
Optimization of Protoplast Isolation and Transformation for a Pilot Study of Genome Editing in Peanut by Targeting the Allergen Gene Ara h 2

 , ,
, ,
Abstract
:1. Introduction
2. Results
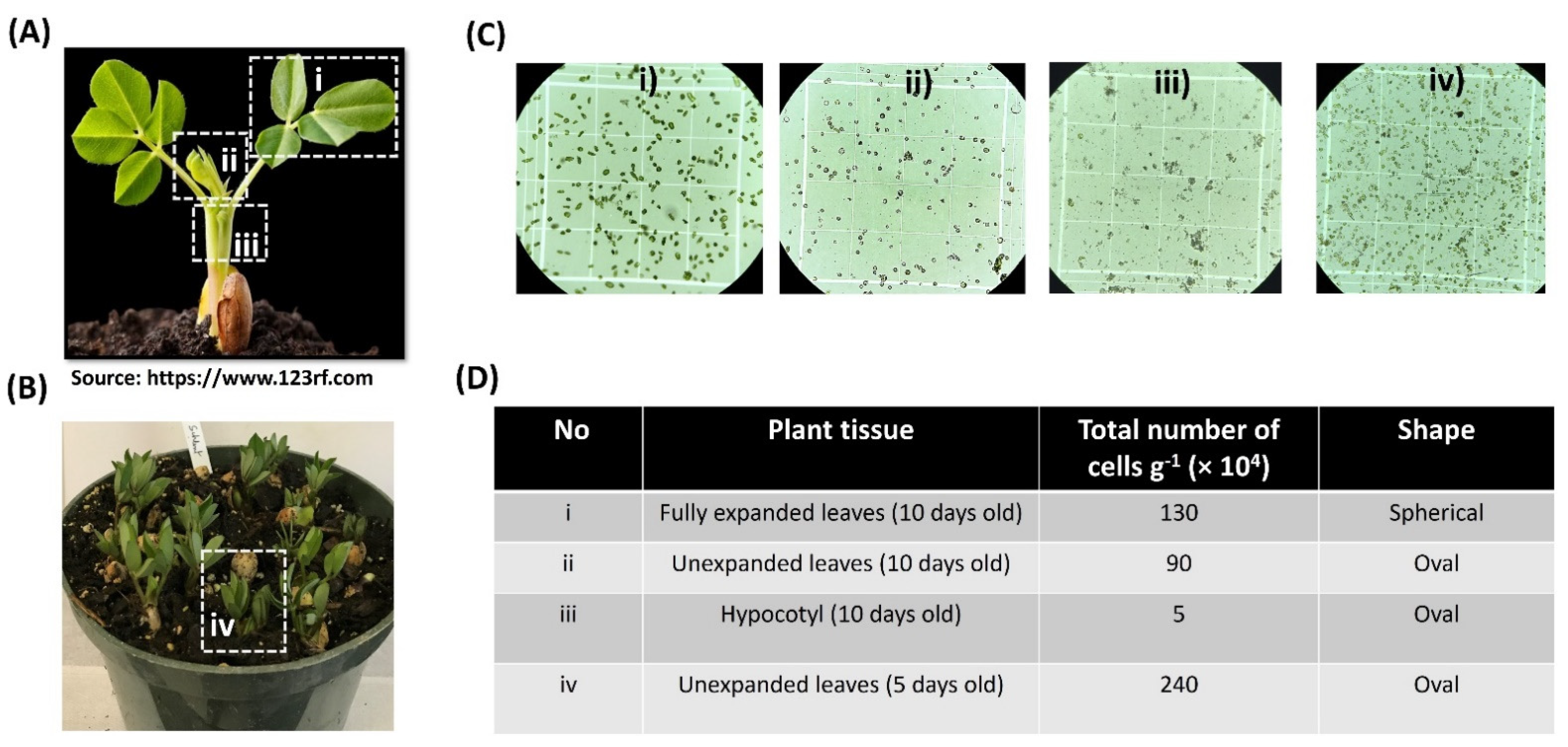
2.1. An Efficient Method of Protoplast Isolation from Peanut Seedlings
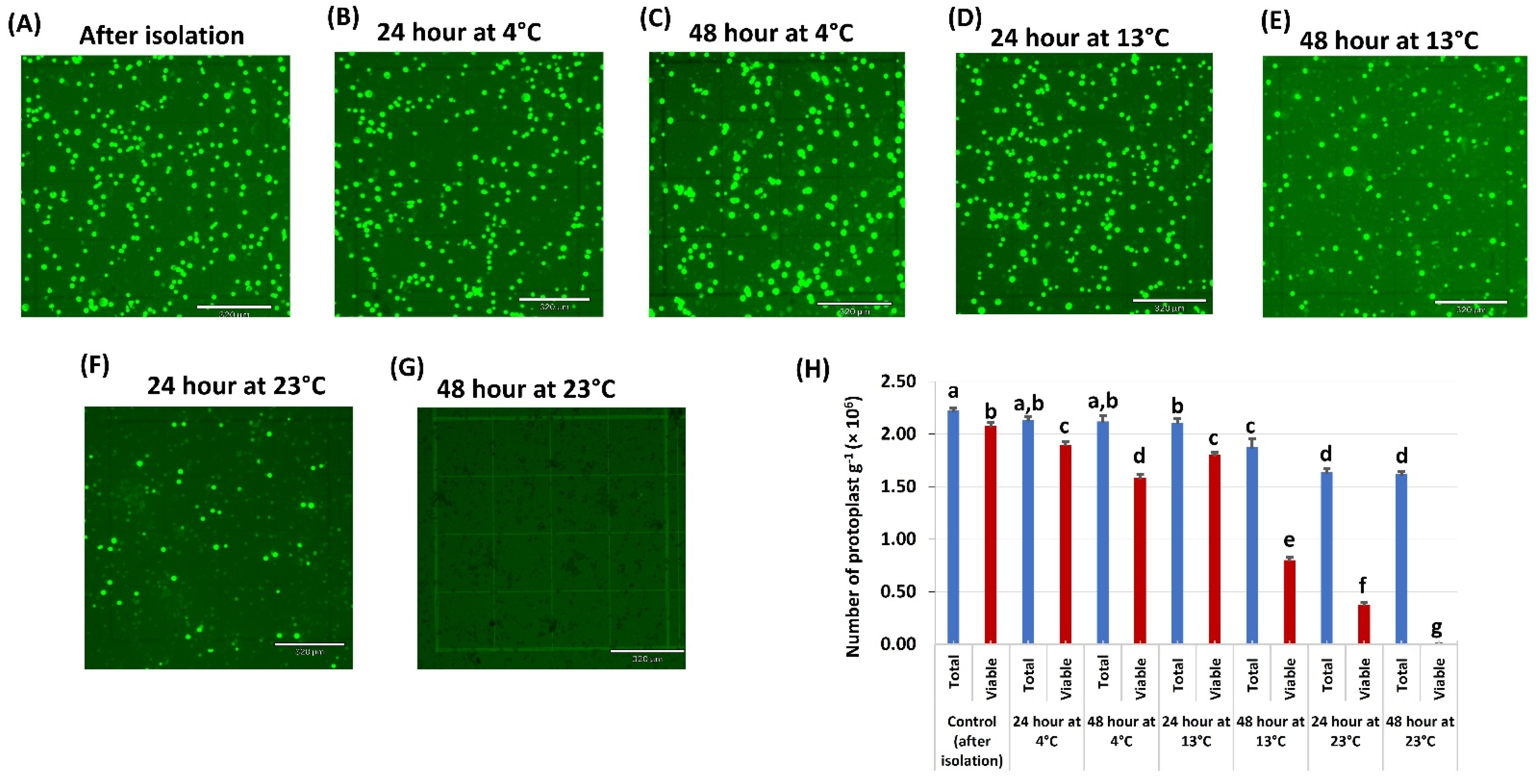
2.2. Temperature Effect on Protoplast Viability and Testing Constitutive Promoters
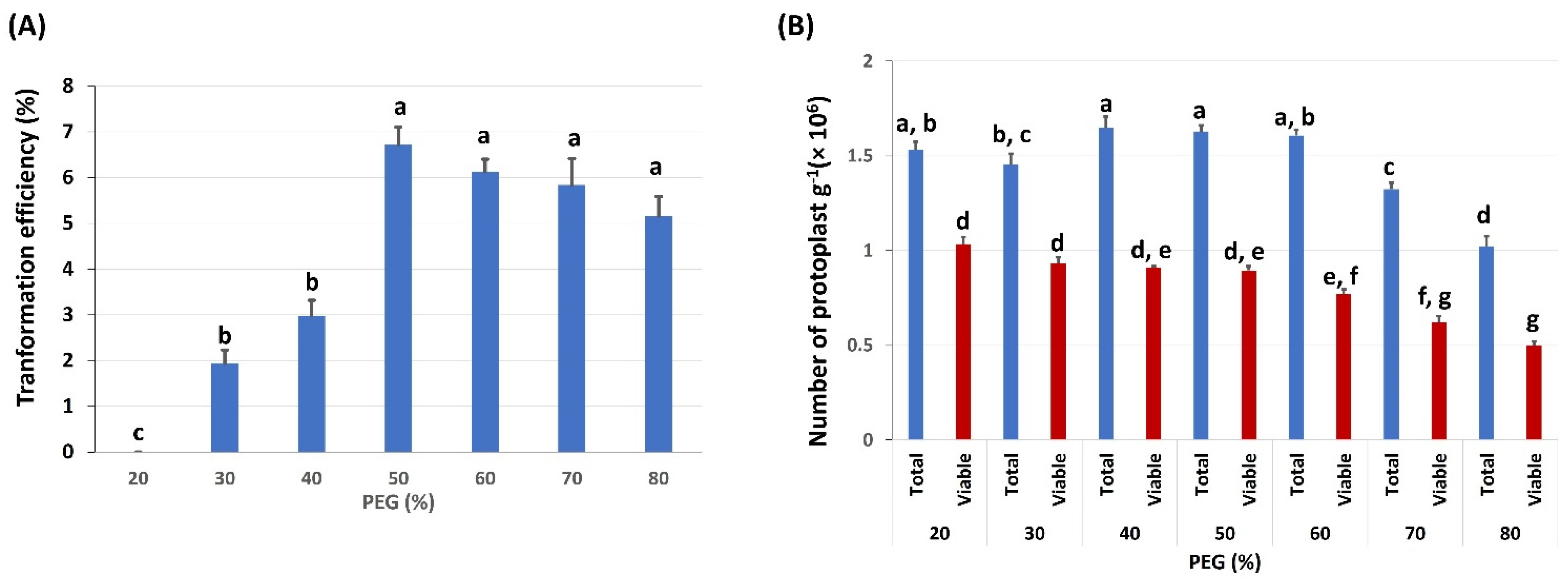
2.3. Effects of PEG Concentration on Protoplast Transformation Efficiency and Viability
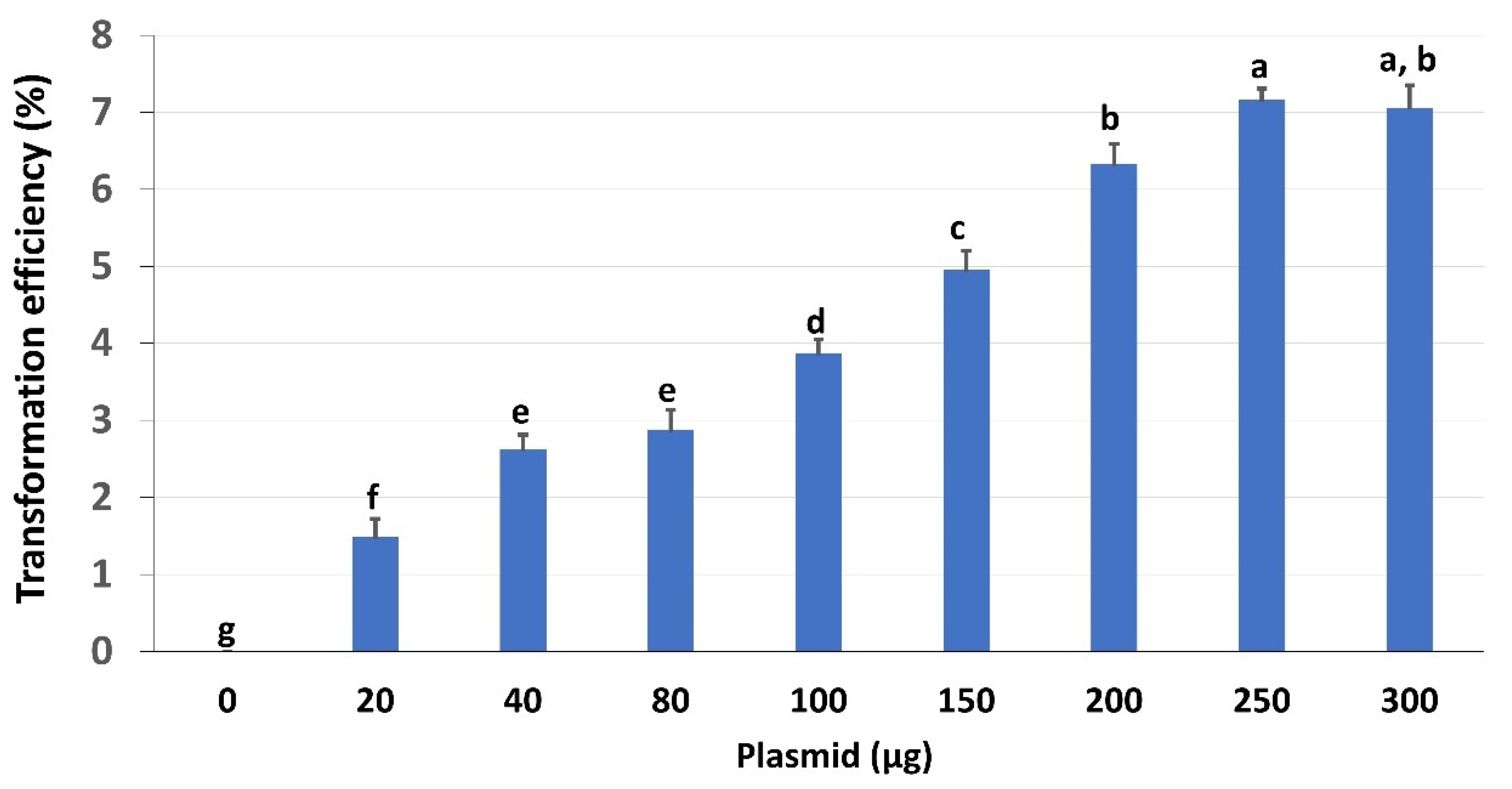
2.4. Effects of Plasmid Concentrations on Transformation Efficiency
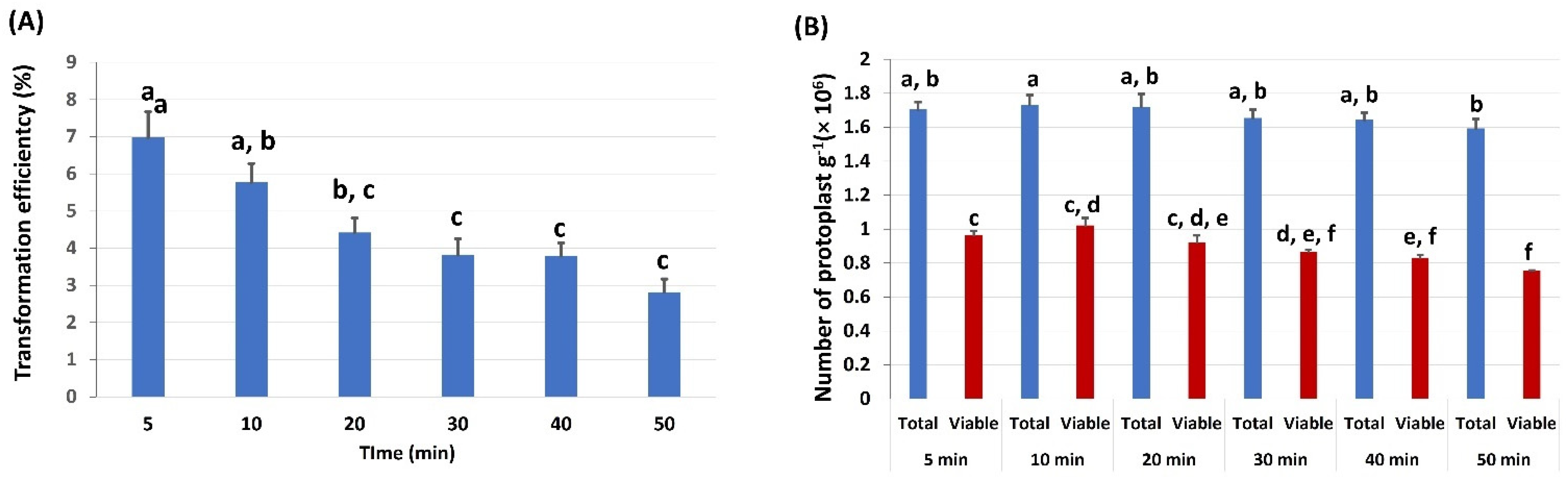
2.5. Effects of PEG Incubation Time on Protoplast Transformation Efficiency
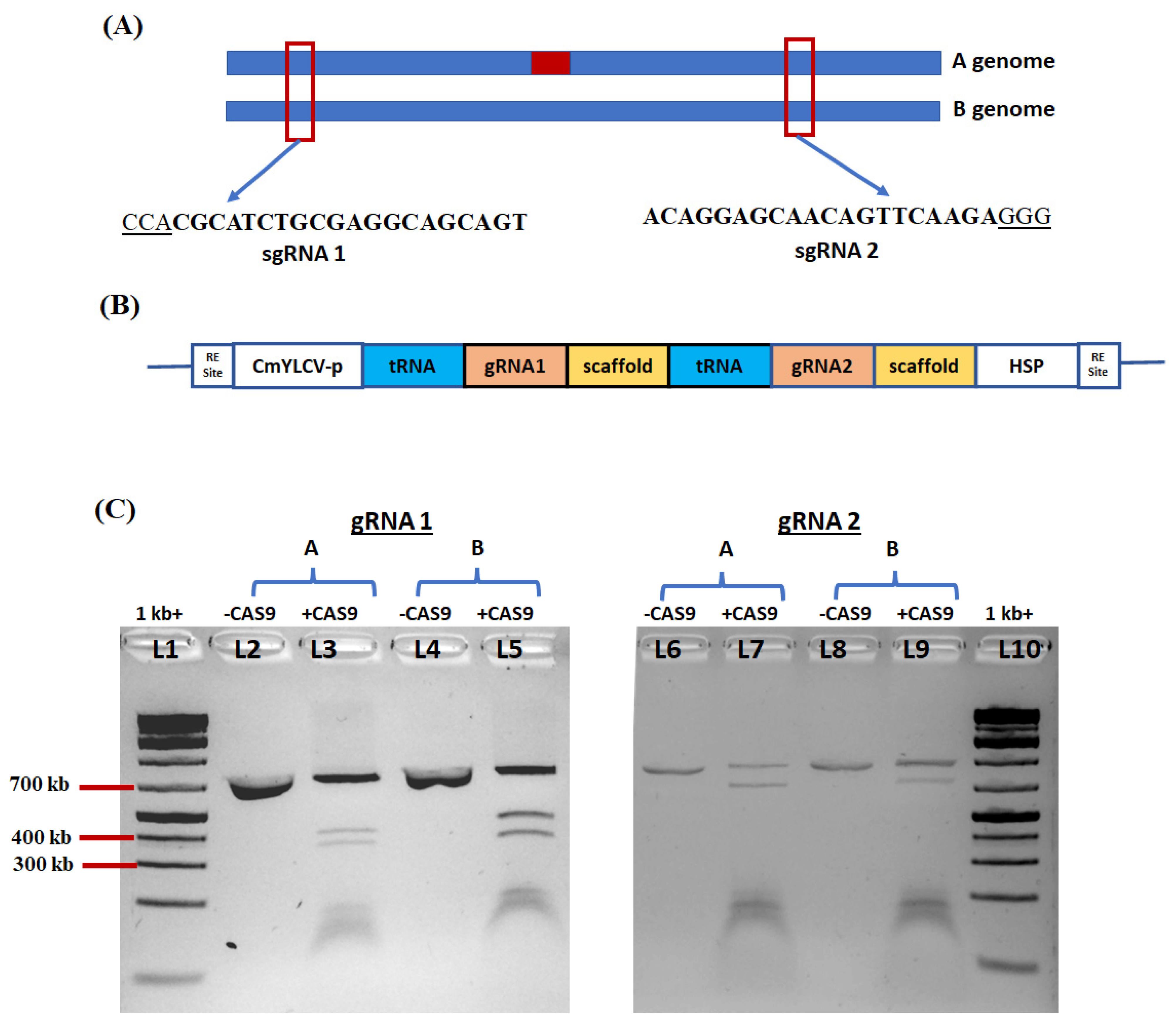
2.6. Selection of DNA Sequence of Ara h 2 Gene Target and Vector Construction
2.7. In Vitro Test of sgRNA Efficiency
2.8. Editing of Ara h 2 Gene in Peanut Protoplasts
3. Discussion
4. Materials and Methods
4.1. Plant Material
4.2. Plasmid Preparation and Constructs
4.3. In Vitro Efficiency Test of sgRNAs
4.4. Protoplast Isolation from Peanut
4.5. Protoplast Counting and Viability Test
4.6. Protoplast Transfection
4.7. Deep Amplicon Sequencing
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Janila, P.; Variath, M.T.; Pandey, M.K.; Desmae, H.; Motagi, B.N.; Okori, P.; Manohar, S.S.; Rathnakumar, A.L.; Radhakrishnan, T.; Liao, B.; et al. Genomic tools in groundnut breeding program: Status and perspectives. Front. Plant Sci. 2016, 7, 2004–2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, F.M.; Harrison, K.; Armitage, A.D.; Simkin, A.J.; Harrison, R.J. CRISPR/Cas9-mediated mutagenesis of phytoene desaturase in diploid and octoploid strawberry. Plant Methods 2019, 15, 45. [Google Scholar] [CrossRef] [Green Version]
- Bertioli, D.J.; Jenkins, J.; Clevenger, J.; Dudchenko, O.; Gao, D.; Seijo, G.; Leal-Bertioli, S.C.M.; Ren, L.; Farmer, A.D.; Pandey, M.K.; et al. The genome sequence of segmental allotetraploid peanut Arachis hypogaea. Nat. Genet. 2019, 51, 877–884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhuang, W.; Chen, H.; Yang, M.; Wang, J.; Pandey, M.K.; Zhang, C.; Chang, W.C.; Zhang, L.; Zhang, X.; Tang, R.; et al. The genome of cultivated peanut provides insight into legume karyotypes, polyploid evolution and crop domestication. Nat. Genet. 2019, 51, 865–876. [Google Scholar] [CrossRef] [PubMed]
- Dash, S.; Cannon, E.K.S.; Kalberer, S.R.; Farmer, A.D.; Cannon, S.B. PeanutBase and Other Bioinformatic Resources for Peanut; Elsevier: Amsterdam, The Netherlands, 2016; ISBN 9781630670382. [Google Scholar]
- Advances in Genetics and Genomics for Sustainable Peanut Production. In Sustainable Agriculture and New Biotechnologies, 1st ed.; CRC Press: Boca Raton, FL, USA, 2011; pp. 341–367.
- Pandey, M.K.; Monyo, E.; Ozias-Akins, P.; Liang, X.; Guimarães, P.; Nigam, S.N.; Upadhyaya, H.D.; Janila, P.; Zhang, X.; Guo, B.; et al. Advances in Arachis genomics for peanut improvement. Biotechnol. Adv. 2012, 30, 639–651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandey, M.K.; Pandey, A.K.; Kumar, R.; Nwosu, C.V.; Guo, B.; Wright, G.C.; Bhat, R.S.; Chen, X.; Bera, S.K.; Yuan, M.; et al. Translational genomics for achieving higher genetic gains in groundnut. Theor. Appl. Genet. 2020, 133, 1679–1702. [Google Scholar] [CrossRef] [Green Version]
- Stalker, H.T.; Tallury, S.P.; Ozias-Akins, P.; Bertioli, D.; Bertioli, S.C.L. The Value of Diploid Peanut Relatives for Breeding and Genomics. Peanut Sci. 2013, 40, 70–88. [Google Scholar] [CrossRef] [Green Version]
- Liang, Y.; Baring, M.; Wang, S.; Septiningsih, E.M. Mapping QTLs for Leafspot Resistance in Peanut Using SNP-Based Next-Generation Sequencing Markers. Plant Breed. Biotechnol. 2017, 5, 115–122. [Google Scholar] [CrossRef] [Green Version]
- Liang, Y.; Baring, M.R.; Septiningsih, E.M. Mapping of Quantitative Trait Loci for Yield and Grade Related Traits in Peanut (Arachis hypogaeaL.) Using High-Resolution SNP Markers. Plant Breed. Biotechnol. 2018, 6, 454–462. [Google Scholar] [CrossRef]
- Liang, Y.; Cason, J.M.; Baring, M.R.; Septiningsih, E.M. Identification of QTLs associated with Sclerotinia blight resistance in peanut (Arachis hypogaea L.). Genet. Resour. Crop. Evol. 2020, 68, 629–637. [Google Scholar] [CrossRef]
- Ozias-Akins, P.; Cannon, E.K.S.; Cannon, S.B. Genomics Resources for Peanut Improvement. In The Peanut Genome; Springer: Berlin/Heidelberg, Germany, 2017. [Google Scholar]
- Bhat, R.S.; Shirasawa, K.; Sharma, V.; Isobe, S.N.; Hirakawa, H.; Kuwata, C.; Pandey, M.K.; Varshney, R.K.; Gowda, M.V.C. Population Genomics of Peanut. In Population Genomics; Springer: Berlin/Heidelberg, Germany, 2021. [Google Scholar]
- Sun, L.; Hu, R.; Shen, G.; Zhang, H. Genetic Engineering Peanut for Higher Drought- and Salt-Tolerance. Food Nutr. Sci. 2013, 4, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Su, L.; Zeng, X.; Zheng, D.; Hong, L.; Li, L. Agrobacterium rhizogenes-mediated transformation of Arachis hypogaea: An efficient tool for functional study of genes. Biotechnol. Biotechnol. Equip. 2016, 30, 869–878. [Google Scholar] [CrossRef] [Green Version]
- Mehta, R.; Radhakrishnan, T.; Kumar, A.; Yadav, R.; Dobaria, J.R.; Thirumalaisamy, P.P.; Jain, R.K.; Chigurupati, P. Coat protein-mediated transgenic resistance of peanut (Arachis hypogaea L.) to peanut stem necrosis disease through Agrobacterium-mediated genetic transformation. Indian J. Virol. 2013, 24, 205–213. [Google Scholar] [CrossRef]
- Keshavareddy, G.; Rohini, S.; Ramu, S.V.; Sundaresha, S.; Kumar, A.R.V.; Kumar, P.A.; Udayakumar, M. Transgenics in groundnut (Arachis hypogaea L.) expressing cry1AcF gene for resistance to Spodoptera litura (F.). Physiol. Mol. Biol. Plants 2013, 19, 343–352. [Google Scholar] [CrossRef] [Green Version]
- Prasad, K.; Bhatnagar-Mathur, P.; Waliyar, F.; Sharma, K.K. Overexpression of a chitinase gene in transgenic peanut confers enhanced resistance to major soil borne and foliar fungal pathogens. J. Plant Biochem. Biotechnol. 2013, 22, 222–233. [Google Scholar] [CrossRef] [Green Version]
- Banavath, J.N.; Chakradhar, T.; Pandit, V.; Konduru, S.; Guduru, K.K.; Akila, C.S.; Podha, S.; Puli, C.O.R. Stress inducible overexpression of AtHDG11 leads to improved drought and salt stress tolerance in peanut (Arachis hypogaea L.). Front. Chem. 2018, 6, 34. [Google Scholar] [CrossRef] [Green Version]
- Dodo, H.W.; Konan, K.N.; Chen, F.C.; Egnin, M.; Viquez, O.M. Alleviating peanut allergy using genetic engineering: The silencing of the immunodominant allergen Ara h 2 leads to its significant reduction and a decrease in peanut allergenicity. Plant Biotechnol. J. 2008, 6, 135–145. [Google Scholar] [CrossRef]
- Liang, Y.; Biswas, S.; Kim, B.; Bailey-Serres, J.; Septiningsih, E. Improved Transformation and Regeneration of Indica Rice: Disruption of SUB1A as a Test Case via CRISPR-Cas9. Int. J. Mol. Sci. 2021, 22, 6989. [Google Scholar] [CrossRef] [PubMed]
- Molina-Risco, M.; Ibarra, O.; Faion-Molina, M.; Kim, B.; Septiningsih, E.M.; Thomson, M.J. Optimizing Agrobacterium-Mediated Transformation and CRISPR-Cas9 Gene Editing in the tropical japonica Rice Variety Presidio. Int. J. Mol. Sci. 2021, 22, 10909. [Google Scholar] [CrossRef]
- Zhou, H.; Liu, B.; Weeks, D.P.; Spalding, M.H.; Yang, B. Large chromosomal deletions and heritable small genetic changes induced by CRISPR/Cas9 in rice. Nucleic Acids Res. 2014, 42, 10903–10914. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Hung, S.S.C.; Yek, J.; El Wazan, L.; Nguyen, T.; Khan, S.; Lim, S.Y.; Hewitt, A.W.; Wong, R.C.B. A Simple Cloning-free Method to Efficiently Induce Gene Expression Using CRISPR/Cas9. Mol. Ther. Nucleic Acids 2019, 14, 184–191. [Google Scholar] [CrossRef] [Green Version]
- Razzaq, A.; Saleem, F.; Kanwal, M.; Mustafa, G.; Yousaf, S.; Arshad, H.M.I.; Hameed, M.K.; Khan, M.S.; Joyia, F.A. Modern Trends in Plant Genome Editing: An Inclusive Review of the CRISPR/Cas9 Toolbox. Int. J. Mol. Sci. 2019, 20, 4045. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.-S.; Hsu, C.-T.; Yang, L.-H.; Lee, L.-Y.; Fu, J.-Y.; Cheng, Q.-W.; Wu, F.-H.; Hsiao, H.C.-W.; Zhang, Y.; Zhang, R.; et al. Application of protoplast technology to CRISPR/Cas9 mutagenesis: From single-cell mutation detection to mutant plant regeneration. Plant Biotechnol. J. 2017, 16, 1295–1310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sivanandhan, G.; Bae, S.; Sung, C.; Choi, S.-R.; Lee, G.-J.; Lim, Y.-P. Optimization of Protoplast Isolation from Leaf Mesophylls of Chinese Cabbage (Brassica rapa ssp. pekinensis) and Subsequent Transfection with a Binary Vector. Plants 2021, 10, 2636. [Google Scholar] [CrossRef]
- Yue, J.-J.; Yuan, J.-L.; Wu, F.-H.; Yuan, Y.-H.; Cheng, Q.-W.; Hsu, C.-T.; Lin, C.-S. Protoplasts: From Isolation to CRISPR/Cas Genome Editing Application. Front. Genome Ed. 2021, 3, 717017. [Google Scholar] [CrossRef]
- Mueller, G.A.; Maleki, S.J.; Pedersen, L.C. The Molecular Basis of Peanut Allergy. Curr. Allergy Asthma Rep. 2014, 14, 429. [Google Scholar] [CrossRef] [Green Version]
- Oelck, M.M.; Bapat, V.A.; Schieder, O. Protoplast Culture of Three Legumes: Arachis hypogaea, Melilotus officinalis, Trifolium resupinatum. Z. Für Pflanzenphysiol. 1982, 106, 173–177. [Google Scholar] [CrossRef]
- Liu, H.; Ding, Y.; Zhou, Y.; Jin, W.; Xie, K.; Chen, L.L. CRISPR-P 2.0: An Improved CRISPR-Cas9 Tool for Genome Editing in Plants. Mol. Plant 2017, 10, 530–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Yu, G.; Chen, Z.; Han, J.; Hu, Y.; Wang, K. Optimization of protoplast isolation, transformation and its application in sugarcane (Saccharum spontaneum L). Crop. J. 2020, 9, 133–142. [Google Scholar] [CrossRef]
- Wu, F.; Hanzawa, Y. A simple method for isolation of soybean protoplasts and application to transient gene expression analyses. J. Vis. Exp. 2018, 2018, 57258. [Google Scholar] [CrossRef] [PubMed]
- Cheng, N.; Nakata, P.A. Development of a rapid and efficient protoplast isolation and transfection method for chickpea (Cicer arietinum). MethodsX 2020, 7, 101025. [Google Scholar] [CrossRef] [PubMed]
- Taiz, L.; Jones, R.L. The isolation of barley-aleurone protoplasts. Planta 1971, 101, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Reed, K.M.; Bargmann, B.O.R. Protoplast Regeneration and Its Use in New Plant Breeding Technologies. Front. Genome Ed. 2021, 3, 734951. [Google Scholar] [CrossRef]
- Shan, Q.; Wang, Y.; Li, J.; Gao, C. Genome editing in rice and wheat using the CRISPR/Cas system. Nat. Protoc. 2014, 9, 2395–2410. [Google Scholar] [CrossRef]
- Brandt, K.M.; Gunn, H.; Moretti, N.; Zemetra, R.S. A Streamlined Protocol for Wheat (Triticum aestivum) Protoplast Isolation and Transformation with CRISPR-Cas Ribonucleoprotein Complexes. Front. Plant Sci. 2020, 11, 769. [Google Scholar] [CrossRef]
- Masani, M.Y.A.; Noll, G.A.; Parveez, G.K.A.; Sambanthamurthi, R.; Prüfer, D. Efficient Transformation of Oil Palm Protoplasts by PEG-Mediated Transfection and DNA Microinjection. PLoS ONE 2014, 9, e96831. [Google Scholar] [CrossRef] [Green Version]
- Yuan, M.; Zhu, J.; Gong, L.; He, L.; Lee, C.; Han, S.; Chen, C.; He, G. Mutagenesis of FAD2 genes in peanut with CRISPR/Cas9 based gene editing. BMC Biotechnol. 2019, 19, 24. [Google Scholar] [CrossRef]
- Hudzieczek, V.; Cegan, R.; Cermak, T.; Bacovska, N.; Machalkova, Z.; Dolezal, K.; Plihalova, L.; Voytas, D.; Hobza, R.; Vyskot, B. Agrobacterium rhizogenes-mediated transformation of a dioecious plant model Silene latifolia. N. Biotechnol. 2019, 48, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Gelvin, S.B. Agrobacterium—Mediated Plant Transformation: The Biology behind the “Gene-Jockeying” Tool. Microbiol. Mol. Biol. Rev. 2003, 67, 16–37. [Google Scholar] [CrossRef] [Green Version]
- Bortesi, L.; Fischer, R. The CRISPR/Cas9 system for plant genome editing and beyond. Biotechnol. Adv. 2015, 33, 41–52. [Google Scholar] [CrossRef]
- Burow, M.D.; Baring, M.R.; Puppala, N.; Simpson, C.E.; Ayers, J.L.; Cason, J.; Schubert, A.M.; Muitia, A.; López, Y. Registration of ‘Schubert’ Peanut. J. Plant Regist. 2014, 8, 122–126. [Google Scholar] [CrossRef]
- Čermák, T.; Curtin, S.J.; Gil-Humanes, J.; Čegan, R.; Kono, T.J.Y.; Konečná, E.; Belanto, J.J.; Starker, C.G.; Mathre, J.W.; Greenstein, R.L.; et al. A Multipurpose Toolkit to Enable Advanced Genome Engineering in Plants. Plant Cell 2017, 29, 1196–1217. [Google Scholar] [CrossRef] [Green Version]
- Xie, K.; Minkenberg, B.; Yang, Y. Boosting CRISPR/Cas9 multiplex editing capability with the endogenous tRNA-processing system. Proc. Natl. Acad. Sci. USA 2015, 112, 3570–3575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Jarret, R.L.; Demski, J.W. Regeneration of Plants From Protoplasts of Arachis Species (Peanut). In Plant Protoplasts and Genetic Engineering; Springer: Berlin/Heidelberg, Germany, 1995. [Google Scholar]
- Li, X. A Transient Expression Assay Using Arabidopsis Mesophyll Protoplasts. Bio-Protocol 2011, 1, e70. [Google Scholar] [CrossRef]
- Doyle, J. A rapid isolation procedure for small amounts of leaf tissue. Phytochem. Bull. 1987, 19, 11–15. [Google Scholar]
- Texas A&M Institute for Genome Sciences and Society (TIGSS) Lab. Available online: https://genomics.tamu.edu (accessed on 18 December 2021).
- Connelly, J.P.; Pruett-Miller, S.M. CRIS.py: A Versatile and High-throughput Analysis Program for CRISPR-based Genome Editing. Sci. Rep. 2019, 9, 4194. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Plant No | Ara h 2 gRNA Target Region (5′-3′) | Type of Edit | Editing Efficiency |
|---|---|---|---|
| Ara h 2A (genome A) gRNA1 NGS results | |||
| WT | GCTGCCCACGCATCTGCGAGGCAGCAGTGGGAACTCCAA | ||
| S1 | GCTGCCCACGC------TGCGAGGCAGCAGTGGGAACTCCAA | 3 bp deletion | 0.8% |
| S2 | GCTGCCCACG----------GCGAGGCAGCAGTGGGAACTCCAA | 5 bp deletion | 0.37% |
| Ara h 2B (genome B) gRNA1 NGS results | |||
| WT | GCTGCCCACGCATCTGCGAGGCAGCAGTGGGAACTCCAA | ||
| S1 | GCTGCCCACGCATCTGCGAGGCAGCAGTGGGAACTCCAA | No edit | |
| S2 | GCTGCCCACGC--------GCGAGGCAGCAGTGGGAACTCCAA | 4 bp deletion | 0.20% |
| Ara h 2A (genome A)gRNA2 NGS results | |||
| WT | GGGAGGCAACAGGAGCAACAGTTCAAGAGGGAGCTCAG | ||
| S1 | GGGAGGCAACAGGAGCAAC-------------AGAGGGAGCTCAG | 6 bp deletion | 0.14% |
| S2 | GGGAGGCAACAGGAGCAACAG------AAGAGGGAGCTCAG | 3 bp deletion | 0.13% |
| Ara h 2B (genome B) gRNA2 NGS results | |||
| WT | GGGAGGCAACAGGAGCAACAGTTCAAGAGGGAGCTCAG | ||
| S1 | GGGAGGCAACAGGAGCAACAGTTCAAGAGGGAGCTCAG | No edit | |
| S2 | GGGAGGCAACAGGAGCAACAG------AAGAGGGAGCTCAG | 3 bp deletion | 0.16% |
| Solution Name | Composition |
|---|---|
| Enzyme solution | 3% cellulase RS (Yakult, Tokyo, Japan), 0.1% macroenzyme, 0.5% pectinase, 0.4 M Mannitol, 20 mM KCl, and 20 mM MES (pH 5.7), 10 mM CaCl2, 0.1% BSA Special instructions: MES, mannitol, H2O, cellulase RS, macroenzyme, and pectinase were stirred and incubated at 55 °C for 10 min. The solution was cooled to room temperature, and CaCl2 and BSA were added in and gently mixed. |
| W5 solution | 154 mM NaCl, 125 mM CaCl2, 5 mM KCl, and 2 mM MES (pH 5.7) |
| Washing and Incubation Solution (WS1) | 0.5 M Mannitol, 20 mM KCl, and 4 mM MES (pH 5.7) |
| MMG Solution | 0.4 M Mannitol, 15 mM MgCl2, and 4 mM MES (pH 5.7) |
| PEG–CaCl2 solution | 0.2 M Mannitol, 0.1 M CaCl2, and 20–80% PEG 4000 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Biswas, S.; Wahl, N.J.; Thomson, M.J.; Cason, J.M.; McCutchen, B.F.; Septiningsih, E.M. Optimization of Protoplast Isolation and Transformation for a Pilot Study of Genome Editing in Peanut by Targeting the Allergen Gene Ara h 2. Int. J. Mol. Sci. 2022, 23, 837. https://doi.org/10.3390/ijms23020837
Biswas S, Wahl NJ, Thomson MJ, Cason JM, McCutchen BF, Septiningsih EM. Optimization of Protoplast Isolation and Transformation for a Pilot Study of Genome Editing in Peanut by Targeting the Allergen Gene Ara h 2. International Journal of Molecular Sciences. 2022; 23(2):837. https://doi.org/10.3390/ijms23020837
Chicago/Turabian StyleBiswas, Sudip, Nancy J. Wahl, Michael J. Thomson, John M. Cason, Bill F. McCutchen, and Endang M. Septiningsih. 2022. "Optimization of Protoplast Isolation and Transformation for a Pilot Study of Genome Editing in Peanut by Targeting the Allergen Gene Ara h 2" International Journal of Molecular Sciences 23, no. 2: 837. https://doi.org/10.3390/ijms23020837