
Hemosiderin Accumulation in Liver Decreases Iron Availability in Tachycardia-Induced Porcine Congestive Heart Failure Model

 , , , , , , and
, , , , , , and
Abstract
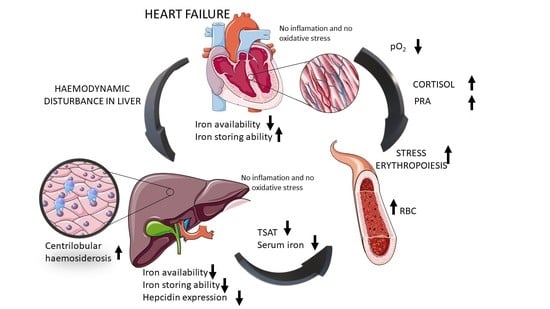
:
1. Introduction
2. Results
2.1. RV Pacing Results in a Deterioration of LV Function, Neurohormonal Activation, LV Dilatation, Heart Hypertrophy and Hepatomegaly
2.2. Paced Pigs Reveal Indices of Enhanced Erythropoiesis and ID, but with Red Blood Cells Remaining Normocytic, and Normochromic
2.3. HF Development Leads to Hepatocellular Injury with Preserved Hepatic Synthesis Capacity and Excretory Functions as Well as to Mild Systemic Oxidative Stress without Inflammation
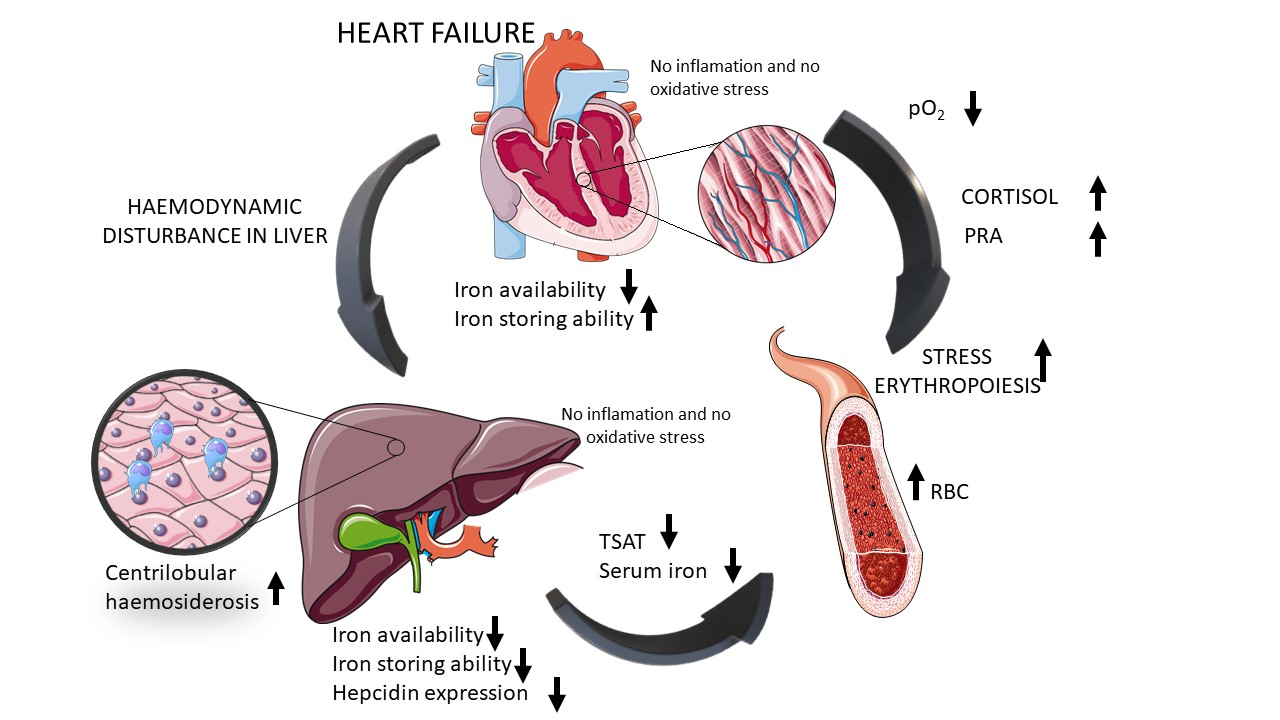
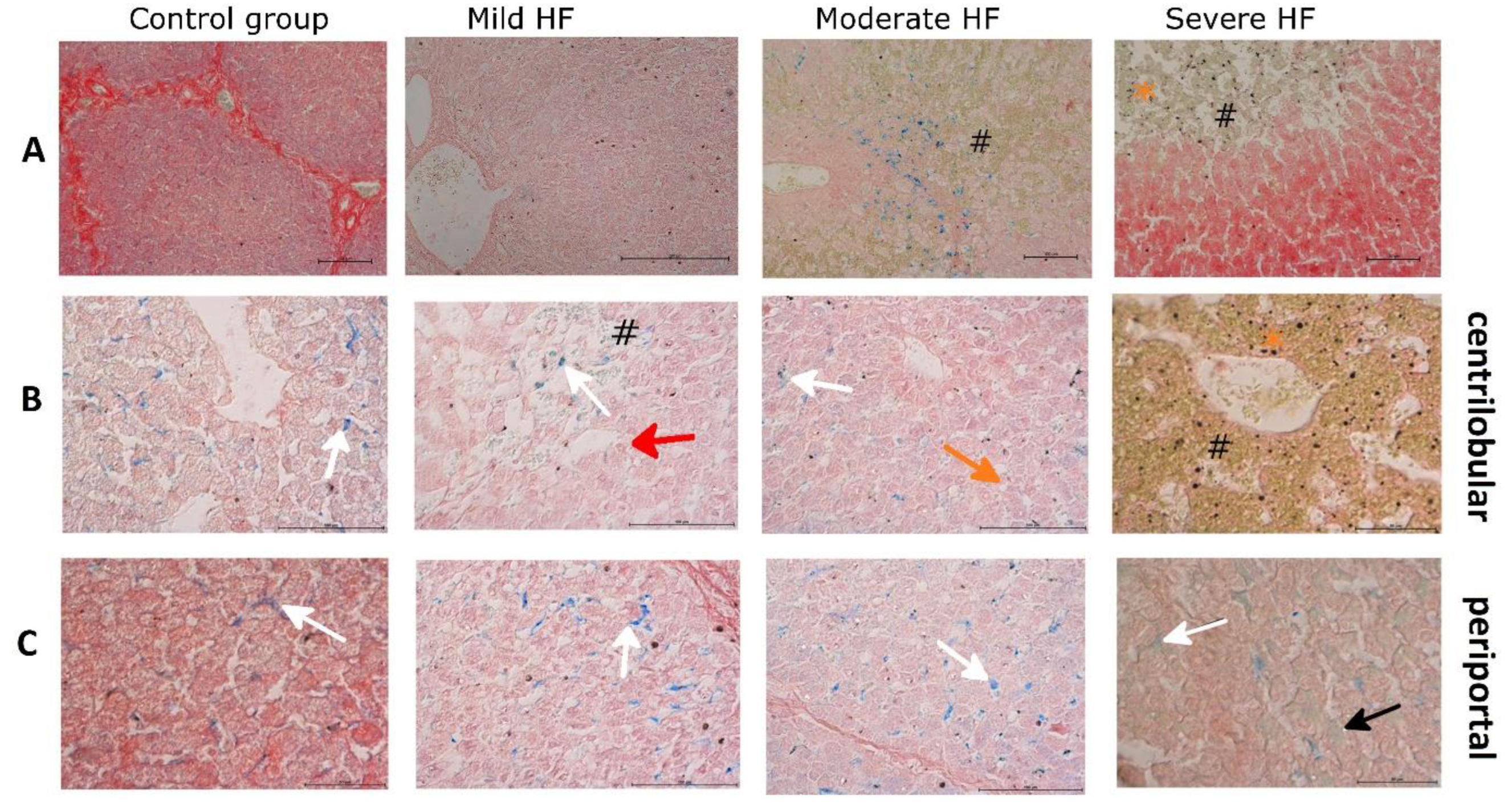
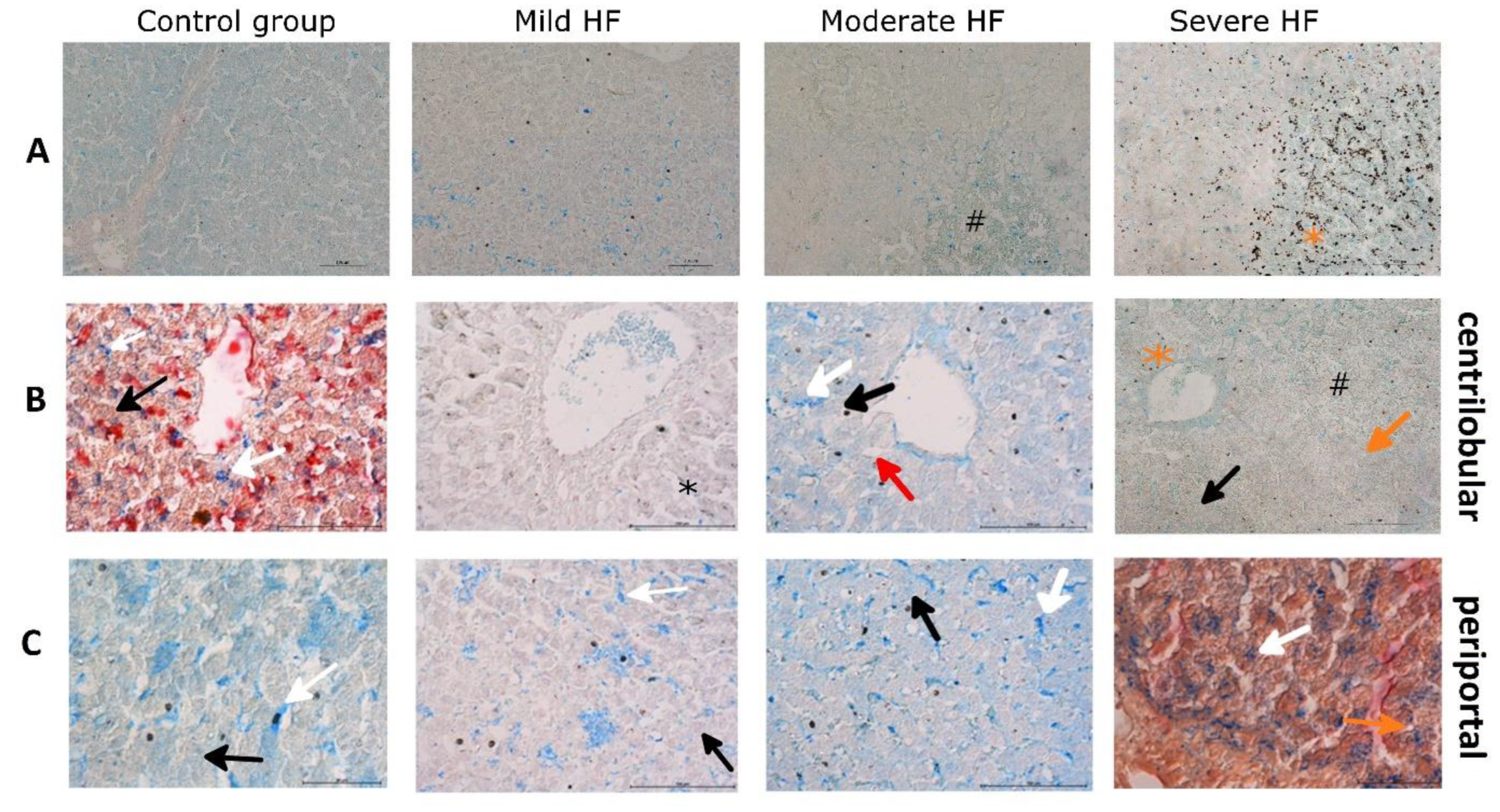
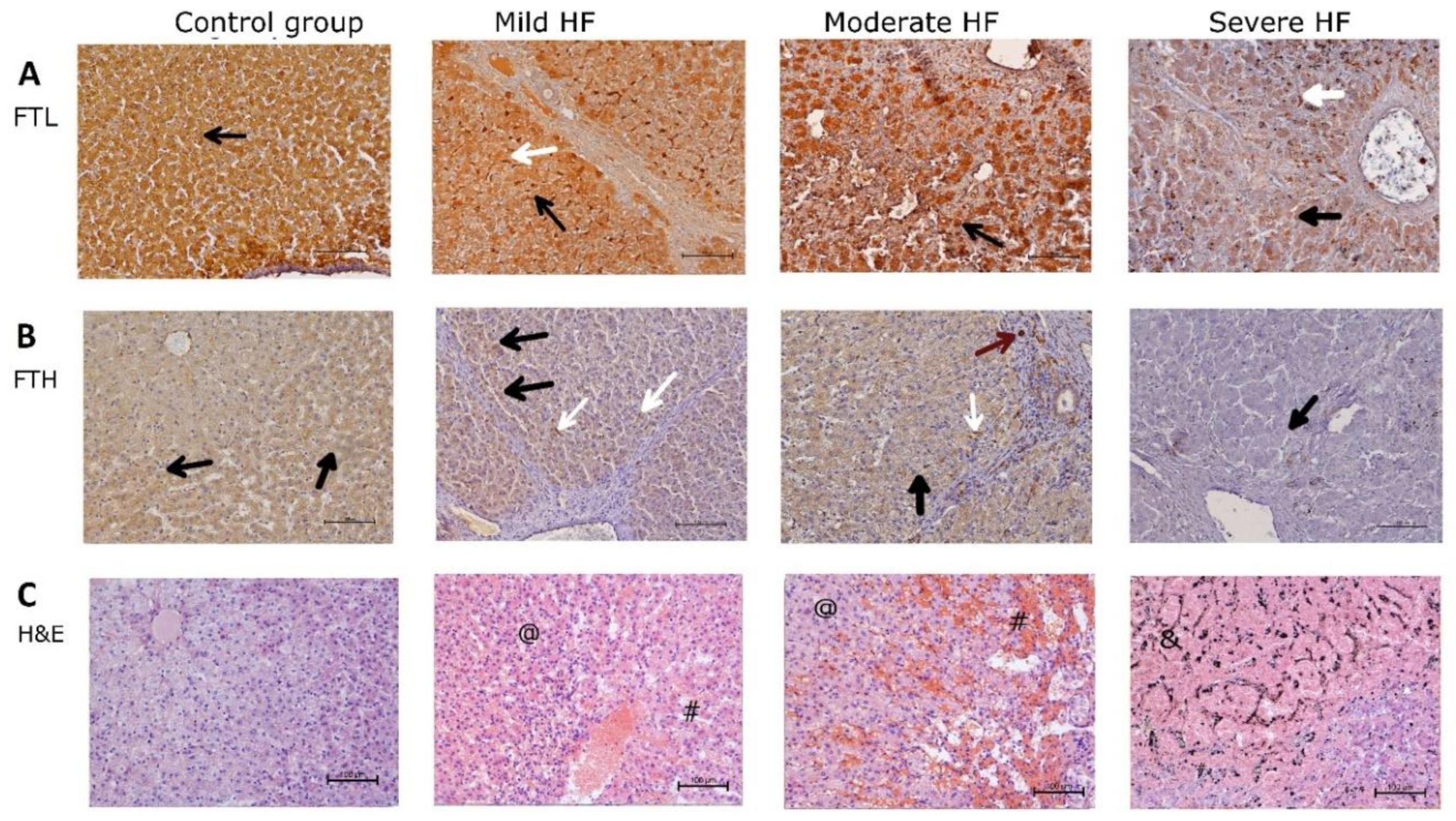
2.4. HF Development through Centrilobular Sinusoidal Congestion Leads to Accumulation of Iron-Overloaded Kupffer Cells and Centrilobular Multiple Hemosiderin Deposits, then, Hepatic Cords Atrophy with a Concomitant Drop of Kupffer Cells and Massive Hemosiderin Deposits
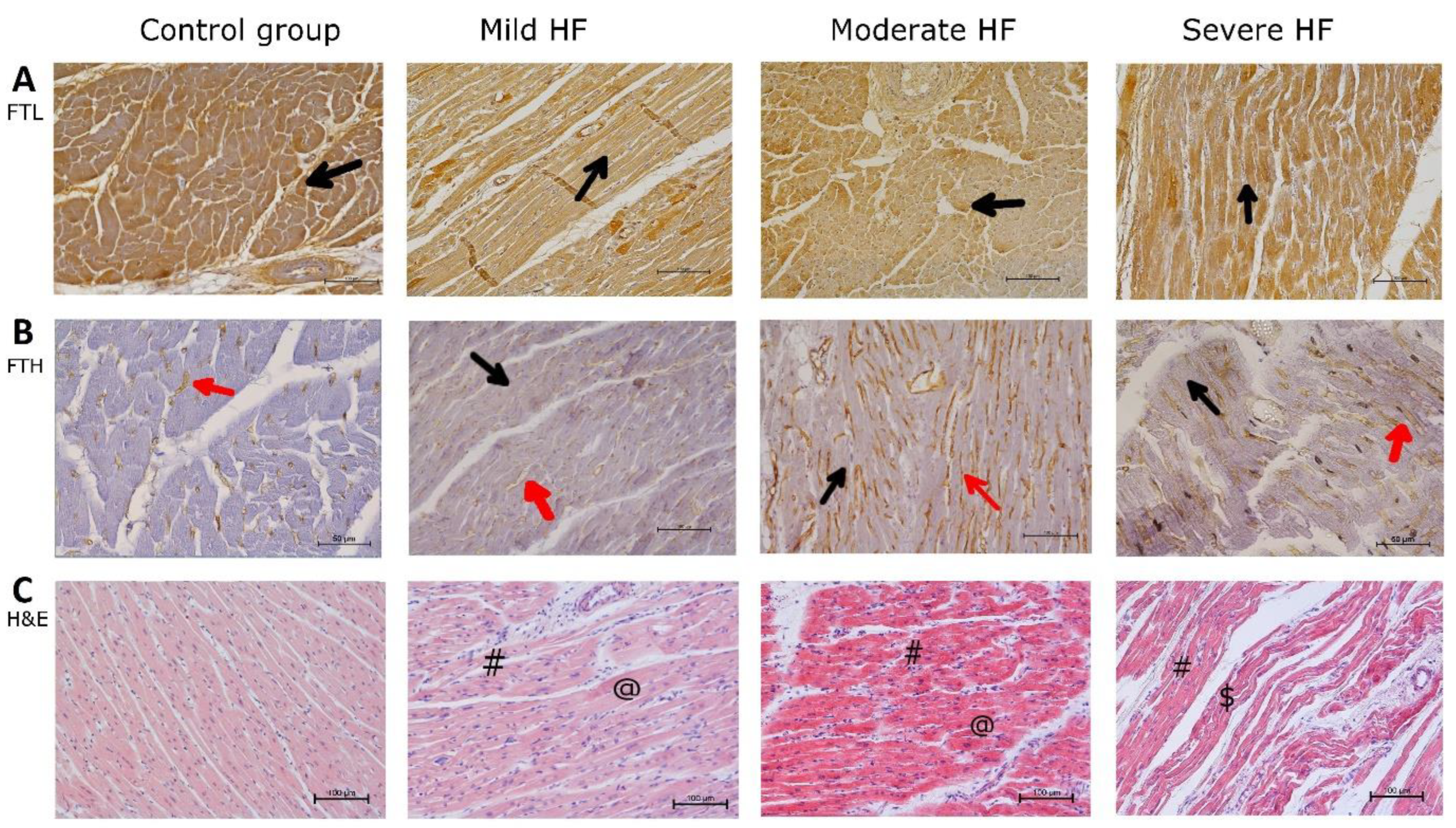
2.5. Porcine Myocardium Reveals Ultrastructural Changes Typical for HF
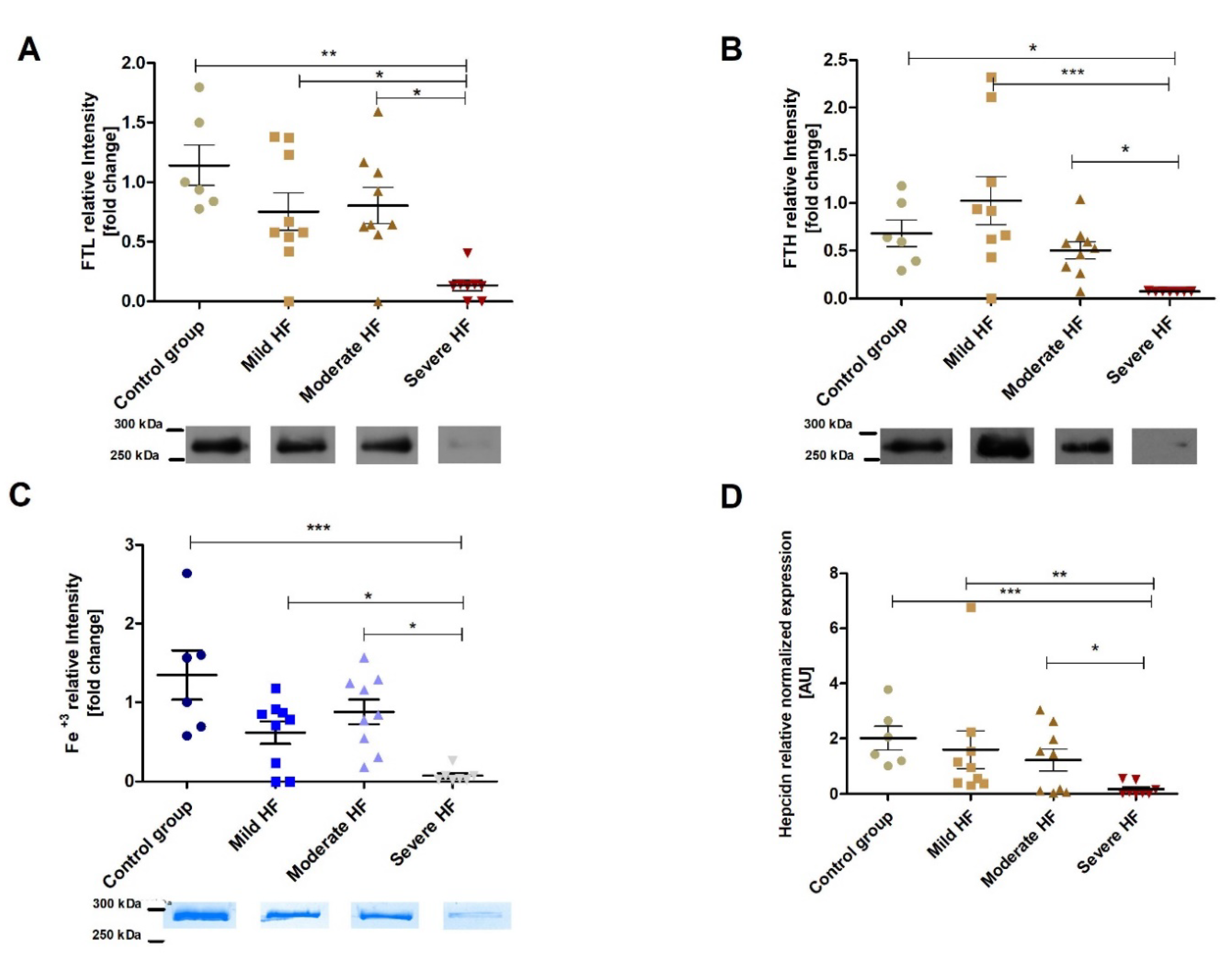
2.6. In the Liver the Ferritin-Bound Fe3+ (Iron Stores), and Assembled Ferritin (Ability to Store Iron) Are Decreasing along with HF Development with Concomitant Hepcidin Suppression
2.7. Increase of Myocardial Assembled Ferritin in HF Is Accompanied with Reduction of Ferritin-Bound Fe3+
3. Discussion
4. Materials and Methods
4.1. Study Approval
4.2. Experimental Design
4.3. Echocardiography
4.4. Neurohormonal Activation and Serum Iron Status
4.5. Cytokine Measurement
4.6. Histological Analyses
4.7. Non-Heme Iron Histochemistry
4.8. Immunohistochemistry
4.9. Ferritin Isolation
4.10. SDS-PAGE, Prussian Blue Reaction, Western Blotting
4.11. RNA Isolation and Real-Time PCR
4.12. Aconitase Activity
4.13. Measurement of Malonyldialdehyde Level
4.14. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Klip, I.T.; Comin-Colet, J.; Voors, A.A.; Ponikowski, P.; Enjuanes, C.; Banasiak, W.; Lok, D.J.; Rosentryt, P.; Torrens, A.; Polonski, L.; et al. Iron Deficiency in Chronic Heart Failure: An International Pooled Analysis. Am. Heart J. 2013, 165, 575–582. [Google Scholar] [CrossRef] [PubMed]
- Ponikowski, P.; Voors, A.A.; Anker, S.D.; Bueno, H.; Cleland, J.G.F.; Coats, A.J.S.; Falk, V.; González-Juanatey, J.R.; Harjola, V.-P.; Jankowska, E.A.; et al. 2016 ESC Guidelines for the Diagnosis and Treatment of Acute and Chronic Heart Failure. Eur. J. Heart Fail. 2016, 18, 891–975. [Google Scholar] [CrossRef]
- McDonagh, T.; Damy, T.; Doehner, W.; Lam, C.S.P.; Sindone, A.; van der Meer, P.; Cohen-Solal, A.; Kindermann, I.; Manito, N.; Pfister, O.; et al. Screening, Diagnosis and Treatment of Iron Deficiency in Chronic Heart Failure: Putting the 2016 European Society of Cardiology Heart Failure Guidelines into Clinical Practice. Eur. J. Heart Fail. 2018, 20, 1664–1672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponikowski, P.; Kirwan, B.A.; Anker, S.D.; McDonagh, T.; Dorobantu, M.; Drozdz, J.; Fabien, V.; Filippatos, G.; Göhring, U.M.; Keren, A.; et al. Ferric Carboxymaltose for Iron Deficiency at Discharge after Acute Heart Failure: A Multicentre, Double-Blind, Randomised, Controlled Trial. Lancet 2020, 396, 1895–1904. [Google Scholar] [CrossRef]
- Kobak, K.A.; Radwańska, M.; Dzięgała, M.; Kasztura, M.; Josiak, K.; Banasiak, W.; Ponikowski, P.; Jankowska, E.A. Structural and Functional Abnormalities in Iron-Depleted Heart. Heart Fail. Rev. 2019, 24, 269–277. [Google Scholar] [CrossRef] [Green Version]
- Gutierrez, K.; Dicks, N.; Glanzner, W.G.; Agellon, L.B.; Bordignon, V. Efficacy of the Porcine Species in Biomedical Research. Front. Genet. 2015, 6, 293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasławski, R.; Pasławska, U.; Szuba, A.; Nicpoń, J. Swine as a Model of Experimental Atherosclerosis. Adv. Clin. Exp. Med. 2011, 20, 211–215. [Google Scholar]
- Paslawska, U.; Gajek, J.; Kiczak, L.; Noszczyk-Nowak, A.; Skrzypczak, P.; Bania, J.; Tomaszek, A.; Zacharski, M.; Sambor, I.; Dziegiel, P.; et al. Development of Porcine Model of Chronic Tachycardia-Induced Cardiomyopathy. Int. J. Cardiol. 2011, 153, 36–41. [Google Scholar] [CrossRef]
- Allen, L.A.; Felker, G.M.; Pocock, S.; McMurray, J.J.V.; Pfeffer, M.A.; Swedberg, K.; Wang, D.; Yusuf, S.; Michelson, E.L.; Granger, C.B. Liver Function Abnormalities and Outcome in Patients with Chronic Heart Failure: Data from the Candesartan in Heart Failure: Assessment of Reduction in Mortality and Morbidity (CHARM) Program. Eur. J. Heart Fail. 2009, 11, 170–177. [Google Scholar] [CrossRef]
- Vollmar, B.; Menger, M.D. The Hepatic Microcirculation: Mechanistic Contributions and Therapeutic Targets in Liver Injury and Repair. Physiol. Rev. 2009, 89, 1269–1339. [Google Scholar] [CrossRef]
- Giallourakis, C.C.; Rosenberg, P.M.; Friedman, L.S. The Liver in Heart Failure. Clin. Liver Dis. 2002, 6, 947–967. [Google Scholar] [CrossRef]
- Koehne de Gonzalez, A.K.; Lefkowitch, J.H. Heart Disease and the Liver: Pathologic Evaluation. Gastroenterol. Clin. N. Am. 2017, 46, 421–435. [Google Scholar] [CrossRef]
- Anderson, E.R.; Shah, Y.M. Iron Homeostasis in the Liver. Compr. Physiol. 2013, 3, 315–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barton, J.C.; Edwards, C.Q.; Phatak, P.D.; Britton, R.S.; Bacon, B.R. (Eds.) Handbook of Iron Overload Disorders; Cambridge University Press: Cambridge, UK, 2010; ISBN 9780511777035. [Google Scholar]
- Poelzl, G.; Auer, J. Cardiohepatic Syndrome. Curr. Heart Fail. Rep. 2015, 12, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Harrison, P.M.; Arosio, P. The Ferritins: Molecular Properties, Iron Storage Function and Cellular Regulation. Biochim. Biophys. Acta (BBA)-Bioenerg. 1996, 1275, 161–203. [Google Scholar] [CrossRef] [Green Version]
- Ganz, T. Hepcidin—A Regular of Intestinal Iron Absorption and Iron Recycling by Macrophages. Best Pract. Res. Clin. Haematol. 2005, 18, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Ginzburg, Y.Z. Hepcidin-Ferroportin Axis in Health and Disease. Vitam. Horm. 2019, 110, 17–45. [Google Scholar] [CrossRef]
- Correale, M.; Tarantino, N.; Petrucci, R.; Tricarico, L.; Laonigro, I.; Di Biase, M.; Brunetti, N.D. Liver Disease and Heart Failure: Back and Forth. Eur. J. Intern. Med. 2018, 48, 25–34. [Google Scholar] [CrossRef]
- Raja, K.; Thung, S.N.; Fiel, M.I.; Chang, C. Drug-Induced Steatohepatitis Leading to Cirrhosis: Long-Term Toxicity of Amiodarone Use. Semin. Liver Dis. 2009, 29, 423–428. [Google Scholar] [CrossRef]
- Kim, I.H.; Kisseleva, T.; Brenner, D.A. Aging and Liver Disease. Curr. Opin. Gastroenterol. 2015, 31, 184–191. [Google Scholar] [CrossRef] [Green Version]
- Racine-Samson, L.; Scoazec, J.Y.; D’Errico, A.; Fiorentino, M.; Christa, L.; Moreau, A.; Roda, C.; Grigioni, W.F.; Feldmann, G. The Metabolic Organization of the Adult Human Liver: A Comparative Study of Normal, Fibrotic, and Cirrhotic Liver Tissue. Hepatology 1996, 24, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Biegus, J.; Hillege, H.L.; Postmus, D.; Valente, M.A.E.; Bloomfield, D.M.; Cleland, J.G.F.; Cotter, G.; Davison, B.A.; Dittrich, H.C.; Fiuzat, M.; et al. Abnormal Liver Function Tests in Acute Heart Failure: Relationship with Clinical Characteristics and Outcome in the PROTECT Study. Eur. J. Heart Fail. 2016, 18, 830–839. [Google Scholar] [CrossRef] [Green Version]
- Samsky, M.D.; Dunning, A.; DeVore, A.D.; Schulte, P.J.; Starling, R.C.; Wilson Tang, W.H.; Armstrong, P.W.; Ezekowitz, J.A.; Butler, J.; McMurray, J.J.; et al. Liver Function Tests in Patients with Acute Heart Failure and Associated Outcomes: Insights from ASCEND-HF. Eur. J. Heart Fail. 2016, 18, 424–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, W.; He, X.; Wu, D.; Xue, R.; Dong, B.; Owusu-Agyeman, M.; Zhao, J.; Cai, L.; You, Z.; Dong, Y.; et al. Prognostic Implication of Liver Function Tests in Heart Failure With Preserved Ejection Fraction Without Chronic Hepatic Diseases: Insight From TOPCAT Trial. Front. Cardiovasc. Med. 2021, 8, 330. [Google Scholar] [CrossRef]
- Holmes-Hampton, G.P.; Tong, W.H.; Rouault, T.A. Biochemical and Biophysical Methods for Studying Mitochondrial Iron Metabolism, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2014; Volume 547, ISBN 9780128014158. [Google Scholar]
- Meguro, R.; Asano, Y.; Odagiri, S.; Li, C.; Iwatsuki, H.; Shoumura, K. Nonheme-Iron Histochemistry for Light and Electron Microscopy: A Historical, Theoretical and Technical Review. Arch. Histol. Cytol. 2007, 70, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Muckenthaler, M.U.; Rivella, S.; Hentze, M.W.; Galy, B. A Red Carpet for Iron Metabolism. Cell 2017, 168, 344–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, W.H.; Rouault, T.A. Metabolic Regulation of Citrate and Iron by Aconitases: Role of Iron-Sulfur Cluster Biogenesis. BioMetals 2007, 20, 549–564. [Google Scholar] [CrossRef] [PubMed]
- Crooks, D.R.; Maio, N.; Lane, A.N.; Jarnik, M.; Higashi, R.M.; Haller, R.G.; Yang, Y.; Fan, T.W.M.; Marston Linehan, W.; Rouault, T.A. Acute Loss of Iron–Sulfur Clusters Results in Metabolic Reprogramming and Generation of Lipid Droplets in Mammalian Cells. J. Biol. Chem. 2018, 293, 8297–8311. [Google Scholar] [CrossRef] [Green Version]
- Knutson, M.D.; Oukka, M.; Koss, L.M.; Aydemir, F.; Wessling-Resnick, M. Iron Release from Macrophages after Erythrophagocytosis Is Up-Regulated by Ferroportin 1 Overexpression and down-Regulated by Hepcidin. Proc. Natl. Acad. Sci. USA 2005, 102, 1324–1328. [Google Scholar] [CrossRef] [Green Version]
- Theurl, I.; Hilgendorf, I.; Nairz, M.; Tymoszuk, P.; Haschka, D.; Asshoff, M.; He, S.; Gerhardt, L.M.S.; Holderried, T.A.W.; Seifert, M.; et al. On-Demand Erythrocyte Disposal and Iron Recycling Requires Transient Macrophages in the Liver. Nat. Med. 2016, 22, 945–951. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Shander, A.; Sazama, K. Clinical Consequences of Iron Overload from Chronic Red Blood Cell Transfusions, Its Diagnosis, and Its Management by Chelation Therapy. Transfusion 2010, 50, 1144–1155. [Google Scholar] [CrossRef]
- Koorts, A.M.; Viljoen, M. Ferritin and Ferritin Isoforms I: Structure-Function Relationships, Synthesis, Degradation and Secretion. Arch. Physiol. Biochem. 2007, 113, 30–54. [Google Scholar] [CrossRef] [Green Version]
- Miyazaki, E.; Kato, J.; Kobune, M.; Okumura, K.; Sasaki, K.; Shintani, N.; Arosio, P.; Niitsu, Y. Denatured H-Ferritin Subunit Is a Major Constituent of Haemosiderin in the Liver of Patients with Iron Overload. Gut 2002, 50, 413–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volpe, M.; Tritto, C.; Testa, U.; Rao, M.A.E.; Martucci, R.; Mirante, A.; Enea, I.; Russo, R.; Rubattu, S.; Condorelli, G.L.; et al. Blood Levels of Erythropoietin in Congestive Heart Failure and Correlation with Clinical, Hemodynamic, and Hormonal Profiles. Am. J. Cardiol. 1994, 74, 468–473. [Google Scholar] [CrossRef]
- Wen, D.; Boissel, J.P.R.; Tracy, T.E.; Gruninger, R.H.; Mulcahy, L.S.; Czelusniak, J.; Goodman, M.; Bunn, H.F. Erythropoietin Structure-Function Relationships: High Degree of Sequence Homology among Mammals. Blood 1993, 82, 1507–1516. [Google Scholar] [CrossRef] [Green Version]
- Zemella, A.; Thoring, L.; Hoffmeister, C.; Šamalíková, M.; Ehren, P.; Wüstenhagen, D.A.; Kubick, S. Cell-Free Protein Synthesis as a Novel Tool for Directed Glycoengineering of Active Erythropoietin. Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Hattangadi, S.M.; Wong, P.; Zhang, L.; Flygare, J.; Lodish, H.F. From Stem Cell to Red Cell: Regulation of Erythropoiesis at Multiple Levels by Multiple Proteins, RNAs, and Chromatin Modifications. Blood 2011, 118, 6258–6268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roth, M.-P.; Meynard, D.; Coppin, H. Regulators of Hepcidin Expression. In Vitamins and Hormones; Elsevier Inc.: Amsterdam, The Netherlands, 2019; pp. 101–129. ISBN 9780128178423. [Google Scholar]
- Peyrin-Biroulet, L.; Williet, N.; Cacoub, P. Guidelines on the Diagnosis and Treatment of Iron Deficiency across Indications: A Systematic Review. Am. J. Clin. Nutr. 2015, 102, 1585–1594. [Google Scholar] [CrossRef] [Green Version]
- Von Haehling, S.; Ebner, N.; Evertz, R.; Ponikowski, P.; Anker, S.D. Iron Deficiency in Heart Failure. JACC Heart Fail. 2019, 7, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Maeder, M.T.; Khammy, O.; Dos Remedios, C.; Kaye, D.M. Myocardial and Systemic Iron Depletion in Heart Failure: Implications for Anemia Accompanying Heart Failure. J. Am. Coll. Cardiol. 2011, 58, 474–480. [Google Scholar] [CrossRef] [Green Version]
- Melenovsky, V.; Petrak, J.; Mracek, T.; Benes, J.; Borlaug, B.A.; Nuskova, H.; Pluhacek, T.; Spatenka, J.; Kovalcikova, J.; Drahota, Z.; et al. Myocardial Iron Content and Mitochondrial Function in Human Heart Failure: A Direct Tissue Analysis. Eur. J. Heart Fail. 2017, 19, 522–530. [Google Scholar] [CrossRef]
- Cohn, J.N.; Ferrari, R.; Sharpe, N. Cardiac Remodeling-Concepts and Clinical Implications: A Consensus Paper from an International Forum on Cardiac Remodeling. J. Am. Coll. Cardiol. 2000, 35, 569–582. [Google Scholar] [CrossRef] [Green Version]
- Abe, H.; Semba, H.; Takeda, N. The Roles of Hypoxia Signaling in the Pathogenesis of Cardiovascular Diseases. J. Atheroscler. Thromb. 2017, 24, 884–894. [Google Scholar] [CrossRef] [Green Version]
- Ghafourian, K.; Shapiro, J.S.; Goodman, L.; Ardehali, H. Iron and Heart Failure: Diagnosis, Therapies, and Future Directions. JACC Basic Transl. Sci. 2020, 5, 300–313. [Google Scholar] [CrossRef]
- Hartupee, J.; Mann, D.L. Neurohormonal Activation in Heart Failure with Reduced Ejection Fraction. Nat. Rev. Cardiol. 2016, 14, 30–38. [Google Scholar] [CrossRef] [Green Version]
- Tajima, S.; Ikeda, Y.; Enomoto, H.; Imao, M.; Horinouchi, Y.; Izawa-Ishizawa, Y.; Kihira, Y.; Miyamoto, L.; Ishizawa, K.; Tsuchiya, K.; et al. Angiotensin II Alters the Expression of Duodenal Iron Transporters, Hepatic Hepcidin, and Body Iron Distribution in Mice. Eur. J. Nutr. 2015, 54, 709–719. [Google Scholar] [CrossRef] [PubMed]
- Tajes, M.; Díez-López, C.; Enjuanes, C.; Moliner, P.; Ferreiro, J.L.; Garay, A.; Jiménez-Marrero, S.; Yun, S.; Sosa, S.G.; Alcoberro, L.; et al. Neurohormonal Activation Induces Intracellular Iron Deficiency and Mitochondrial Dysfunction in Cardiac Cells. Cell Biosci. 2021, 11, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Moliner, P.; Enjuanes, C.; Tajes, M.; Cainzos-Achirica, M.; Lupón, J.; Garay, A.; Jimenez-Marrero, S.; Yun, S.; Farré, N.; Cladellas, M.; et al. Association between Norepinephrine Levels and Abnormal Iron Status in Patients with Chronic Heart Failure: Is Iron Deficiency More than a Comorbidity? J. Am. Heart Assoc. 2019, 8, e010887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toblli, J.E.; Di Gennaro, F.; Rivas, C. Changes in Echocardiographic Parameters in Iron Deficiency Patients with Heart Failure and Chronic Kidney Disease Treated with Intravenous Iron. Heart Lung Circ. 2015, 24, 686–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiczak, L.; Tomaszek, A.; Pasławska, U.; Bania, J.; Noszczyk-Nowak, A.; Skrzypczak, P.; Pasławski, R.; Zacharski, M.; Janiszewski, A.; Kuropka, P.K.P.; et al. Sex Differences in Porcine Left Ventricular Myocardial Remodeling Due to Right Ventricular Pacing. Biol. Sex Differ. 2015, 6, 32. [Google Scholar] [CrossRef] [Green Version]
- Zacharski, M.; Tomaszek, A.; Kiczak, L.; Ugorski, M.; Bania, J.; Pasławska, U.; Rybinska, I.; Jankowska, E.A.; Janiszewski, A.; Ponikowski, P. Catabolic/Anabolic Imbalance Is Accompanied by Changes of Left Ventricular Steroid Nuclear Receptor Expression in Tachycardia-Induced Systolic Heart Failure in Male Pigs Steroid Hormones in a Pig Model of Heart Failure. J. Card. Fail. 2021, 27, 682–692. [Google Scholar] [CrossRef] [PubMed]
- Tomaszek, A.; Kiczak, L.; Bania, J.; Paslawska, U.; Zacharski, M.; Janiszewski, A.; Noszczyk-Nowak, A.; Dziegiel, P.; Kuropka, P.; Ponikowski, P.; et al. Increased Gene Expression of Catecholamine-Synthesizing Enzymes in Adrenal Glands Contributes to High Circulating Catecholamines in Pigs with Tachycardia-Induced Cardiomyopathy. J. Physiol. Pharmacol. 2015, 66, 227–231. [Google Scholar] [PubMed]
- Paslawska, U.; Kiczak, L.; Bania, J.; Paslawski, R.; Janiszewski, A.; Dzięgiel, P.; Zacharski, M.; Tomaszek, A.; Michlik, K. Inducible NO Synthase Is Constitutively Expressed in Porcine Myocardium and Its Level Decreases along with Tachycardia-Induced Heart Failure. Cardiovasc. Pathol. 2016, 25, 3–11. [Google Scholar] [CrossRef]
- Sheehan, D.C.; Hrapchak, B.B. Theory and Practice of Histotechnology, 2nd ed.; Mosby, Incorporated: St. Louis, MI, USA, 1980; ISBN 0801645735. [Google Scholar]
- Carson, F. Histotechnology: A Self-Instructional Text, 1st ed.; ASCP Press: Chicago, IL, USA, 1991. [Google Scholar]
- Orchard, G.E. Pigments and Minerals. In Bancroft’s Theory and Practice of Histological Techniques; Elsevier Inc.: Amsterdam, The Netherlands, 2013. [Google Scholar]
- Krishna, M. Role of Special Stains in Diagnostic Liver Pathology. Clin. Liver Dis. 2013, 2, 8–10. [Google Scholar] [CrossRef]
- Bezkorovainy, A. Chemistry and Biology of Iron Storage. In Biochemistry of Nonheme Iron; Springer: Boston, MA, USA, 1980; pp. 207–269. [Google Scholar]
- Gabrio, B.W.; Shoden, A.; Finch, C.A. Quantitative Fractionation of Tissue Ferritin and Hemosiderin. J. Biol. Chem. 1953, 204, 815–821. [Google Scholar] [CrossRef]
- Bertrand, M.L.; Harris, D.C. Insensitivity of the Ferritin Iron Core to Heat Treatment. Experientia 1979, 35, 300–301. [Google Scholar] [CrossRef]
- Laemmli, U.K. Cleavage of Structural Proteins during the Assembly of the Head of Bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Netto, L.E.S.; Stadtman, E.R. The Iron-Catalyzed Oxidation of Dithiothreitol Is a Biphasic Process: Hydrogen Peroxide Is Involved in the Initiation of a Free Radical Chain of Reactions. Arch. Biochem. Biophys. 1996, 333, 233–242. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Controls (n = 6) | Mild HF (n = 9) | Moderate HF (n = 9) | Severe HF (n = 8) | Spearman Correlatory Rank Coefficients R with p, for All Animals | |
|---|---|---|---|---|---|
| Hematological parameters | |||||
| RBC (×1012/L) | 5.62 ± 0.51 | 5.88 ± 0.47 | 5.86 ± 0.83 | 6.72 ± 1.09 * | R = 0.38, p = 0.03 |
| R-RBC (%) | 86.4 ± 7.5 | 96.2 ± 6.3 * | 99.2 ± 7.3 ** | 110.2 ± 19 * | R = 0.57, p = 0.006 |
| Hb (mmol/L) | 5.96 ± 0.40 | 5.81 ± 0.39 | 5.96 ± 0.82 | 7.09 ± 1.48 | R = 0.33, p = 0.07 |
| R-Hb (%) | 92.2 ± 6.3 | 98.6 ± 5.8 | 101.1 ± 7.3 * | 120.1 ± 23 * | R = 0.67, p = 0.00007 |
| HCT (%) | 30.9 ± 1.6 | 31.0 ± 2.2 | 32.1 ± 3.9 | 37.4 ± 7.9 | R = 0.39, p = 0.03 |
| R-HCT (%) | 92.3 ± 7.9 | 97.0 ± 6.2 | 103.5 ± 8.4 * | 122.7 ± 27 * | R = 0.62, p = 0.0002 |
| WBC (×109/L) | 15.90 ± 6.13 | 14.00 ± 3.44 | 11.70 ± 2.29 | 9.00 ± 1.26 ** | R = −0.63, p = 0.0001 |
| PLT (×109/L) | 303 ± 103 | 235 ± 46 | 188 ± 60 * | 219 ± 48 | R = −0.35, p = 0.05 |
| MCV (fL) | 54.5 ± 2.43 | 52.8 ± 1.98 | 53.7 ± 2.61 | 54.4 ± 2.88 | R = 0.02, p = 0.89 |
| MCH (fmol) | 1.03 ± 0.06 | 0.99 ± 0.05 | 1.01 ± 0.08 | 1.04 ± 0.07 | R = 0.09, p = 0.60 |
| MCHC (mmol/L) | 18.8 ± 0.9 | 18.7 ± 0.8 | 18.7 ± 0.6 | 18.9 ± 0.7 | R = −0.02, p = 0.87 |
| Iron status | |||||
| Serum iron (μg/dL) | 126 ± 21 | 100 ± 23 | 106 ± 20 | 74 ± 23 ** | R = −0.52, p = 0.002 |
| TSAT (%) # | 39.1 ± 6,9 | 31.9 ± 7.9 | 30.1 ± 13 | 22.0 ± 7.5 * | R = −0.57, p = 0.01 |
| TIBC (μg/dL) # | 343 ± 29 | 315 ± 42 | 416 ± 103 | 412 ± 83 | R = 0.44, p = 0.06 |
| Serum ferritin (ng/mL) | 1.98 ± 2.17 | 1.88 ± 3.04 | 0.94 ± 0.78 | 1.54 ± 0.46 | R = 0.16, p = 0.38 |
| Controls (n = 6) | Mild HF (n = 9) | Moderate HF (n = 9) | Severe HF (n = 8) | Spearman Correlatory Rank Coefficients R with p, for All Animals | |
|---|---|---|---|---|---|
| Biochemical parameters | |||||
| Serum albumin (g/L) | 36.5 ± 3.0 | 34.7 ± 2.7 | 37.0 ± 2.6 | 31.8 ± 3.1 | R = −0.27, p = 0.17 |
| Total serum protein (g/L) | 61.5 ± 5.6 | 57.2 ± 3.9 | 56.7 ± 4.4 | 59.6 ± 7.0 | R = −0.02, p = 0.9 |
| Total serum bilirubin (μmol/L) | 1.62 ± 1.32 | 1.83 ± 1.62 | 2.47 ± 2.45 | 4.01 ± 5.21 | R = 0.29, p = 0.11 |
| AST (U/L) | 21.33 ± 11.08 | 21.56 ± 8.99 | 29.38 ± 12.25 | 35.33 ± 12.25 | R = 0.42, p = 0.02 |
| ALT (U/L) | 51.3 ± 17.4 | 41.6 ± 5.4 | 39.5 ± 21.1 | 32.3 ± 15.9 | R = −0.32, p = 0.08 |
| AST/ALT ratio | 0.43 ± 0.21 | 0.52 ± 0.22 | 1.04 ± 0.95 | 1.88 ± 1.72 * | R = 0.54, p = 0.002 |
| Glucose (mmol/L) | 4.73 ± 0.71 | 5.39 ± 0.63 | 5.39 ± 0.91 | 5.86 ± 0.97 * | R = 0.37, p = 0.03 |
| Triglyceride (mmole/L) | 0.27 ± 0.07 | 0.29 ± 0.09 | 0.36 ± 0.08 | 0.43 ± 0.13 | R = 0.47, p = 0.006 |
| Inflammatory and oxidative stress status | |||||
| Serum IL-1β (pg/mL) | 2.53 ± 2.93 | 4.04 ± 3.34 | 8.96 ± 9.7 | 5.09 ± 4.37 | R = 0.24, p = 0.19 |
| Serum IL-6 (pg/mL) | 1.61 ± 2.15 | 6.02 ± 4.78 | 7.55 ± 7.97 | 7.89 ± 7.07 * | R = 0.31, p = 0.08 |
| Serum TNF-α (pg/mL) | 56.2 ± 27.8 | 73.2 ± 30.7 | 78.1 ± 46.1 | 64.4 ± 33.1 | R = 0.07, p = 0.69 |
| Serum MDA (μmol/L) | 6.79 ± 2.42 | 6.19 ± 1.58 | 8.92 ± 6.70 | 8.46 ± 2.09 | R = 0.36, p = 0.04 |
| Liver aconitase activity in mitochondrial fraction (mU/mg of protein) | 2.42 ± 1.03 | 3.59 ± 0.83 | 1.68 ± 1.10 | 1.39 ± 0.99 | R = −0.50, p = 0.004 |
| Liver aconitase activity in cytoplasmic fraction (mU/mg of protein) | 7.08 ± 3.69 | 10.03 ± 3.50 | 6.90 ± 2.07 | 3.92 ± 3.00 | R = −0.40, p = 0.02 |
| Liver MDA (nmol/mg of tissue) | 0.07 ± 0.06 | 0.09 ± 0.08 | 0.07 ± 0.05 | 0.1 ± 0.06 | R = 0.12, p = 0.50 |
| LV aconitase activity in mitochondrial fraction (mU/mg of protein) | 0.36 ± 0.56 | 0.36 ± 0.30 | 0.99 ± 1.56 | 0.52 ± 1.05 | R = −0.05, p = 0.77 |
| LV aconitase activity in cytoplasmic fraction (mU/mg of protein) | 0.21 ± 0.17 | 0.09 ± 0.09 | 0.28 ± 0.34 | 0.44 ± 0.42 | R = 0.33, p = 0.07 |
| LV MDA (nmol/mg of tissue) | 0.05 ± 0.02 | 0.04 ± 0.01 | 0.03 ± 0.01 | 0.04 ± 0.01 | R = −0.22, p = 0.30 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kasztura, M.; Kiczak, L.; Pasławska, U.; Bania, J.; Janiszewski, A.; Tomaszek, A.; Zacharski, M.; Noszczyk-Nowak, A.; Pasławski, R.; Tabiś, A.; et al. Hemosiderin Accumulation in Liver Decreases Iron Availability in Tachycardia-Induced Porcine Congestive Heart Failure Model. Int. J. Mol. Sci. 2022, 23, 1026. https://doi.org/10.3390/ijms23031026
Kasztura M, Kiczak L, Pasławska U, Bania J, Janiszewski A, Tomaszek A, Zacharski M, Noszczyk-Nowak A, Pasławski R, Tabiś A, et al. Hemosiderin Accumulation in Liver Decreases Iron Availability in Tachycardia-Induced Porcine Congestive Heart Failure Model. International Journal of Molecular Sciences. 2022; 23(3):1026. https://doi.org/10.3390/ijms23031026
Chicago/Turabian StyleKasztura, Monika, Liliana Kiczak, Urszula Pasławska, Jacek Bania, Adrian Janiszewski, Alicja Tomaszek, Maciej Zacharski, Agnieszka Noszczyk-Nowak, Robert Pasławski, Aleksandra Tabiś, and et al. 2022. "Hemosiderin Accumulation in Liver Decreases Iron Availability in Tachycardia-Induced Porcine Congestive Heart Failure Model" International Journal of Molecular Sciences 23, no. 3: 1026. https://doi.org/10.3390/ijms23031026