
Role of GALNT2 on Insulin Sensitivity, Lipid Metabolism and Fat Homeostasis


1
Research Unit of Diabetes and Endocrine Diseases, Fondazione IRCCS Casa Sollievo della Sofferenza, San Giovanni Rotondo (FG), 71013 Foggia, Italy
2
Department of Experimental Medicine, Sapienza University, 00161 Rome, Italy
*
Authors to whom correspondence should be addressed.
Int. J. Mol. Sci. 2022, 23(2), 929; https://doi.org/10.3390/ijms23020929
Submission received: 20 December 2021
/
Revised: 11 January 2022
/
Accepted: 13 January 2022
/
Published: 15 January 2022
(This article belongs to the Special Issue Insulin Signaling and Fat Homeostasis in Metabolic and Neurodegenerative Disorders)
{kind=link}
Abstract
:O-linked glycosylation, the greatest form of post-translational modifications, plays a key role in regulating the majority of physiological processes. It is, therefore, not surprising that abnormal O-linked glycosylation has been related to several human diseases. Recently, GALNT2, which encodes the GalNAc-transferase 2 involved in the first step of O-linked glycosylation, has attracted great attention as a possible player in many highly prevalent human metabolic diseases, including atherogenic dyslipidemia, type 2 diabetes and obesity, all clustered on the common ground of insulin resistance. Data available both in human and animal models point to GALNT2 as a molecule that shapes the risk of the aforementioned abnormalities affecting diverse protein functions, which eventually cause clinically distinct phenotypes (a typical example of pleiotropism). Pathways linking GALNT2 to dyslipidemia and insulin resistance have been partly identified, while those for type 2 diabetes and obesity are yet to be understood. Here, we will provide a brief overview on the present knowledge on GALNT2 function and dysfunction and propose novel insights on the complex pathogenesis of the aforementioned metabolic diseases, which all impose a heavy burden for patients, their families and the entire society.
1. Introduction
O-linked glycosylation, the greatest diverse form of post-translational modifications, affects the folding, stability, processing and trafficking of most secreted proteins [1], thus playing a key role in regulating many physiological processes. It is, therefore, not surprising that abnormal O-glycosylation has been related to several human diseases [2]. GALNT2 encodes the polypeptide N-acetyl-galactosaminyl-transferase 2 (ppGalNAc-T2, also referred to as GalNAc-T2) [3,4], which transfers N-acetyl galactosamine to the serine/threonine hydroxyl group of specific acceptor peptide motifs [5,6,7] in the first step of O-glycosylation. In the last few years, GALNT2 has attracted great attention as a possible player in many human metabolic abnormalities that all share the common ground of insulin resistance, including atherogenic dyslipidemia [8,9,10,11,12,13,14], type 2 diabetes [15,16] and obesity [17,18]. After very briefly introducing insulin resistance, its role in several highly prevalent human diseases and how abnormal insulin signaling may cause it, we will offer to the readers an overview on what is known about GALNT2 function and dysfunction, with the aim of providing novel insights on the complex pathogenesis of the aforementioned metabolic diseases.
2. Insulin Resistance and Metabolic Diseases
Insulin resistance is a complex and highly prevalent metabolic disorder, which is mainly due to the inability of multiple insulin target sites, including but not limited to skeletal muscle, liver, and adipose tissue, to respond to insulin. It is widely accepted that insulin resistance represents a common pathogenic ground for many additional disorders and highly prevalent diseases, comprising atherogenic dyslipidemia [19], type 2 diabetes, obesity and hypertension [19,20], all clustering in metabolic syndrome [21] and in the strictly related cardiovascular disease and kidney dysfunction [19,22,23,24,25], which are important and established causes of morbidity and mortality worldwide [26]. In all, insulin resistance is one of the main causes of suffering for many patients and their relatives while imposing a heavy burden on healthcare systems and the whole society. The heavy, vast, heterogeneous and deleterious impact of insulin resistance is not surprising considering the central role insulin is known to play in orchestrating several interwoven signals, constantly occurring in the body, which coordinate the ability of a variety of organs to sense and supplement the availability of energy molecules. Unraveling the several and complex molecular abnormalities underlying insulin resistance in various target tissues is an essential prerequisite for tackling the whole constellation comprising the aforementioned cardiometabolic abnormalities and is, therefore, timely needed.
Over the last few decades, several possible pathophysiological mechanisms leading to insulin-resistant individuals eventually developing the aforementioned metabolic and vascular disorders have been addressed. Just as an example, many studies have demonstrated that when pancreatic beta-cells fail to provide compensatory insulin secretion (chronic compensatory hyperinsulinemia response) [19,27], insulin resistance leads to a progressive loss of beta-cell mass and/or function and, thus, to a further impairment of insulin secretion. This altered interplay between insulin sensitivity and secretion, in turn, leads to and/or exacerbates lipid and carbohydrate substrate imbalance and is instrumental for the development not only of type 2 diabetes, but also atherogenic dyslipidemia (characterized by high triglycerides and low high-density lipoprotein (HDL)-cholesterol levels) and all the additional abnormalities of the constellation described above [19,27,28,29].
The intertwined risk factors shaping the risk of developing insulin resistance also draw a complex scenario that has yet to be fully clarified. In general, it is widely accepted that over both genetic predisposition and environmental factors, obesity and adipose tissue dysfunction are major movers of insulin resistance [30,31]. It is also well established that insulin resistance is the major, and probably the only, link between overweight/obesity and the numerous adverse clinical outcomes related to adiposity excess, including the aforementioned metabolic syndrome, cardiovascular disease, kidney dysfunction, fatty liver disease and also some cancers [32]. Keeping focused on adipose tissue homeostasis, which is one of the main objectives of this article, it is of note that adiposity excess has been largely demonstrated to eventually cause dysfunctional and insulin-resistant adipose tissue, which, in turn, as plasma lipid levels increase, leads to visceral and ectopic fat accumulation [33]. However, it is also true, and very interesting indeed, that adipose tissue dysfunctionalities have also been described in non-obese insulin-resistant individuals [34,35,36,37], thus leaving open the possibility that the relationships between insulin resistance and adipose tissue dysfunction are binary, with each of the two players representing the possible cause or possible effect of this association. Thus, understanding the mechanisms underlying adipose tissue dysfunction is also a prerequisite for tackling the whole constellation of abnormalities which recognize insulin resistance as a common pathogenic ground and is, therefore, an urgent need.
The first step of insulin action involves its binding to the insulin receptor at the plasma membrane, which consists of two extracellular α and two transmembrane β subunits [38,39,40]. Following the binding of insulin, the receptor’s β subunit becomes tyrosine phosphorylated and then activated [38,39,40]. This activation stimulates the receptor’s intrinsic tyrosine kinase activity, which catalyzes the phosphorylation of other proteins. These proteins include several insulin receptor substrates (IRS1, IRS2, IRS3 and IRS4) [38,39,40], which act as “docking molecules” for Src-Homology 2 domain-containing proteins, eventually enabling downstream signal transmission [38,39,40]. The effects of insulin on glucose metabolism are mediated by IRS1, which activates phosphatidyl inositol-3-kinase, which then produces phosphatidyl-inositol-3,4,5-triphosphate (PIP3) [38,39,40]. PIP3, in turn, activates the serine/threonine protein kinase beta Akt2, the central mediator of insulin’s effects on glucose and lipid metabolism, including glucose uptake and the synthesis of glycogen and fatty acids [38,39,40]. Several genes encode molecules that inhibit this pathway by various mechanisms, including a direct interaction with the insulin receptor (ectonucleotide pyrophosphatase/phosphodiesterase 1, ENPP1) [41,42,43,44,45,46] or Akt2 (tribbles pseudokinase 3, TRIB3) [47], tyrosine dephosphorylation of the insulin receptor (protein tyrosine phosphatase non-receptor type 1, PTPN1), serine phosphorylation of IRS1 (tumor necrosis factor, TNF and interleukin 6, IL6) and 5′ dephosphorylation of PIP3 (inositol polyphosphate phosphatase-like 1, INPPL1; also known as SH2-containing inositol phosphatase 2, SHIP2) [38,39,40,48,49]. In some individuals, insulin resistance and related traits are likely to be caused by abnormalities in this complex network that is modulated by both stimulatory and inhibitory molecules [38,39,48,49]. In addition, some studies point to insulin resistance at the level of the pancreatic β cells as an important determinant of impaired insulin secretion [50,51], the best predictor of future loss of glucose homeostasis [52,53]. Overall, abnormal insulin signaling, through a combined effect on both insulin resistance and deficient insulin secretion, becomes a key factor in the pathogenesis of type 2 diabetes, atherogenic dyslipidemia, metabolic syndrome and all related metabolic and cardiovascular abnormalities. Although our knowledge has improved over the last three decades [40], several gaps on the precise molecular events leading to the failure of insulin signaling still remain to be understood. Some novel cues linking insulin signaling, insulin resistance, atherogenic dyslipidemia and adipose tissue dysfunction will be provided in the following part of this article.
3. GALNT2 Gene, Protein Function and Targets
GALNT2, located on chromosome 1q42.13 [4], belongs to a large family of homologous genes coding in humans for 20 different glycosyltransferases all involved in O-glycosylation [54]. As indicated by intron/exon positioning analyses [54], GALNT2 likely shares a common ancestor with the other GALNT genes [54] and exhibits more than 98% sequence similarity among species [3]. Of note, phylogenetic analyses suggest that the enzymatic function of ppGalNAc-T2 has also been highly conserved throughout evolution [55,56].
ppGalNAc-T2 participates in the initiation step of O-linked glycosylation [57,58] by covalently linking N-acetyl-D-galactosamine (GalNAc) to the hydroxyl group of serine or threonine on specific peptide acceptor motifs [5,6,7]. It is of note that in contrast to the other isoenzymes of the same family, which show distinct non-redundant substrate specificity [59,60,61], ppGalNAc-T2 serves a broad spectrum of target peptides [57], thus modifying a large number of substrates and contributing enormously to widen protein diversity [57]. In addition, GALNT2 is expressed in nearly all cell types and tissues [62] and contributes to the function of several proteins in an organ-specific manner [9,54,63,64,65,66,67,68,69,70]. Based on these premises, it is not unexpected that GALNT2/ppGalNAc-T2 is considered by far the major contributor of the whole O-glycoproteome [57].
Only a few ppGalNAc-T2-targets have been identified so far. The most studied are angiopoietin-like protein 3 (ANGPTL3) [71,72,73], and apolipoprotein C3 (ApoC-III) [9,74], both impairing triglycerides (TG) clearance through inhibition of lipoprotein lipase (LPL) [75,76,77,78,79] and phospholipid transfer protein (PLTP) [9] that transfers phospholipids from TG-rich proteins to HDLs [80,81], which all point GALNT2 as a main actor on lipid metabolism. In addition, ppGalNAc-T2 glycosylates and affects turnover, steady-state levels and dimerization [64,65,66,67] of membrane receptors involved in several energy metabolic pathways and cell growth (i.e., β1-adrenergic [64], epidermal growth factor (EGF) [65,66], insulin-like growth factor 1 (IGF-1) receptors [67]), thus potentially affecting downstream events. Finally, ppGalNAc-T2 modulates the release of TNF-α [70], a central player of chronic inflammation, adipose cell metabolism and insulin resistance [82]. Based on these findings, it is conceivable that changes in GALNT2/ppGalNAc-T2 expression and/or function may contribute to shaping the risk of several metabolic abnormalities characterized by energy substrate imbalance, systemic low-grade inflammation and vasculature damages.
4. GALNT2 and Insulin Sensitivity
Evidence on the role of GALNT2 as a mediator of insulin sensitivity comes from our studies on human liver cells (HepG2) as well as on mouse fibroblasts (3T3L1) during their differentiation into mature adipocytes [83,84]. The reduction in GALNT2 expression in HepG2 cells impairs insulin signaling and action, as indicated by the reduced ability to stimulate insulin receptor, IRS-1 and protein kinase beta Akt2 phosphorylation [83] and to down-regulate the gluconeogenetic enzyme phosphoenolpyruvate carboxykinase (PEPCK) [83]. Coherently, data in pre-adipocytes show that GALNT2 over-expression significantly improves insulin signaling and, in turn, stimulates adipocyte maturation and enlargement [84]. Unfortunately, the mechanism through which GALNT2 affects insulin sensitivity is not yet known. In this regard, it is of note that in both HepG2 and 3T3L1 cells, the expression of GALNT2 is inversely correlated with that of ENPP1, a well-established negative modulator of insulin signaling [41,42,43,44,45,46]. This correlation makes it possible to hypothesize that ENPP1 down-regulation is likely to mediate, at least partly, the insulin-sensitizing effect of GALNT2. Whether ENPP1 is a primary ppGalNAc-T2 target or its modulation is secondary to upstream O-glycosylation events is not yet known. Taken as a whole, our data on two different insulin responsive cells do suggest that GALNT2 is a positive modulator of insulin sensitivity.
A role of GALNT2 in modulating insulin sensitivity is also suggested by preliminary genetic studies indicating that GALNT2 variability is associated with HOMA-insulin resistance index in women with polycystic ovary syndrome [85].
5. GALNT2 and Atherogenic Dyslipidemia
As said before, three main players in lipid metabolism have been validated as non-redundant substrates of ppGalNAc-T2. These are ANGPTL3 [71,72,73] and ApoC-III [9,74], which affect TG clearance, and PLTP [9] that acts on HDL-C clearance. Therefore, it is not surprising that studies on gene variability, gene expression changes and loss-of-function (LOF) mutations have pointed to GALNT2 as a shaper of both serum HDL-C and TG levels [8,9,10,11,12,13,14].
Extreme conditions of GALNT2 loss of function (LOF), ranging from humans (homozygous individuals carrying the GALNT2 p.Phe104Ser or p.Gln289 * mutations) to genetically modified nonhuman primates and mice, affect HDL-C consistently across species [9,86]. Notably, GALNT2-LOF also increased TG levels in monkeys and mice but not in humans [9], thus proposing that GALNT2 modulates lipid metabolism in a species-specific manner.
Additionally, fine tuning of GALNT2 affects lipid metabolism. In detail, data from genome-wide studies highlight that the common rs4846914 SNP at the GALNT2 locus, which is associated with lower GALNT2 expression in human liver [9], is also associated with increased TG and lower HDL-C levels [14], a combination known as atherogenic dyslipidemia, which is a marker of insulin resistance and an established risk factor for coronary artery disease (CAD) [81,87]. Along the same line, data in peripheral white blood cells (PWBC) from individuals with a wide range of metabolic clinical conditions show that GALNT2 expression levels are correlated directly with HDL-C and inversely with TG serum levels [10]. Of note, GALNT2 mRNA levels remain significantly associated with TG but no longer with HDL-C in conditional analyses including both lipid fractions, thus suggesting the association with HDL-C is mainly mediated by the very much expected strong and inverse correlation between HDL-C and TG levels [88]. Finally, the GALNT2 promoter is hyper-methylated, a proxy of gene down-regulation, in patients with CAD and low HDL-C levels [19,89].
6. GALNT2, Type 2 Diabetes and Hyperglycemia
Studies in human PWBC show that GALNT2 levels are reduced in obese patients with type 2 diabetes as compared to obese patients with no diabetes and even more to non-obese control individuals [15]. These human data resemble those reporting GALNT2 down-regulation in liver of Goto-Kakizaki diabetic rats as compared to their normoglycemic counterparts [16]. The biology underlying these associations (e.g., is GALNT2 down-regulation causing hyperglycemia or, vice versa, is hyperglycemia causing GALNT2 down-regulation?) is clearly indicated by data in human cultured monocytes, where a high glucose concentration causes GALNT2 down-regulation [15]. These data together with those on the direct relationship between GALNT2 expression and insulin signaling [83,84] make it possible to hypothesize that hyperglycemia-induced GALNT2 down-regulation is part of the mechanism underlying hyperglycemia-induced insulin resistance [90].
7. GALNT2 and Obesity
Given the well-established role of insulin sensitivity in promoting fat mass expansion, it is not surprising that GALNT2 over-expression in pre-adipocytes, by improving insulin signaling, stimulates adipocyte maturation [84] and leads to enlarged mature adipocytes characterized by small and clustered LD [91]. However, it cannot be excluded that GALNT2 also affects adipogenesis independently of insulin signaling, possibly mediating coordinated changes in key genes affecting adipocyte maturation (such as Adig, Retn, Nr1h3, Ucp1, Slc2a4, Lipe, Fgf2 and Tcs22d3) [84]. Of note, GALNT2 expression is increased in subcutaneous adipose tissue from obese individuals [17], thus making it possible to hypothesize a cause–effect relationship, with GALNT2 expression changes contributing to the increase in adiposity. This hypothesis is further supported by models of GALNT2-LOF mutations in both cattle [18] and rodents [86] which, in addition to severe congenital abnormalities, show reduced body weight as compared to their wild-type counterparts [18,86].
8. Conclusions
Taken altogether, the data that we have reported here are quite consistent with the contribution of GALNT2 to several highly prevalent metabolic abnormalities sharing the common ground of insulin resistance, namely atherogenic dyslipidemia, type 2 diabetes and obesity. Unfortunately, they leave open the question on which mechanisms underlie some of the effects of GALNT2 on the aforementioned diseases. Pathways through which GALNT2 impairs lipid metabolism and insulin resistance have been partly identified, while those connecting GALNT2 to hyperglycemia/type 2 diabetes and obesity are only scarcely understood. With this caveat in mind, it is possible to envision a scenario in which GALNT2 operates through various mechanisms (Figure 1). The simplest, and probably too simple, picture is with GALNT2 changes inducing insulin resistance, which is pathogenic for virtually all the metabolic disorders mentioned above. Conversely, in a more complex and probably more realistic scenario, alternative pathogenic pathways, uniquely affecting different metabolic abnormalities, can be envisioned. This second hypothesis is facilitated by the knowledge that different degrees of GALNT2 changes affect differently specific protein functions in an organ-specific manner, thus causing clinically distinct phenotypes (pleiotropism) [88]. A clear paradigm of this pleiotropic effect comes from data showing that while mild changes in ppGalNAc-T2 activity have been associated only with dyslipidemia [9,10,13,14], very rare and severe LOF homozygous mutations, other than heavily impairing lipid metabolism [9,86,92], cause the multisystem GALNT2-congenital disorder of glycosylation (CDG2T), which severely affects the central nervous system, muscle function, immunity control, several aspects of the endocrine system, and coagulation [18,86].
Overall, we must admit that, unfortunately, no single scenario is available within a solid framework for the many effects exerted by GALNT2 on the cluster of metabolic abnormalities, all sharing the common ground of insulin resistance. Several data indicate that they may actually represent different effects with different mechanisms (e.g., pleiotropism), although it is possible that, for at least some of them, a pathogenic link mediated by abnormal insulin signaling and insulin resistance does actually exist.
A further lack of knowledge concerns the possibility of modulating GALNT2 with the drugs currently in use for other purposes (i.e., drug repositioning). To the best of our knowledge, there are no in vivo or in vitro studies that have specifically investigated the effect of any drug on GALNT2 expression or ppGalNAc-T2 enzyme activity. Given the potential role of GALNT2 on the aforementioned severe and highly prevalent disorders, studies addressing the potential role of drugs already available today on GALNT2 would certainly be useful and are, therefore, timely.
In all, several studies both in humans and animal models point to GALNT2 as a contributing molecule to highly prevalent metabolic abnormalities, including atherogenic dyslipidemia, type 2 diabetes and obesity, which all impose a heavy burden for patients, their families and society as a whole. Conversely, our knowledge of the intimate mechanisms through which GALNT2 exerts these effects is still poor. A full understanding of GALNT2 function and dysfunction is, therefore, mandatory and will certainly improve our comprehension of the aforementioned metabolic diseases. Such knowledge gain is a prerequisite for discovering new pathogenic targets to be addressed with new therapies, so as to hopefully ameliorate the strategies currently available to treat patients with related insulin-resistance abnormalities.
Author Contributions
Conceptualization, A.A., A.M., R.D.P. and V.T.; data curation, A.A; writing—original draft preparation, A.A., A.M., R.D.P. and V.T.; writing—review and editing, A.A. and R.D.P.; supervision, R.D.P. and V.T.; funding acquisition, A.M., R.D.P. and V.T. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Italian Ministry of Health Ricerca Corrente: RC2021 and RC2022 (A.M., R.D.P., V.T.).
Acknowledgments
We would like to thank Marco Giorgio Baroni (University of L’Aquila, Department of Clinical Medicine), for inviting us to write this review.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| ANGPTL3 | angiopoietin-like protein 3 |
| ApoC-III | apolipoprotein C3 |
| CAD | coronary artery disease |
| CDG2T | congenital disorder of glycosylation |
| ENPP1 | ectonucleotide pyrophosphatase/phosphodiesterase 1 |
| GalNAc | N-acetyl-D-galactosamine |
| GalNAc-T2 | polypeptide N-acetyl-galactosaminyl-transferase 2 |
| HDL | high-density lipoprotein |
| HOMA | homeostasis model assessment |
| IRS | insulin receptor substrates |
| LOF | loss of function |
| LPL | lipoprotein lipase |
| PEPCK | phosphoenolpyruvate carboxykinase |
| PIP3 | phosphatidyl-inositol-3,4,5-triphosphate |
| PLTP | phospholipid transfer protein |
| PTPN1 | protein tyrosine phosphatase non-receptor type 1 |
| PWBC | peripheral white blood cells |
| SH2 | Src homology 2 |
| SHIP2 | SH2-containing inositol phosphatase 2 |
| TG | triglycerides |
| TRIB3 | tribbles pseudokinase 3 |
References
- Lira-Navarrete, E.; de Las Rivas, M.; Compañón, I.; Pallarés, M.C.; Kong, Y.; Iglesias-Fernández, J.; Bernardes, G.J.; Peregrina, J.M.; Rovira, C.; Bernadó, P.; et al. Dynamic interplay between catalytic and lectin domains of GalNAc-transferases modulates protein O-glycosylation. Nat. Commun. 2015, 6, 6937. [Google Scholar] [CrossRef] [Green Version]
- Reily, C.; Stewart, T.J.; Renfrow, M.B.; Novak, J. Glycosylation in health and disease. Nat. Rev. Nephrol. 2019, 15, 346–366. [Google Scholar] [CrossRef] [PubMed]
- White, T.; Bennett, E.P.; Takio, K.; Sørensen, T.; Bonding, N.; Clausen, H. Purification and cDNA cloning of a human UDP-N-acetyl-alpha-D-galactosamine:polypeptide N-acetylgalactosaminyltransferase. J. Biol. Chem. 1995, 270, 24156–24165. [Google Scholar] [CrossRef] [Green Version]
- Bennett, E.P.; Weghuis, D.O.; Merkx, G.; van Kessel, A.G.; Eiberg, H.; Clausen, H. Genomic organization and chromosomal localization of three members of the UDP-N-acetylgalactosamine: Polypeptide N-acetylgalactosaminyltransferase family. Glycobiology 1998, 8, 547–555. [Google Scholar] [CrossRef] [Green Version]
- Pisano, A.; Redmond, J.W.; Williams, K.L.; Gooley, A.A. Glycosylation sites identified by solid-phase Edman degradation: O-linked glycosylation motifs on human glycophorin A. Glycobiology 1993, 3, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Hansen, J.E.; Lund, O.; Nielsen, J.O.; Brunak, S. O-GLYCBASE: A revised database of O-glycosylated proteins. Nucleic Acids Res. 1996, 24, 248–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, R.; Birch, H.; Rapacki, K.; Brunak, S.; Hansen, J.E. O-GLYCBASE version 4.0: A revised database of O-glycosylated proteins. Nucleic Acids Res. 1999, 27, 370–372. [Google Scholar] [CrossRef] [PubMed]
- Roman, T.S.; Marvelle, A.F.; Fogarty, M.P.; Vadlamudi, S.; Gonzalez, A.J.; Buchkovich, M.L.; Huyghe, J.R.; Fuchsberger, C.; Jackson, A.U.; Wu, Y.; et al. Multiple Hepatic Regulatory Variants at the GALNT2 GWAS Locus Associated with High-Density Lipoprotein Cholesterol. Am. J. Hum. Genet. 2015, 97, 801–815. [Google Scholar] [CrossRef] [Green Version]
- Khetarpal, S.A.; Schjoldager, K.T.; Christoffersen, C.; Raghavan, A.; Edmondson, A.C.; Reutter, H.M.; Ahmed, B.; Ouazzani, R.; Peloso, G.M.; Vitali, C.; et al. Loss of Function of GALNT2 Lowers High-Density Lipoproteins in Humans, Nonhuman Primates, and Rodents. Cell Metab. 2016, 24, 234–245. [Google Scholar] [CrossRef] [Green Version]
- Marucci, A.; Mangiacotti, D.; Trischitta, V.; Di Paola, R. GALNT2 mRNA levels are associated with serum triglycerides in humans. Endocrine 2016, 53, 331–334. [Google Scholar] [CrossRef]
- Willer, C.J.; Sanna, S.; Jackson, A.U.; Scuteri, A.; Bonnycastle, L.L.; Clarke, R.; Heath, S.C.; Timpson, N.J.; Najjar, S.S.; Stringham, H.M.; et al. Newly identified loci that influence lipid concentrations and risk of coronary artery disease. Nat. Genet. 2008, 40, 161–169. [Google Scholar] [CrossRef] [Green Version]
- Willer, C.J.; Mohlke, K.L. Finding genes and variants for lipid levels after genome-wide association analysis. Curr. Opin. Lipidol. 2012, 23, 98–103. [Google Scholar] [CrossRef] [Green Version]
- Teslovich, T.M.; Musunuru, K.; Smith, A.V.; Edmondson, A.C.; Stylianou, I.M.; Koseki, M.; Pirruccello, J.P.; Ripatti, S.; Chasman, D.I.; Willer, C.J.; et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature 2010, 466, 707–713. [Google Scholar] [CrossRef]
- Kathiresan, S.; Melander, O.; Guiducci, C.; Surti, A.; Burtt, N.P.; Rieder, M.J.; Cooper, G.M.; Roos, C.; Voight, B.F.; Havulinna, A.S.; et al. Six new loci associated with blood low-density lipoprotein cholesterol, high-density lipoprotein cholesterol or triglycerides in humans. Nat. Genet. 2008, 40, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Marucci, A.; di Mauro, L.; Menzaghi, C.; Prudente, S.; Mangiacotti, D.; Fini, G.; Lotti, G.; Trischitta, V.; Di Paola, R. GALNT2 expression is reduced in patients with Type 2 diabetes: Possible role of hyperglycemia. PLoS ONE 2013, 8, e70159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almon, R.R.; DuBois, D.C.; Lai, W.; Xue, B.; Nie, J.; Jusko, W.J. Gene expression analysis of hepatic roles in cause and development of diabetes in Goto-Kakizaki rats. J. Endocrinol. 2009, 200, 331–346. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.H.; Nair, S.; Rousseau, E.; Allison, D.B.; Page, G.P.; Tataranni, P.A.; Bogardus, C.; Permana, P.A. Microarray profiling of isolated abdominal subcutaneous adipocytes from obese vs non-obese Pima Indians: Increased expression of inflammation-related genes. Diabetologia 2005, 48, 1776–1783. [Google Scholar] [CrossRef] [Green Version]
- Reynolds, E.G.M.; Neeley, C.; Lopdell, T.J.; Keehan, M.; Dittmer, K.; Harland, C.S.; Couldrey, C.; Johnson, T.J.J.; Tiplady, K.; Worth, G.; et al. Non-additive association analysis using proxy phenotypes identifies novel cattle syndromes. Nat. Genet. 2021, 53, 949–954. [Google Scholar] [CrossRef]
- Reaven, G.M. Banting lecture 1988. Role of insulin resistance in human disease. Diabetes 1988, 37, 1595–1607. [Google Scholar] [CrossRef]
- Ferrannini, E.; Buzzigoli, G.; Bonadonna, R.; Giorico, M.A.; Oleggini, M.; Graziadei, L.; Pedrinelli, R.; Brandi, L.; Bevilacqua, S. Insulin resistance in essential hypertension. N. Engl. J. Med. 1987, 317, 350–357. [Google Scholar] [CrossRef] [PubMed]
- Eckel, R.H.; Alberti, K.G.; Grundy, S.M.; Zimmet, P.Z. The metabolic syndrome. Lancet 2010, 375, 181–183. [Google Scholar] [CrossRef]
- Hedblad, B.; Nilsson, P.; Engström, G.; Berglund, G.; Janzon, L. Insulin resistance in non-diabetic subjects is associated with increased incidence of myocardial infarction and death. Diabet. Med. 2002, 19, 470–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, P.W.; D’Agostino, R.B.; Parise, H.; Sullivan, L.; Meigs, J.B. Metabolic syndrome as a precursor of cardiovascular disease and type 2 diabetes mellitus. Circulation 2005, 112, 3066–3072. [Google Scholar] [CrossRef] [PubMed]
- Wannamethee, S.G.; Shaper, A.G.; Lennon, L.; Morris, R.W. Metabolic syndrome vs Framingham Risk Score for prediction of coronary heart disease, stroke, and type 2 diabetes mellitus. Arch. Intern. Med. 2005, 165, 2644–2650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, J.E.; do Carmo, J.M.; da Silva, A.A.; Wang, Z.; Hall, M.E. Obesity, kidney dysfunction and hypertension: Mechanistic links. Nat. Rev. Nephrol. 2019, 15, 367–385. [Google Scholar] [CrossRef]
- WHO. Noncommunicable Diseases, Progress Monitor 2017; WHO: Geneva, Switzerland, 2017. [Google Scholar]
- Kahn, C.R. Banting Lecture. Insulin action, diabetogenes, and the cause of type II diabetes. Diabetes 1994, 43, 1066–1084. [Google Scholar] [CrossRef]
- Martin, B.C.; Warram, J.H.; Krolewski, A.S.; Bergman, R.N.; Soeldner, J.S.; Kahn, C.R. Role of glucose and insulin resistance in development of type 2 diabetes mellitus: Results of a 25-year follow-up study. Lancet 1992, 340, 925–929. [Google Scholar] [CrossRef]
- Weyer, C.; Bogardus, C.; Mott, D.M.; Pratley, R.E. The natural history of insulin secretory dysfunction and insulin resistance in the pathogenesis of type 2 diabetes mellitus. J. Clin. Investig. 1999, 104, 787–794. [Google Scholar] [CrossRef] [PubMed]
- Ghaben, A.L.; Scherer, P.E. Adipogenesis and metabolic health. Nat. Rev. Mol. Cell Biol. 2019, 20, 242–258. [Google Scholar] [CrossRef]
- Smith, U.; Kahn, B.B. Adipose tissue regulates insulin sensitivity: Role of adipogenesis, de novo lipogenesis and novel lipids. J. Intern. Med. 2016, 280, 465–475. [Google Scholar] [CrossRef] [Green Version]
- Reaven, G.M. Insulin resistance: The link between obesity and cardiovascular disease. Med. Clin. N. Am. 2011, 95, 875–892. [Google Scholar] [CrossRef] [PubMed]
- Rutkowski, J.M.; Stern, J.H.; Scherer, P.E. The cell biology of fat expansion. J. Cell Biol. 2015, 208, 501–512. [Google Scholar] [CrossRef] [Green Version]
- Vatier, C.; Vantyghem, M.C.; Storey, C.; Jéru, I.; Christin-Maitre, S.; Fève, B.; Lascols, O.; Beltrand, J.; Carel, J.C.; Vigouroux, C.; et al. Monogenic forms of lipodystrophic syndromes: Diagnosis, detection, and practical management considerations from clinical cases. Curr. Med. Res. Opin. 2019, 35, 543–552. [Google Scholar] [CrossRef] [PubMed]
- Van Maldergem, L.; Magré, J.; Khallouf, T.E.; Gedde-Dahl, T.; Delépine, M.; Trygstad, O.; Seemanova, E.; Stephenson, T.; Albott, C.S.; Bonnici, F.; et al. Genotype-phenotype relationships in Berardinelli-Seip congenital lipodystrophy. J. Med. Genet. 2002, 39, 722–733. [Google Scholar] [CrossRef]
- Gomes, K.B.; Pardini, V.C.; Ferreira, A.C.; Fernandes, A.P. Phenotypic heterogeneity in biochemical parameters correlates with mutations in AGPAT2 or Seipin genes among Berardinelli-Seip congenital lipodystrophy patients. J. Inherit. Metab. Dis. 2005, 28, 1123–1131. [Google Scholar] [CrossRef]
- Knebel, B.; Müller-Wieland, D.; Kotzka, J. Lipodystrophies-Disorders of the Fatty Tissue. Int. J. Mol. Sci. 2020, 21, 8778. [Google Scholar] [CrossRef]
- Cohen, P. The twentieth century struggle to decipher insulin signalling. Nat. Rev. Mol. Cell Biol. 2006, 7, 867–873. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, C.M.; Emanuelli, B.; Kahn, C.R. Critical nodes in signalling pathways: Insights into insulin action. Nat. Rev. Mol. Cell Biol. 2006, 7, 85–96. [Google Scholar] [CrossRef]
- Saltiel, A.R. Insulin signaling in health and disease. J. Clin. Investig. 2021, 131, e142241. [Google Scholar] [CrossRef] [PubMed]
- Roberts, F.; Zhu, D.; Farquharson, C.; Macrae, V.E. ENPP1 in the Regulation of Mineralization and Beyond. Trends Biochem. Sci. 2019, 44, 616–628. [Google Scholar] [CrossRef]
- Maddux, B.A.; Sbraccia, P.; Kumakura, S.; Sasson, S.; Youngren, J.; Fisher, A.; Spencer, S.; Grupe, A.; Henzel, W.; Stewart, T.A. Membrane glycoprotein PC-1 and insulin resistance in non-insulin-dependent diabetes mellitus. Nature 1995, 373, 448–451. [Google Scholar] [CrossRef]
- Maddux, B.A.; Goldfine, I.D. Membrane glycoprotein PC-1 inhibition of insulin receptor function occurs via direct interaction with the receptor alpha-subunit. Diabetes 2000, 49, 13–19. [Google Scholar] [CrossRef] [Green Version]
- Maddux, B.; Chang, Y.; Accili, D.; McGuinness, O.; Youngren, J.; Goldfine, I. Overexpression of the insulin receptor inhibitor PC-1/ENPP1 induces insulin resistance and hyperglycemia. Am. J. Physiol. Endocrinol. Metab. 2006, 290, E746–E749. [Google Scholar] [CrossRef] [Green Version]
- Di Paola, R.; Caporarello, N.; Marucci, A.; Dimatteo, C.; Iadicicco, C.; Del Guerra, S.; Prudente, S.; Sudano, D.; Miele, C.; Parrino, C.; et al. ENPP1 affects insulin action and secretion: Evidences from in vitro studies. PLoS ONE 2011, 6, e19462. [Google Scholar] [CrossRef] [PubMed]
- Dimatteo, C.; Marucci, A.; Palazzo, A.; Cisternino, C.; Marsano, R.M.; Trischitta, V.; Di Paola, R. Role of somatomedin-B-like domains on ENPP1 inhibition of insulin signaling. Biochim. Biophys. Acta 2013, 1833, 552–558. [Google Scholar] [CrossRef] [Green Version]
- Prudente, S.; Sesti, G.; Pandolfi, A.; Andreozzi, F.; Consoli, A.; Trischitta, V. The mammalian tribbles homolog TRIB3, glucose homeostasis, and cardiovascular diseases. Endocr. Rev. 2012, 33, 526–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saltiel, A.R. Putting the brakes on insulin signaling. N. Engl. J. Med. 2003, 349, 2560–2562. [Google Scholar] [CrossRef]
- Goldfine, I.D.; Maddux, B.A.; Youngren, J.F.; Reaven, G.; Accili, D.; Trischitta, V.; Vigneri, R.; Frittitta, L. The role of membrane glycoprotein plasma cell antigen 1/ectonucleotide pyrophosphatase phosphodiesterase 1 in the pathogenesis of insulin resistance and related abnormalities. Endocr. Rev. 2008, 29, 62–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aspinwall, C.A.; Qian, W.J.; Roper, M.G.; Kulkarni, R.N.; Kahn, C.R.; Kennedy, R.T. Roles of insulin receptor substrate-1, phosphatidylinositol 3-kinase, and release of intracellular Ca2+ stores in insulin-stimulated insulin secretion in beta-cells. J. Biol. Chem. 2000, 275, 22331–22338. [Google Scholar] [CrossRef] [Green Version]
- Kulkarni, R.N.; Brüning, J.C.; Winnay, J.N.; Postic, C.; Magnuson, M.A.; Kahn, C.R. Tissue-specific knockout of the insulin receptor in pancreatic beta cells creates an insulin secretory defect similar to that in type 2 diabetes. Cell 1999, 96, 329–339. [Google Scholar] [CrossRef] [Green Version]
- Lyssenko, V.; Almgren, P.; Anevski, D.; Perfekt, R.; Lahti, K.; Nissén, M.; Isomaa, B.; Forsen, B.; Homström, N.; Saloranta, C.; et al. Predictors of and longitudinal changes in insulin sensitivity and secretion preceding onset of type 2 diabetes. Diabetes 2005, 54, 166–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdul-Ghani, M.A.; Williams, K.; DeFronzo, R.A.; Stern, M. What is the best predictor of future type 2 diabetes? Diabetes Care 2007, 30, 1544–1548. [Google Scholar] [CrossRef] [Green Version]
- Bennett, E.P.; Mandel, U.; Clausen, H.; Gerken, T.A.; Fritz, T.A.; Tabak, L.A. Control of mucin-type O-glycosylation: A classification of the polypeptide GalNAc-transferase gene family. Glycobiology 2012, 22, 736–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwientek, T.; Bennett, E.P.; Flores, C.; Thacker, J.; Hollmann, M.; Reis, C.A.; Behrens, J.; Mandel, U.; Keck, B.; Schäfer, M.A.; et al. Functional conservation of subfamilies of putative UDP-N-acetylgalactosamine:polypeptide N-acetylgalactosaminyltransferases in Drosophila, Caenorhabditis elegans, and mammals. One subfamily composed of l(2)35Aa is essential in Drosophila. J. Biol. Chem. 2002, 277, 22623–22638. [Google Scholar] [CrossRef] [Green Version]
- Kaneko, M.N.S.; Narimatsu, H.; Saitou, N. The evolutionary history of glycosyltransferase genes. Trends Glycosci. Glycotechnol. 2000, 13, 147–155. [Google Scholar] [CrossRef]
- Schjoldager, K.T.; Narimatsu, Y.; Joshi, H.J.; Clausen, H. Global view of human protein glycosylation pathways and functions. Nat. Rev. Mol. Cell Biol. 2020, 21, 729–749. [Google Scholar] [CrossRef]
- Steentoft, C.; Vakhrushev, S.Y.; Joshi, H.J.; Kong, Y.; Vester-Christensen, M.B.; Schjoldager, K.T.; Lavrsen, K.; Dabelsteen, S.; Pedersen, N.B.; Marcos-Silva, L.; et al. Precision mapping of the human O-GalNAc glycoproteome through SimpleCell technology. EMBO J. 2013, 32, 1478–1488. [Google Scholar] [CrossRef] [Green Version]
- Schjoldager, K.T.; Joshi, H.J.; Kong, Y.; Goth, C.K.; King, S.L.; Wandall, H.H.; Bennett, E.P.; Vakhrushev, S.Y.; Clausen, H. Deconstruction of O-glycosylation—Gal NA c-T isoforms direct distinct subsets of the O-glycoproteome. EMBO Rep. 2015, 16, 1713–1722. [Google Scholar] [CrossRef] [Green Version]
- Narimatsu, Y.; Joshi, H.J.; Schjoldager, K.T.; Hintze, J.; Halim, A.; Steentoft, C.; Nason, R.; Mandel, U.; Bennett, E.P.; Clausen, H.; et al. Exploring regulation of protein O-glycosylation in isogenic human HEK293 cells by differential O-glycoproteomics. Mol. Cell. Proteom. 2019, 18, 1396–1409. [Google Scholar] [CrossRef] [PubMed]
- Bagdonaite, I.; Pallesen, E.M.H.; Ye, Z.; Vakhrushev, S.Y.; Marinova, I.N.; Nielsen, M.I.; Kramer, S.H.; Pedersen, S.F.; Joshi, H.J.; Bennett, E.P.; et al. O-glycan initiation directs distinct biological pathways and controls epithelial differentiation. EMBO Rep. 2020, 21, 1–17. [Google Scholar] [CrossRef]
- Fagerberg, L.; Hallström, B.M.; Oksvold, P.; Kampf, C.; Djureinovic, D.; Odeberg, J.; Habuka, M.; Tahmasebpoor, S.; Danielsson, A.; Edlund, K.; et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell Proteom. 2014, 13, 397–406. [Google Scholar] [CrossRef] [Green Version]
- Lackman, J.J.; Goth, C.K.; Halim, A.; Vakhrushev, S.Y.; Clausen, H.; Petäjä-Repo, U.E. Site-specific O-glycosylation of N-terminal serine residues by polypeptide GalNAc-transferase 2 modulates human δ-opioid receptor turnover at the plasma membrane. Cell. Signal. 2018, 42, 184–193. [Google Scholar] [CrossRef] [PubMed]
- Goth, C.K.; Tuhkanen, H.E.; Khan, H.; Lackman, J.J.; Wang, S.; Narimatsu, Y.; Hansen, L.H.; Overall, C.M.; Clausen, H.; Schjoldager, K.T.; et al. Site-specific O-glycosylation by polypeptide N-acetylgalactosaminyltransferase 2 (GalNAc-transferase T2) co-regulates β1-adrenergic receptor N-terminal cleavage. J. Biol. Chem. 2017, 292, 4714–4726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, M.C.; Huang, M.J.; Liu, C.H.; Yang, T.L.; Huang, M.C. GALNT2 enhances migration and invasion of oral squamous cell carcinoma by regulating EGFR glycosylation and activity. Oral Oncol. 2014, 50, 478–484. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.M.; Liu, C.H.; Hu, R.H.; Huang, M.J.; Lee, J.; Chen, C.H.; Huang, J.; Lai, H.S.; Lee, P.H.; Hsu, W.M.; et al. Mucin glycosylating enzyme GALNT2 regulates the malignant character of hepatocellular carcinoma by modifying the EGF receptor. Cancer Res. 2011, 71, 7270–7279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, W.L.; Chou, C.H.; Jeng, Y.M.; Lu, M.Y.; Yang, Y.L.; Jou, S.T.; Lin, D.T.; Chang, H.H.; Lin, K.H.; Hsu, W.M.; et al. GALNT2 suppresses malignant phenotypes through IGF-1 receptor and predicts favorable prognosis in neuroblastoma. Oncotarget 2014, 5, 12247–12259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, W.-T.; Yeh, C.-C.; Liu, S.-Y.; Huang, M.-C.; Lai, I.R. The O-glycosylating enzyme GALNT2 suppresses the malignancy of gastric adenocarcinoma by reducing EGFR activities. Am. J. Cancer Res. 2018, 8, 1739–1751. [Google Scholar]
- Schjoldager, K.T.B.G.; Clausen, H. Site-specific protein O-glycosylation modulates proprotein processing-Deciphering specific functions of the large polypeptide GalNAc-transferase gene family. Biochim. Biophys. Acta-Gen. Subj. 2012, 1820, 2079–2094. [Google Scholar] [CrossRef]
- Goth, C.K.; Halim, A.; Khetarpal, S.A.; Rader, D.J.; Clausen, H.; Schjoldager, K.T.B.G. A systematic study of modulation of ADAM-mediated ectodomain shedding by site-specific O-glycosylation. Proc. Natl. Acad. Sci. USA 2015, 112, 14623–14628. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Zhang, Y.; Zhang, M.; Wang, Y. GALNT2 regulates ANGPTL3 cleavage in cells and in vivo of mice. Sci. Rep. 2020, 10, 1–9. [Google Scholar] [CrossRef]
- Schjoldager, K.T.B.G.; Vester-Christensen, M.B.; Bennett, E.P.; Levery, S.B.; Schwientek, T.; Yin, W.; Blixt, O.; Clausen, H. O-glycosylation modulates proprotein convertase activation of angiopoietin-like protein 3: Possible role of polypeptide GalNAc-transferase-2 in regulation of concentrations of plasma lipids. J. Biol. Chem. 2010, 285, 36293–36303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, H.B.; Wang, J.Y.; Sun, Z.L. ANGPTL3 is part of the machinery causing dyslipidemia majorily via LPL inhibition in mastitis mice. Exp. Mol. Pathol. 2017, 103, 242–248. [Google Scholar] [CrossRef]
- Garner, B.; Merry, A.H.; Royle, L.; Harvey, D.J.; Rudd, P.M.; Thillet, J. Structural elucidation of the N- and O-glycans of human apolipoprotein(a): Role of o-glycans in conferring protease resistance. J. Biol. Chem. 2001, 276, 22200–22208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kersten, S. Angiopoietin-like 3 in lipoprotein metabolism. Nat. Rev. Endocrinol. 2017, 13, 731–739. [Google Scholar] [CrossRef]
- Xu, Y.X.; Redon, V.; Yu, H.; Querbes, W.; Pirruccello, J.; Liebow, A.; Deik, A.; Trindade, K.; Wang, X.; Musunuru, K.; et al. Role of angiopoietin-like 3 (ANGPTL3) in regulating plasma level of low-density lipoprotein cholesterol. Atherosclerosis 2018, 268, 196–206. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Soffer, G.; Sztalryd, C.; Horenstein, R.B.; Holleran, S.; Matveyenko, A.; Thomas, T.; Nandakumar, R.; Ngai, C.; Karmally, W.; Ginsberg, H.N.; et al. Effects of APOC3 Heterozygous Deficiency on Plasma Lipid and Lipoprotein Metabolism. Arter. Thromb. Vasc. Biol. 2019, 39, 63–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borén, J.; Packard, C.J.; Taskinen, M.R. The Roles of ApoC-III on the Metabolism of Triglyceride-Rich Lipoproteins in Humans. Front. Endocrinol. 2020, 11, 474. [Google Scholar] [CrossRef] [PubMed]
- Hegele, R.A.; Ban, M.R.; Hsueh, N.; Kennedy, B.A.; Cao, H.; Zou, G.Y.; Anand, S.; Yusuf, S.; Huff, M.W.; Wang, J. A polygenic basis for four classical Fredrickson hyperlipoproteinemia phenotypes that are characterized by hypertriglyceridemia. Hum. Mol. Genet. 2009, 18, 4189–4194. [Google Scholar] [CrossRef] [Green Version]
- Rao, R.; Albers, J.J.; Wolfbauer, G.; Pownall, H.J. Molecular and macromolecular specificity of human plasma phospholipid transfer protein. Biochemistry 1997, 36, 3645–3653. [Google Scholar] [CrossRef] [PubMed]
- Tall, A.R.; Krumholz, S.; Olivecrona, T.; Deckelbaum, R.J. Plasma phospholipid transfer protein enhances transfer and exchange of phospholipids between very low density lipoproteins and high density lipoproteins during lipolysis. J. Lipid Res. 1985, 26, 842–851. [Google Scholar] [CrossRef]
- Vassalli, P. The pathophysiology of tumor necrosis factors. Annu. Rev. Immunol. 1992, 10, 411–452. [Google Scholar] [CrossRef] [PubMed]
- Marucci, A.; Cozzolino, F.; Dimatteo, C.; Monti, M.; Pucci, P.; Trischitta, V.; Di Paola, R. Role of GALNT2 in the modulation of ENPP1 expression, and insulin signaling and action: GALNT2: A novel modulator of insulin signaling. Biochim. Biophys. Acta 2013, 1833, 1388–1395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marucci, A.; Antonucci, A.; De Bonis, C.; Mangiacotti, D.; Scarale, M.G.; Trischitta, V.; Di Paola, R. GALNT2 as a novel modulator of adipogenesis and adipocyte insulin signaling. Int. J. Obes. 2019, 43, 2448–2457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Guan, L.; Liu, H.; Liu, Q.; Fan, P.; Bai, H. GALNT2 Gene Variant rs4846914 Is Associated with Insulin and Insulin Resistance Depending on BMI in PCOS Patients: A Case-Control Study. Reprod. Sci. 2021, 28, 1122–1132. [Google Scholar] [CrossRef] [PubMed]
- Zilmer, M.; Edmondson, A.C.; Khetarpal, S.A.; Alesi, V.; Zaki, M.S.; Rostasy, K.; Madsen, C.G.; Lepri, F.R.; Sinibaldi, L.; Cusmai, R.; et al. Novel congenital disorder of O-linked glycosylation caused by GALNT2 loss of function. Brain 2020, 143, 1114–1126. [Google Scholar] [CrossRef]
- Grundy, S.M. Hypertriglyceridemia, atherogenic dyslipidemia, and the metabolic syndrome. Am. J. Cardiol. 1998, 81, 18B–25B. [Google Scholar] [CrossRef]
- Di Paola, R.; Marucci, A.; Trischitta, V. GALNT2 effect on HDL-cholesterol and triglycerides levels in humans: Evidence of pleiotropy? Nutr. Metab. Cardiovasc. Dis. 2017, 27, 281–282. [Google Scholar] [CrossRef]
- Ghose, S.; Ghosh, S.; Tanwar, V.S.; Tolani, P.; Kutum, R.; Sharma, A.; Bhardwaj, N.; Shamsudheen, K.V.; Verma, A.; Jayarajan, R.; et al. Investigating Coronary Artery Disease methylome through targeted bisulfite sequencing. Gene 2019, 721, 144107. [Google Scholar] [CrossRef]
- Rossetti, L.; Giaccari, A.; DeFronzo, R.A. Glucose toxicity. Diabetes Care 1990, 13, 610–630. [Google Scholar] [CrossRef]
- Antonucci, A.; Marucci, A.; Scarale, M.G.; De Bonis, C.; Mangiacotti, D.; Trischitta, V.; Di Paola, R. Morphological and molecular characterization of GALNT2-mediated adipogenesis. Int. J. Obes. 2021, 45, 1362–1366. [Google Scholar] [CrossRef]
- Tietjen, I.; Hovingh, G.K.; Singaraja, R.R.; Radomski, C.; Barhdadi, A.; McEwen, J.; Chan, E.; Mattice, M.; Legendre, A.; Franchini, P.L.; et al. Segregation of LIPG, CETP, and GALNT2 mutations in Caucasian families with extremely high HDL cholesterol. PLoS ONE 2012, 7, e37437. [Google Scholar] [CrossRef] [Green Version]
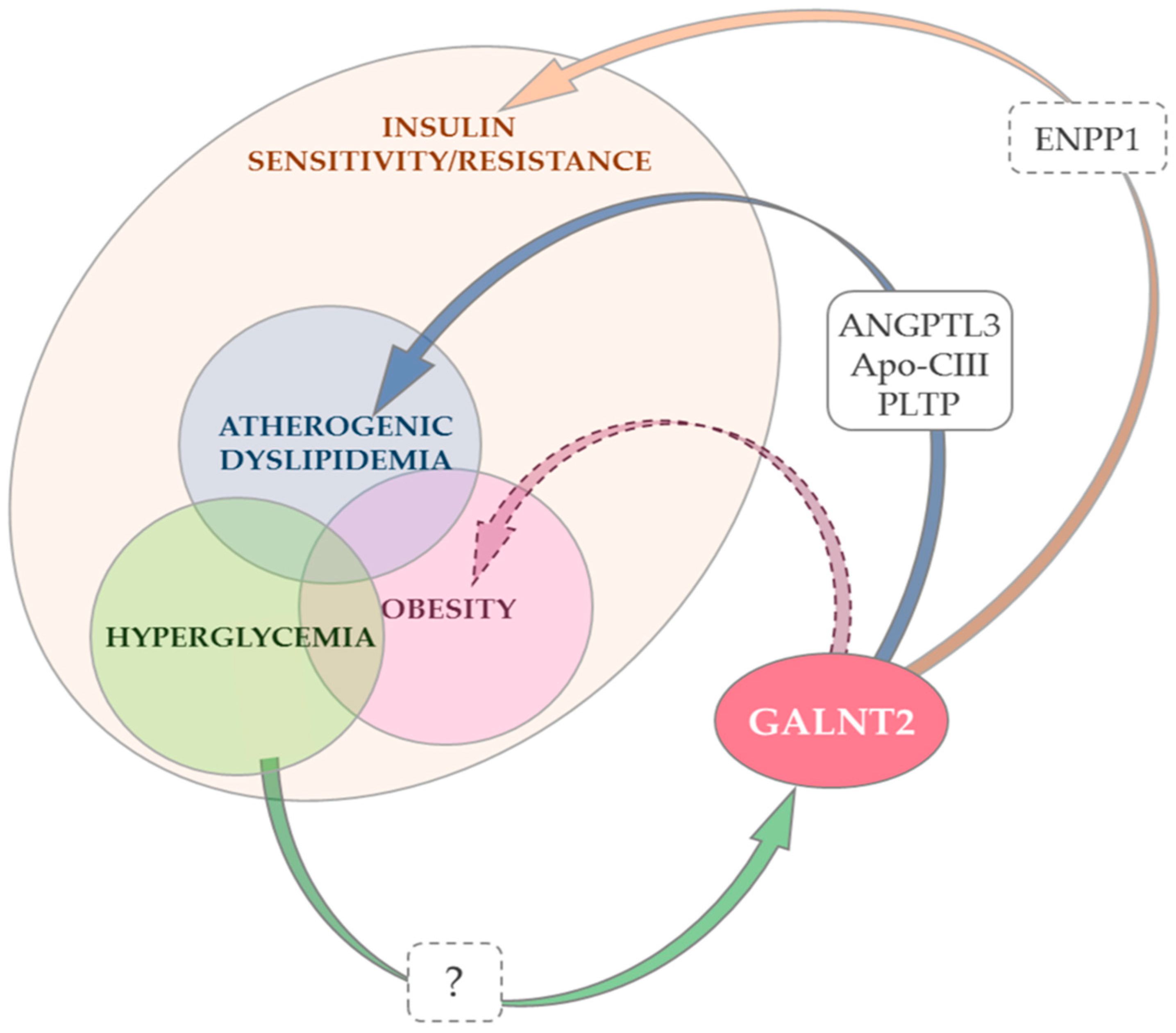
Figure 1.
Changes in GALNT2 expression and/or function and insulin resistance, atherogenic dyslipidemia and obesity. Notes: GALNT2 promotes insulin action by down-regulating ectonucleotide pyrophosphatase/phosphodiesterase 1 (ENPP1) [83], a negative modulator of insulin signaling [41,42,43,44,45,46] (orange arrow). Whether ENPP1 is a primary ppGalNAc-T2 target or its down-regulation is secondary to upstream O-glycosylation events is unknown (dashed box). The role of GALNT2 down-regulation in atherogenic dyslipidemia (low HDL-Cholesterol and/or high triglycerides) is best understood. This effect is mediated by the O-glycosylation of three established targets of ppGalNAc-T2 [9] (the product of GALNT2) (blue arrow). Although no data are currently available, it cannot be excluded that some of the deleterious effects of GALNT2 down-regulation on dyslipidemia are mediated by the concurrent deleterious effect on insulin signaling which, in turn, causes insulin resistance and low HDL-cholesterol/high-triglyceride levels [19]. Experimental data suggest that the effect of GALNT2 on adipocyte maturation, adipogenesis and eventually obesity is at least partly due to its positive modulation of insulin signaling [84]. Whether GALNT2 also affects adipogenesis through other mechanisms, possibly mediating coordinated changes in key genes affecting adipocyte maturation [84], is an alternative possibility that deserves further studies to be addressed (purple dashed arrow). Finally, a high glucose concentration decreases GALNT2 expression and this may play a role in hyperglycemia-induced insulin resistance (glucose toxicity) [15]. The mechanisms through which this down-regulation occurs are not known (green arrow, dashed box).
Figure 1.
Changes in GALNT2 expression and/or function and insulin resistance, atherogenic dyslipidemia and obesity. Notes: GALNT2 promotes insulin action by down-regulating ectonucleotide pyrophosphatase/phosphodiesterase 1 (ENPP1) [83], a negative modulator of insulin signaling [41,42,43,44,45,46] (orange arrow). Whether ENPP1 is a primary ppGalNAc-T2 target or its down-regulation is secondary to upstream O-glycosylation events is unknown (dashed box). The role of GALNT2 down-regulation in atherogenic dyslipidemia (low HDL-Cholesterol and/or high triglycerides) is best understood. This effect is mediated by the O-glycosylation of three established targets of ppGalNAc-T2 [9] (the product of GALNT2) (blue arrow). Although no data are currently available, it cannot be excluded that some of the deleterious effects of GALNT2 down-regulation on dyslipidemia are mediated by the concurrent deleterious effect on insulin signaling which, in turn, causes insulin resistance and low HDL-cholesterol/high-triglyceride levels [19]. Experimental data suggest that the effect of GALNT2 on adipocyte maturation, adipogenesis and eventually obesity is at least partly due to its positive modulation of insulin signaling [84]. Whether GALNT2 also affects adipogenesis through other mechanisms, possibly mediating coordinated changes in key genes affecting adipocyte maturation [84], is an alternative possibility that deserves further studies to be addressed (purple dashed arrow). Finally, a high glucose concentration decreases GALNT2 expression and this may play a role in hyperglycemia-induced insulin resistance (glucose toxicity) [15]. The mechanisms through which this down-regulation occurs are not known (green arrow, dashed box).

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Antonucci, A.; Marucci, A.; Trischitta, V.; Di Paola, R. Role of GALNT2 on Insulin Sensitivity, Lipid Metabolism and Fat Homeostasis. Int. J. Mol. Sci. 2022, 23, 929. https://doi.org/10.3390/ijms23020929
AMA Style
Antonucci A, Marucci A, Trischitta V, Di Paola R. Role of GALNT2 on Insulin Sensitivity, Lipid Metabolism and Fat Homeostasis. International Journal of Molecular Sciences. 2022; 23(2):929. https://doi.org/10.3390/ijms23020929
Chicago/Turabian StyleAntonucci, Alessandra, Antonella Marucci, Vincenzo Trischitta, and Rosa Di Paola. 2022. "Role of GALNT2 on Insulin Sensitivity, Lipid Metabolism and Fat Homeostasis" International Journal of Molecular Sciences 23, no. 2: 929. https://doi.org/10.3390/ijms23020929
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.