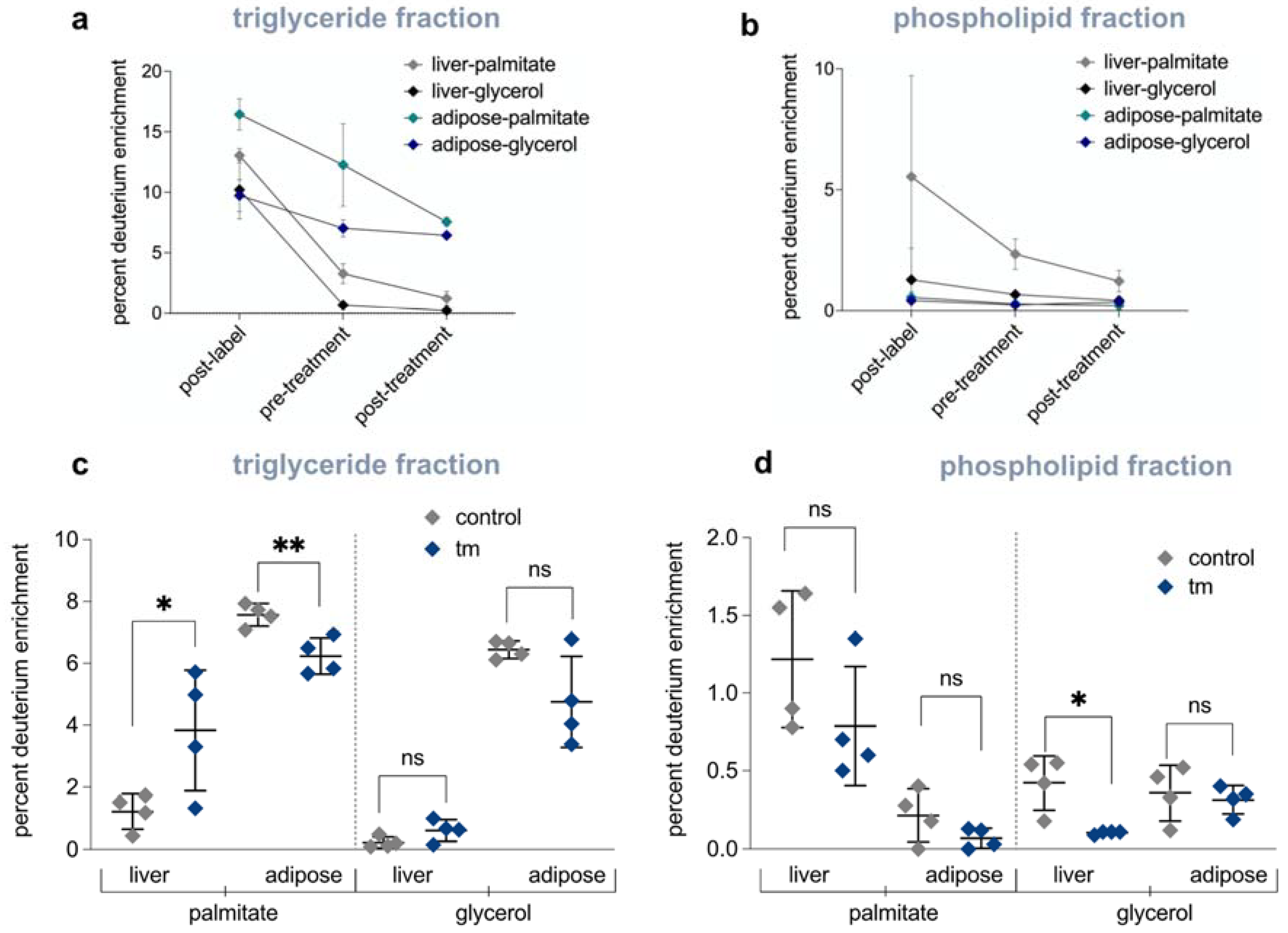
2.6. Mobilization of Fatty Acids from Adipose Tissue to Liver Is Increased under UPRER Conditions
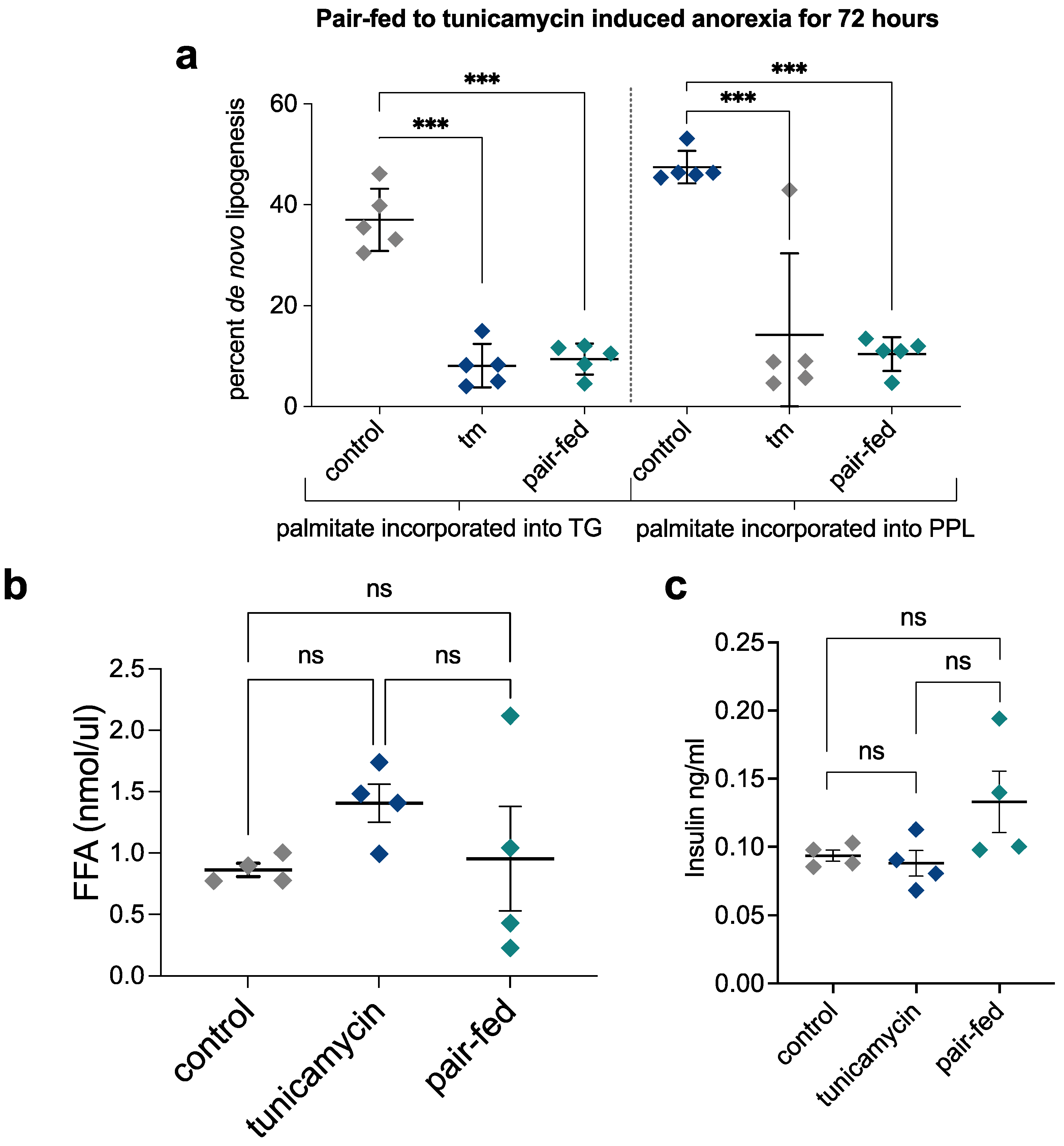
2.7. Pair-Feeding and Measurement of Plasma Insulin and Free Fatty Acid Concentrations
3. Discussion
4. Methods
4.1. Animals
4.2. Deuterated Water Labeling and Tunicamycin Treatment in Mice
4.3. Deuterated Water Labeling and Tunicamycin Treatment in Mice: Pre-Label of Adipose Tissue Triglycerides
4.4. Body Water Enrichment Analysis
4.5. Tissue Preparation for Liquid Chromatography-Mass Spectrometry (LC-MS)
4.6. Liquid Chromatography-Mass Spectrometry (LC-MS) Analysis
4.7. Calculation of Fractional Replacement (f) and Replacement Rate Constant (k) for Individual Proteins
4.8. Statistical Analysis
4.9. KEGG Pathway Analysis
4.10. Tissue Preparation for Gas Chromatography-Mass Spectrometry (GC-MS)
4.11. Gas Chromatography-Mass Spectrometry (GC-MS) Analysis
4.12. Calculation of De Novo Lipogenesis (DNL) and Cholesterol Synthesis
4.13. RNAseq
4.14. Electron Microscopy
4.15. Hematoxylin and Eosin Staining
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Frakes, A.E.; Dillin, A. The UPRER: Sensor and Coordinator of Organismal Homeostasis. Mol. Cell 2017, 66, 761–771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Kaufman, R.J. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature 2016, 529, 326–335. [Google Scholar] [CrossRef]
- Salvadó, L.; Palomer, X.; Barroso, E.; Vázquez-Carrera, M. Targeting endoplasmic reticulum stress in insulin resistance. Trends Endocrinol. Metab. 2015, 26, 438–448. [Google Scholar] [CrossRef]
- Fu, S.; Watkins, S.M.; Hotamisligil, G.S. The role of endoplasmic reticulum in hepatic lipid homeostasis and stress signaling. Cell Metab. 2012, 15, 623–634. [Google Scholar] [CrossRef] [Green Version]
- Cnop, M.; Foufelle, F.; Velloso, L.A. Endoplasmic reticulum stress, obesity and diabetes. Trends Mol. Med. 2012, 18, 59–68. [Google Scholar] [CrossRef]
- Rutkowski, D.T.; Wu, J.; Back, S.-H.; Callaghan, M.U.; Ferris, S.P.; Iqbal, J.; Clark, R.; Miao, H.; Hassler, J.R.; Fornek, J.; et al. UPR pathways combine to prevent hepatic steatosis caused by ER stress-mediated suppression of transcriptional master regulators. Dev. Cell 2008, 15, 829–840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rutkowski, D.T. Liver function and dysfunction—a unique window into the physiological reach of ER stress and the unfolded protein response. FEBS J. 2019, 286, 356–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hotamisligil, G.S. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 2010, 140, 900–917. [Google Scholar] [CrossRef] [Green Version]
- Bertolotti, A.; Zhang, Y.; Hendershot, L.M.; Harding, H.P.; Ron, D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2000, 2, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Walter, P.; Ron, D. The Unfolded Protein Response: From Stress Pathway to Homeostatic Regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [Green Version]
- Holmes, W.E.; Angel, T.E.; Li, K.W.; Hellerstein, M.K. Dynamic Proteomics: In Vivo Proteome-Wide Measurement of Protein Kinetics Using Metabolic Labeling. Methods Enzymol. 2015, 561, 219–276. [Google Scholar]
- Fang, D.-L.; Wan, Y.; Shen, W.; Cao, J.; Sun, Z.-X.; Yu, H.-H.; Zhang, Q.; Cheng, W.-H.; Chen, J.; Ning, B. Endoplasmic reticulum stress leads to lipid accumulation through upregulation of SREBP-1c in normal hepatic and hepatoma cells. Mol. Cell. Biochem. 2013, 381, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Kaufman, R.J. The role of ER stress in lipid metabolism and lipotoxicity. J. Lipid Res. 2016, 57, 1329–1338. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.-S.; Mendez, R.; Heng, H.H.; Yang, Z.-Q.; Zhang, K. Pharmacological ER stress promotes hepatic lipogenesis and lipid droplet formation. Am. J. Transl. Res. 2012, 4, 102–113. [Google Scholar] [PubMed]
- DeZwaan-McCabe, D.; Sheldon, R.; Gorecki, M.C.; Guo, D.-F.; Gansemer, E.; Kaufman, R.J.; Rahmouni, K.; Gillum, M.; Taylor, E.; Teesch, L.M.; et al. ER Stress Inhibits Liver Fatty Acid Oxidation while Unmitigated Stress Leads to Anorexia-Induced Lipolysis and Both Liver and Kidney Steatosis. Cell Rep. 2017, 19, 1794–1806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrema, H.; Zhou, Y.; Zhang, D.; Lee, J.; Hernandez, M.A.S.; Shulman, G.; Ozcan, U. XBP1s Is an Anti-lipogenic Protein. J. Biol. Chem. 2016, 291, 17394–17404. [Google Scholar] [CrossRef] [Green Version]
- Schuck, S.; Prinz, W.A.; Thorn, K.S.; Voss, C.; Walter, P. Membrane expansion alleviates endoplasmic reticulum stress independently of the unfolded protein response. J. Cell Biol. 2009, 187, 525–536. [Google Scholar] [CrossRef] [Green Version]
- Neese, R.A.; Faix, D.; Kletke, C.; Wu, K.; Wang, A.C.; Shackleton, C.H.; Hellerstein, M.K. Measurement of endogenous synthesis of plasma cholesterol in rats and humans using MIDA. Am. J. Physiol. 1993, 264, E136–E147. [Google Scholar] [CrossRef]
- Turner, S.M.; Murphy, E.J.; Neese, R.A.; Antelo, F.; Thomas, T.; Agarwal, A.; Go, C.; Hellerstein, M.K. Measurement of TG synthesis and turnover in vivo by 2H2O incorporation into the glycerol moiety and application of MIDA. Am. J. Physiol. Endocrinol. Metab. 2003, 285, E790–E803. [Google Scholar] [CrossRef] [Green Version]
- Koh, H.-C.E.; Mittendorfer, B. Adipose tissue lipolysis, plasma fatty acids, and glucose homeostasis in people with obesity: New pieces that help solve the puzzle. EBioMedicine 2021, 66, 103311. [Google Scholar] [CrossRef] [PubMed]
- Mohan, S.; Rani, P.M.R.; Brown, L.; Ayyappan, P.; Raghu, K.G. Endoplasmic reticulum stress: A master regulator of metabolic syndrome. Eur. J. Pharmacol. 2019, 860, 172553. [Google Scholar] [CrossRef]
- Vandewynckel, Y.-P.; Laukens, D.; Bogaerts, E.; Paridaens, A.; Bussche, A.V.D.; Verhelst, X.; van Steenkiste, C.; Descamps, B.; Vanhove, C.; Libbrecht, L.; et al. Modulation of the unfolded protein response impedes tumor cell adaptation to proteotoxic stress: A PERK for hepatocellular carcinoma therapy. Hepatol. Int. 2015, 9, 93–104. [Google Scholar] [CrossRef] [Green Version]
- Borgese, N.; Francolini, M.; Snapp, E. Endoplasmic reticulum architecture: Structures in flux. Curr. Opin. Cell Biol. 2006, 18, 358–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daniele, J.R.; Higuchi-Sanabria, R.; Durieux, J.; Monshietehadi, S.; Ramachandran, V.; Tronnes, S.U.; Kelet, N.; Sanchez, M.; Metcalf, M.G.; Garcia, G.; et al. UPRER promotes lipophagy independent of chaperones to extend life span. Sci. Adv. 2020, 6, eaaz1441. [Google Scholar] [CrossRef] [Green Version]
- Yzydorczyk, C.; Li, N.; Rigal, E.; Chehade, H.; Mosig, D.; Armengaud, J.B.; Rolle, T.; Krishnasamy, A.; Orozco, E.; Siddeek, B.; et al. Calorie Restriction in Adulthood Reduces Hepatic Disorders Induced by Transient Postnatal Overfeeding in Mice. Nutrients 2019, 11, 2796. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, S.J.; Madrigal-Matute, J.; Scheibye-Knudsen, M.; Fang, E.F.; Aon, M.; González-Reyes, J.A.; Cortassa, S.; Kaushik, S.; Gonzalez-Freire, M.; Patel, B.; et al. Effects of Sex, Strain, and Energy Intake on Hallmarks of Aging in Mice. Cell Metab. 2016, 23, 1093–1112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sriburi, R.; Jackowski, S.; Mori, K.; Brewer, J.W. XBP1: A link between the unfolded protein response, lipid biosynthesis, and biogenesis of the endoplasmic reticulum. J. Cell Biol. 2004, 167, 35–41. [Google Scholar] [CrossRef] [Green Version]
- Colgan, S.M.; Tang, D.; Werstuck, G.H.; Austin, R.C. Endoplasmic reticulum stress causes the activation of sterol regulatory element binding protein-2. Int. J. Biochem. Cell Biol. 2007, 39, 1843–1851. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.I.; Shankaran, M.; Yoshino, M.; Schweitzer, G.G.; Chondronikola, M.; Beals, J.W.; Okunade, A.L.; Patterson, B.W.; Nyangau, E.; Field, T.; et al. Insulin resistance drives hepatic de novo lipogenesis in nonalcoholic fatty liver disease. J. Clin. Investig. 2020, 130, 1453–1460. [Google Scholar] [CrossRef]
- Lawitz, E.J.; Coste, A.; Poordad, F.; Alkhouri, N.; Loo, N.; McColgan, B.J.; Tarrant, J.M.; Nguyen, T.; Han, L.; Chung, C.; et al. Acetyl-CoA Carboxylase Inhibitor GS-0976 for 12 Weeks Reduces Hepatic De Novo Lipogenesis and Steatosis in Patients With Nonalcoholic Steatohepatitis. Clin. Gastroenterol. Hepatol. 2018, 16, 1983–1991.e3. [Google Scholar] [CrossRef]
- Lambert, J.E.; Ramos-Roman, M.A.; Browning, J.D.; Parks, E.J. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology 2014, 146, 726–735. [Google Scholar] [CrossRef] [PubMed]
- Begley, C.G.; Ioannidis, J.P.A. Reproducibility in science: Improving the standard for basic and preclinical research. Circ. Res. 2015, 116, 116–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Rendleman, J.; Cheng, Z.; Maity, S.; Kastelic, N.; Munschauer, M.; Allgoewer, K.; Teo, G.; Zhang, Y.B.M.; Lei, A.; Parker, B.; et al. New insights into the cellular temporal response to proteostatic stress. eLife 2018, 7, e39054. [Google Scholar] [CrossRef] [PubMed]
- Reid, D.W.; Chen, Q.; Tay, A.S.L.; Shenolikar, S.; Nicchitta, C.V. The unfolded protein response triggers selective mRNA release from the endoplasmic reticulum. Cell 2014, 158, 1362–1374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Beyer, A.; Aebersold, R. On the dependency of cellular protein levels on mRNA abundance. Cell 2016, 165, 535–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenbaum, D.; Colangelo, C.; Williams, K.; Gerstein, M. Comparing protein abundance and mRNA expression levels on a genomic scale. Genome Biol. 2003, 4, 117. [Google Scholar] [CrossRef] [Green Version]
- Maier, T.; Güell, M.; Serrano, L. Correlation of mRNA and protein in complex biological samples. FEBS Lett. 2009, 583, 3966–3973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, T.-C.; Kuo, T.; Dandan, M.; Lee, R.A.; Chang, M.; Villivalam, S.D.; Liao, S.C.; Costello, D.; Shankaran, M.; Mohammed, H.; et al. The role of striated muscle Pik3r1 in glucose and protein metabolism following chronic glucocorticoid exposure. J. Biol. Chem. 2021, 296, 100395. [Google Scholar] [CrossRef]
- Thompson, A.C.S.; Bruss, M.D.; Price, J.C.; Khambatta, C.F.; Holmes, W.E.; Colangelo, M.; Dalidd, M.; Roberts, L.S.; Astle, C.M.; Harrison, D.E.; et al. Reduced in vivo hepatic proteome replacement rates but not cell proliferation rates predict maximum lifespan extension in mice. Aging Cell 2016, 15, 118–127. [Google Scholar] [CrossRef]
- Busch, R.; Neese, R.A.; Awada, M.; Hayes, G.M.; Hellerstein, M.K. Measurement of cell proliferation by heavy water labeling. Nat. Protoc. 2007, 2, 3045–3057. [Google Scholar] [CrossRef] [PubMed]
- Hellerstein, M.K.; Neese, R.A. Mass isotopomer distribution analysis: A technique for measuring biosynthesis and turnover of polymers. Am. J. Physiol. 1992, 263, E988–E1001. [Google Scholar] [CrossRef] [PubMed]
- Hellerstein, M.K.; Neese, R.A. Mass isotopomer distribution analysis at eight years: Theoretical, analytic, and experimental considerations. Am. J. Physiol. 1999, 276, E1146–E1170. [Google Scholar] [CrossRef]
- Hellerstein, M.K.; Neese, R.A.; Letscher, A.; Linfoot, P.; Turner, S. Hepatic glucose-6-phosphatase flux and glucose phosphorylation, cycling, irreversible disposal, and net balance in vivo in rats. Measurement using the secreted glucuronate technique. Metabolism 1997, 46, 1390–1398. [Google Scholar] [CrossRef]
- Hellerstein, M.K.; Christiansen, M.; Kaempfer, S.; Kletke, C.; Wu, K.; Reid, J.S.; Mulligan, K.; Hellerstein, N.S.; Shackleton, C.H. Measurement of de novo hepatic lipogenesis in humans using stable isotopes. J. Clin. Investig. 1991, 87, 1841–1852. [Google Scholar] [CrossRef]
- Hellerstein, M.K.; Schwarz, J.M.; Neese, R.A. Regulation of hepatic de novo lipogenesis in humans. Annu. Rev. Nutr. 1996, 16, 523–557. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ward, C.P.; Peng, L.; Yuen, S.; Chang, M.; Karapetyan, R.; Nyangau, E.; Mohammed, H.; Palacios, H.; Ziari, N.; Joe, L.K.; et al. ER Unfolded Protein Response in Liver In Vivo Is Characterized by Reduced, Not Increased, De Novo Lipogenesis and Cholesterol Synthesis Rates with Uptake of Fatty Acids from Adipose Tissue: Integrated Gene Expression, Translation Rates and Metabolic Fluxes. Int. J. Mol. Sci. 2022, 23, 1073. https://doi.org/10.3390/ijms23031073
Ward CP, Peng L, Yuen S, Chang M, Karapetyan R, Nyangau E, Mohammed H, Palacios H, Ziari N, Joe LK, et al. ER Unfolded Protein Response in Liver In Vivo Is Characterized by Reduced, Not Increased, De Novo Lipogenesis and Cholesterol Synthesis Rates with Uptake of Fatty Acids from Adipose Tissue: Integrated Gene Expression, Translation Rates and Metabolic Fluxes. International Journal of Molecular Sciences. 2022; 23(3):1073. https://doi.org/10.3390/ijms23031073
Chicago/Turabian StyleWard, Catherine P., Lucy Peng, Samuel Yuen, Michael Chang, Rozalina Karapetyan, Edna Nyangau, Hussein Mohammed, Hector Palacios, Naveed Ziari, Larry K. Joe, and et al. 2022. "ER Unfolded Protein Response in Liver In Vivo Is Characterized by Reduced, Not Increased, De Novo Lipogenesis and Cholesterol Synthesis Rates with Uptake of Fatty Acids from Adipose Tissue: Integrated Gene Expression, Translation Rates and Metabolic Fluxes" International Journal of Molecular Sciences 23, no. 3: 1073. https://doi.org/10.3390/ijms23031073