
Reactive Oxygen Species Production Is Responsible for Antineoplastic Activity of Osmium, Ruthenium, Iridium and Rhodium Half-Sandwich Type Complexes with Bidentate Glycosyl Heterocyclic Ligands in Various Cancer Cell Models

 , , , and
, , , and
Abstract
:1. Introduction
2. Results
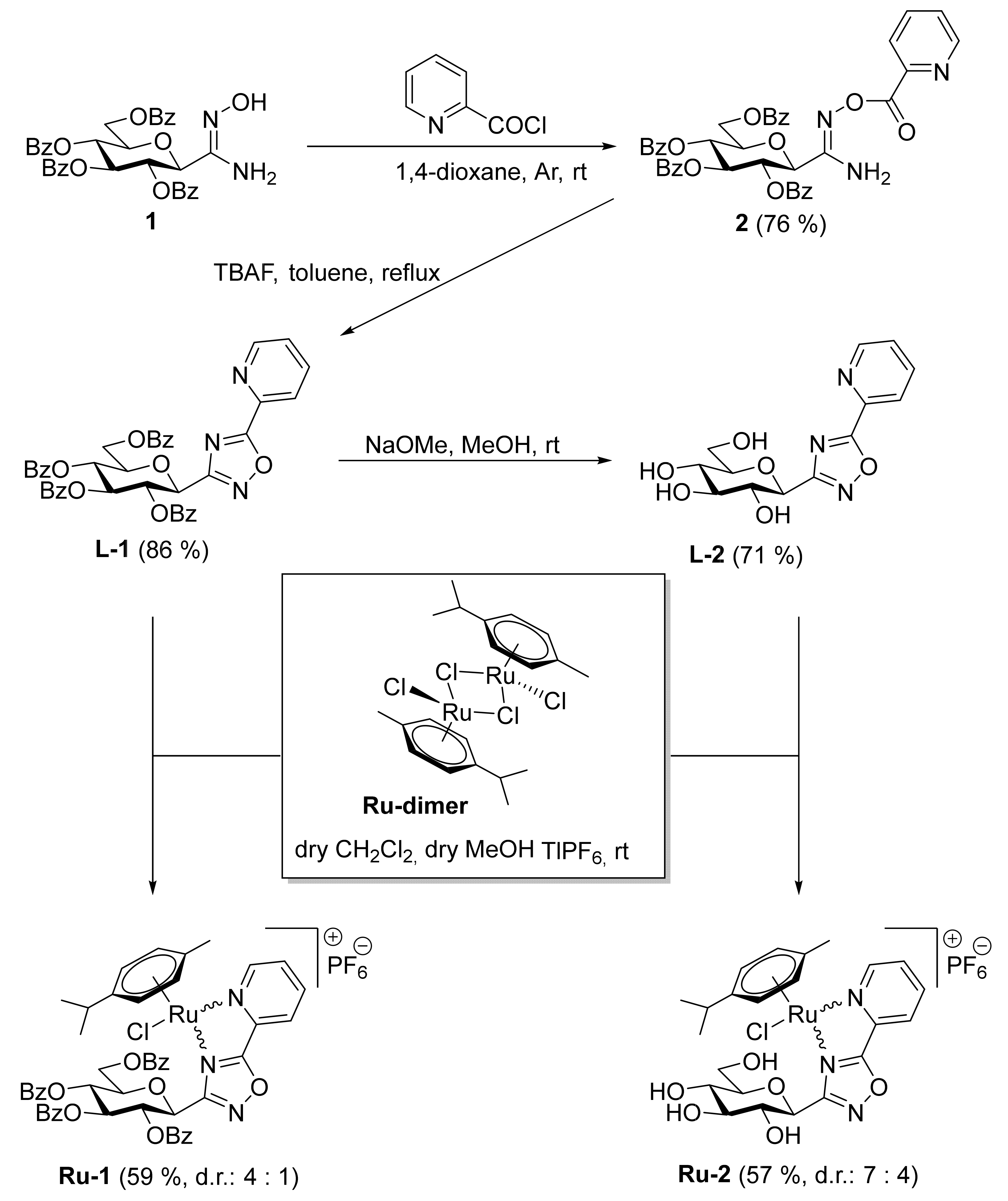
2.1. Chemistry
2.2. Biological Characterization of the Complexes
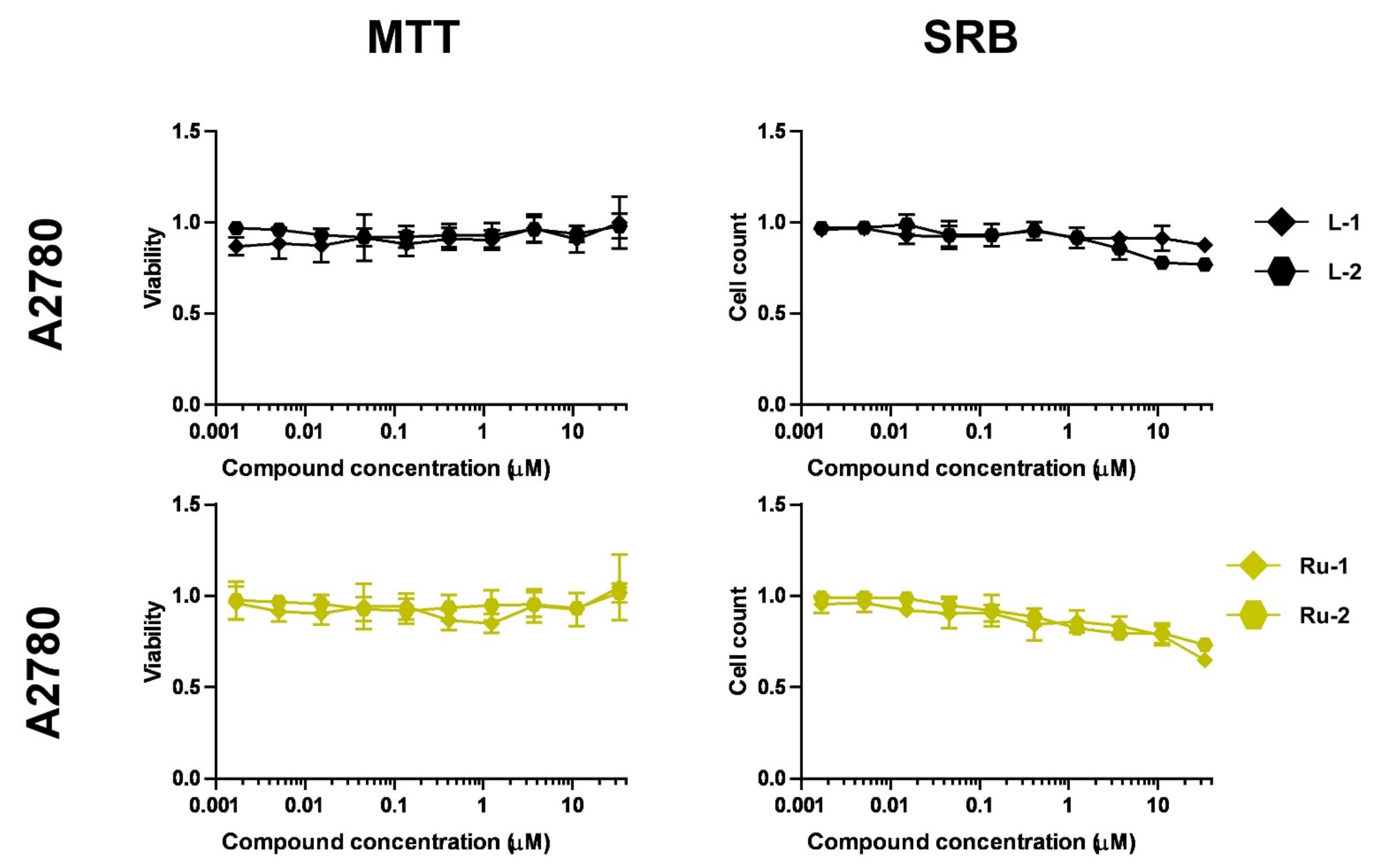
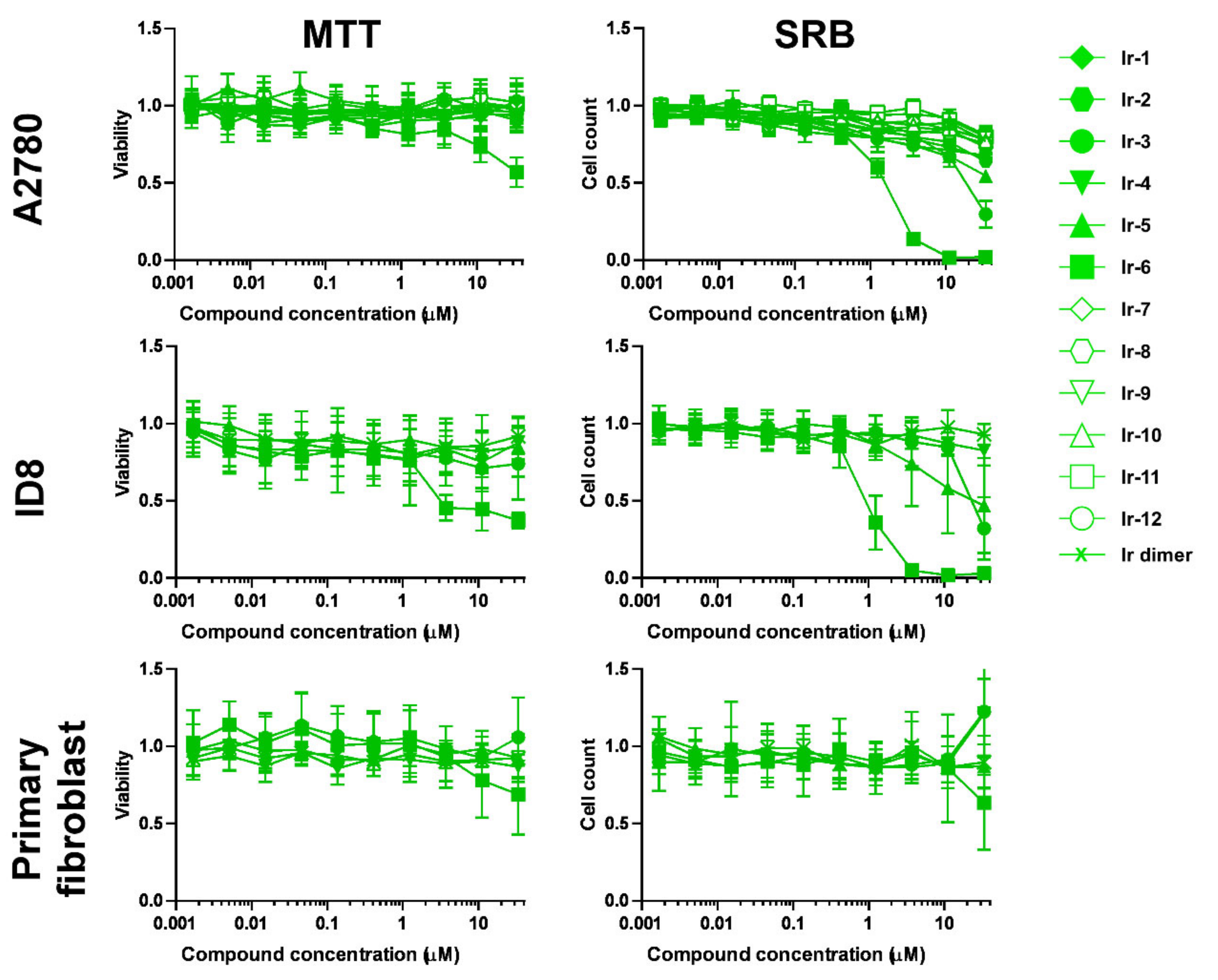
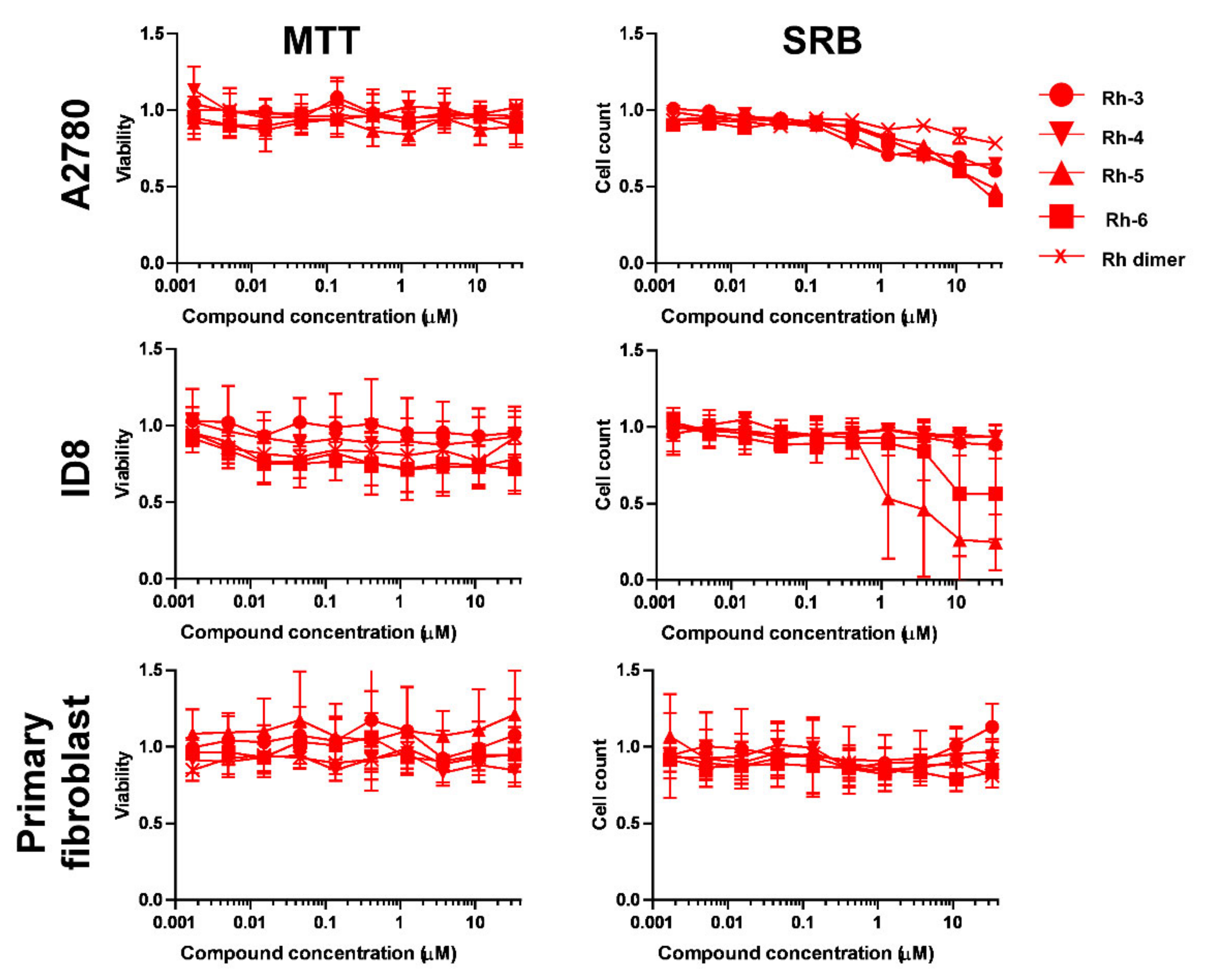
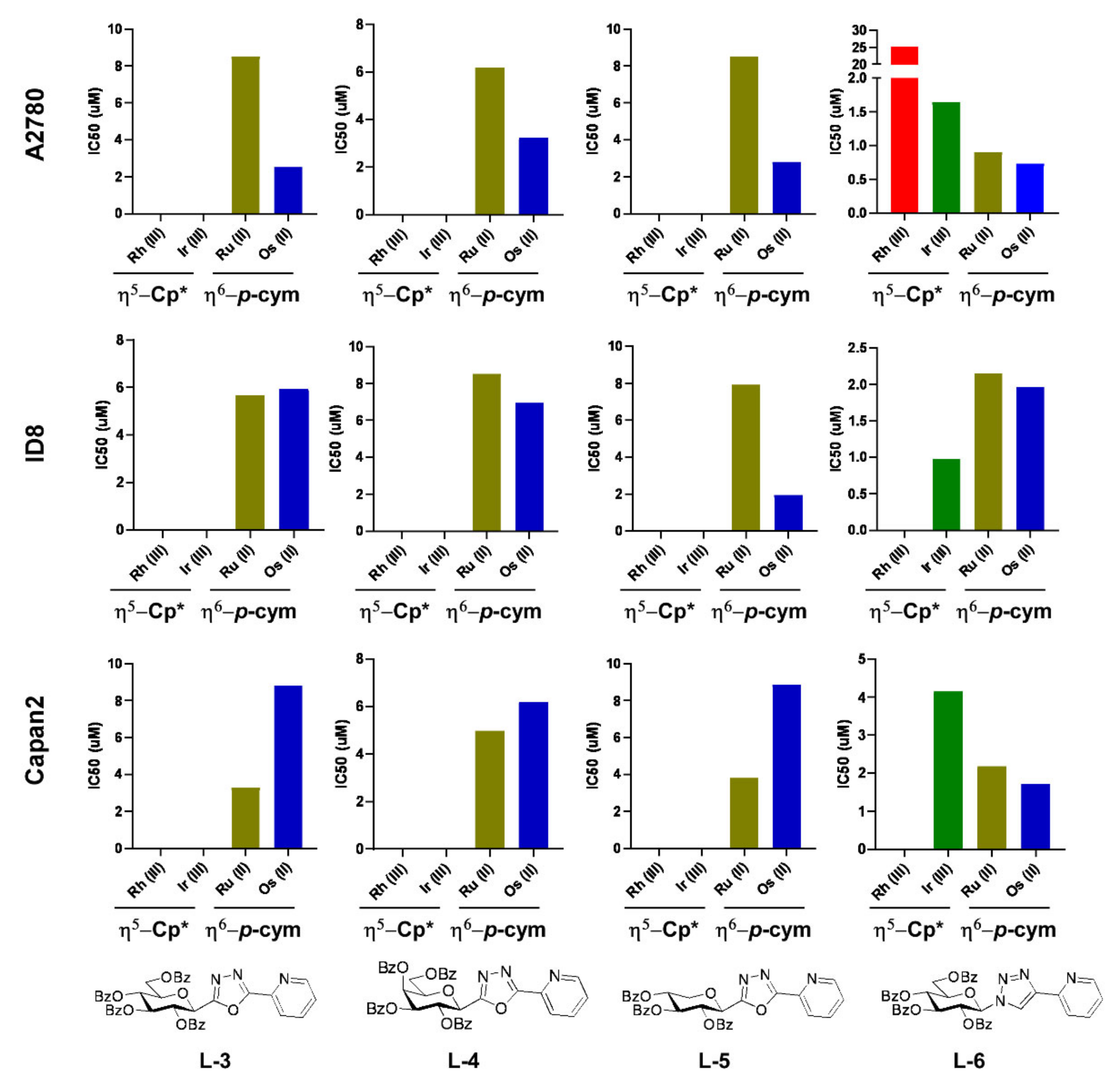
2.2.1. Selection of Biologically Active Complexes
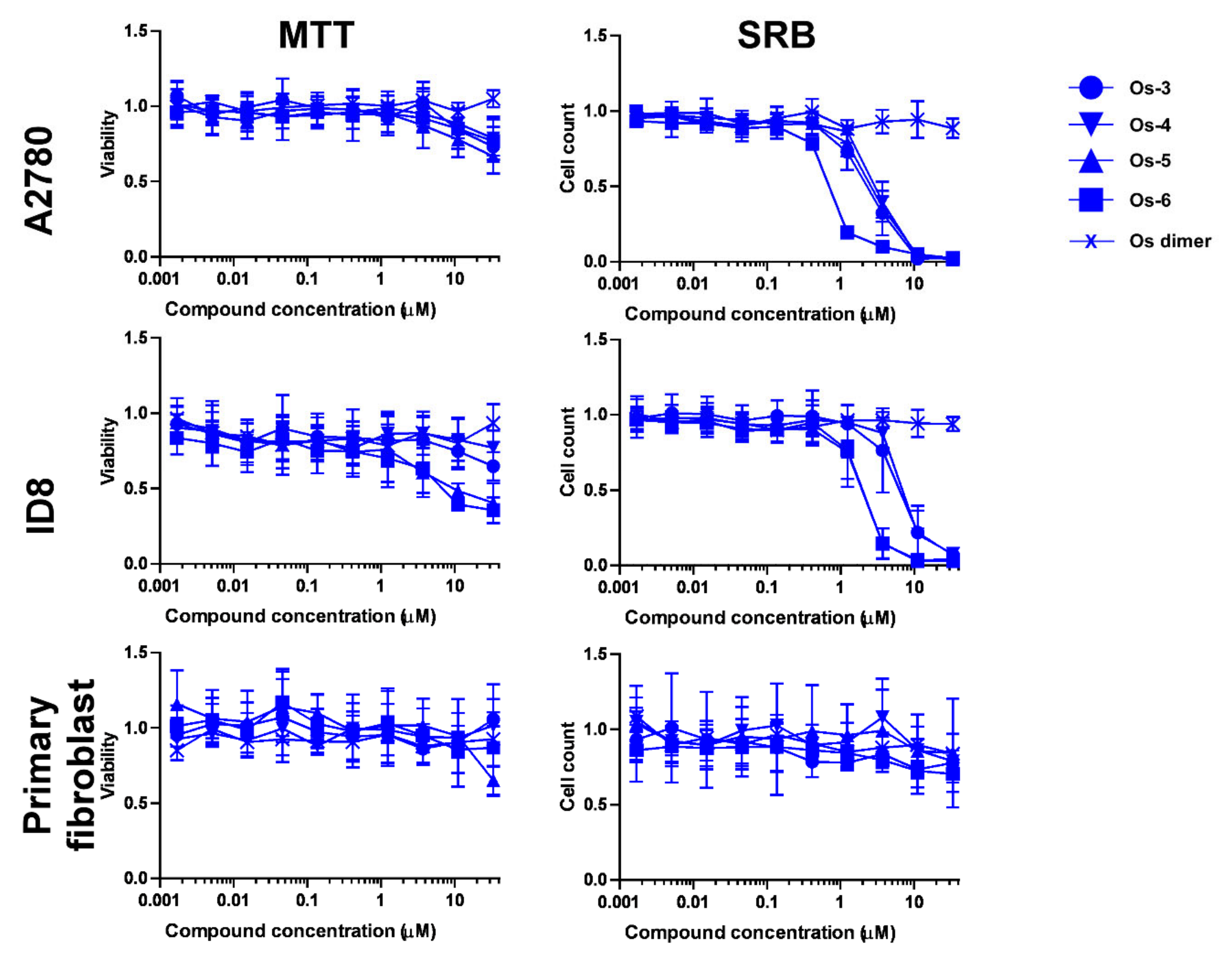
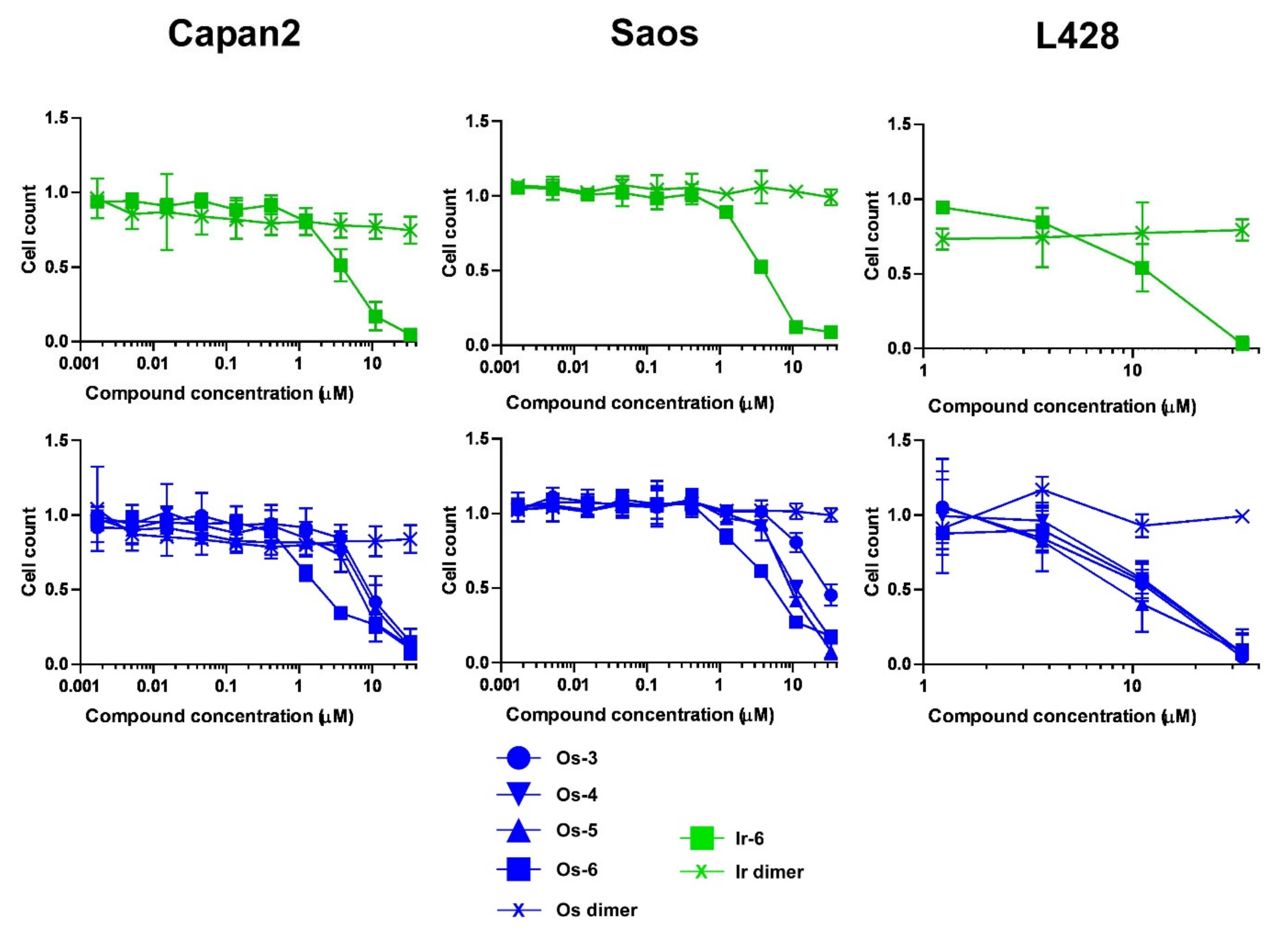
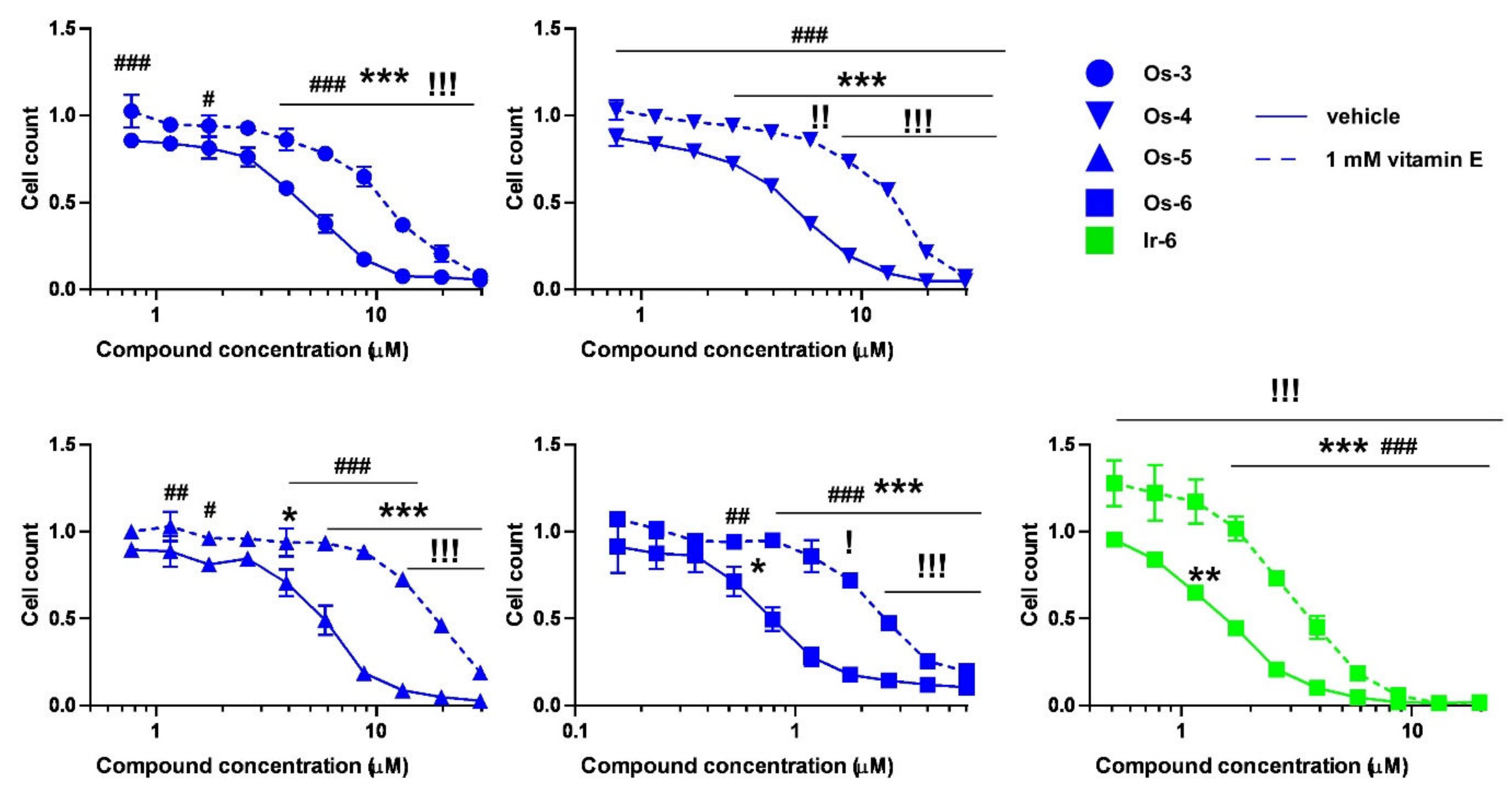
2.2.2. The Bioactive Complexes Are Active on Cell Lines Other than Ovarian Cancer
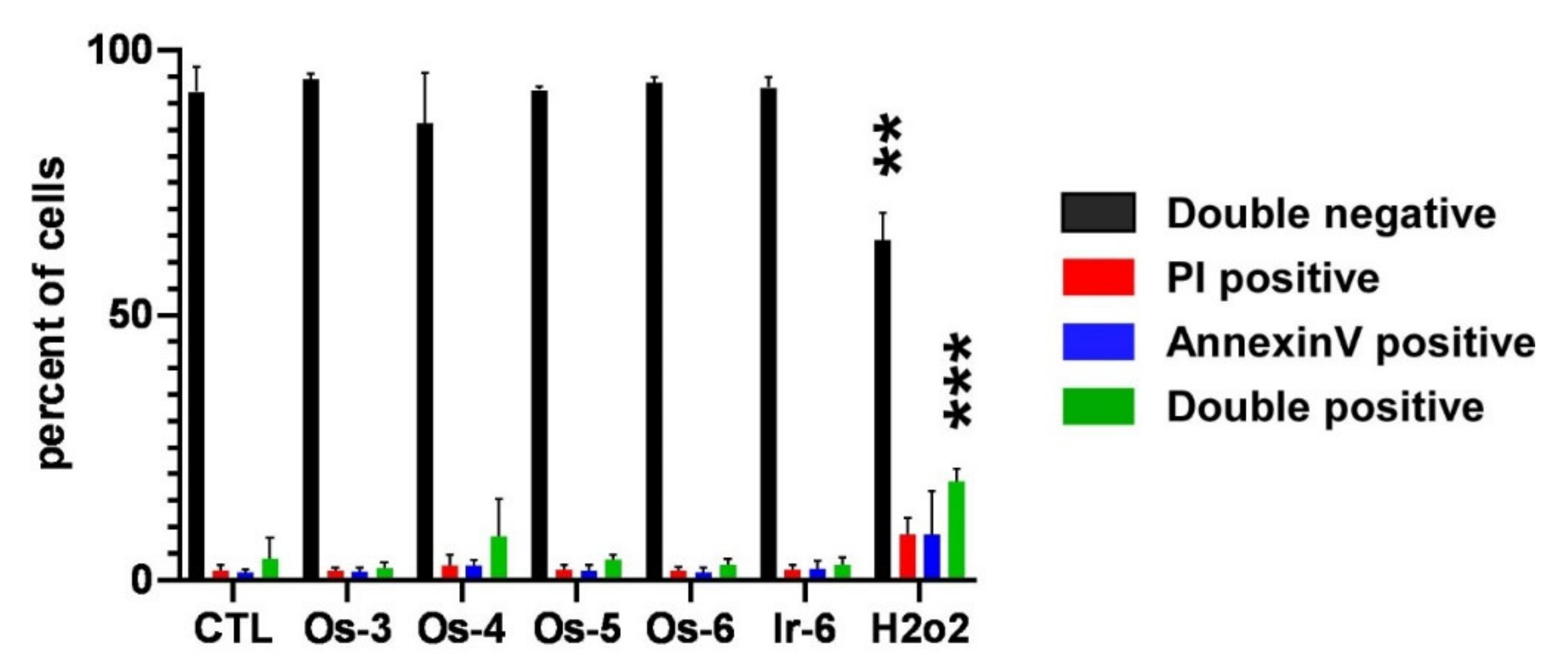
2.2.3. The Active Complexes Induce Oxidative Stress
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Syntheses
5.1.1. General Methods
5.1.2. General Method I for the Synthesis of the [(η6-p-cym)MII(N-N))Cl]PF6 (M = Ru, Os) and [(η5-Cp*)MIII(N-N))Cl]PF6 (M = Ir, Rh)-Type Complexes Containing O-Peracylated Glycosyl Azole Ligands
5.1.3. General Method II for the Formation of the [(η6-p-cym)RuII(N-N))Cl]PF6 and [(η5-Cp*)IrIII(N-N))Cl]PF6 Type Complexes Containing Unprotected Glycosyl Azole Ligands
5.1.4. O-Picolinoyl-C-(2,3,4,6-tetra-O-benzoyl-β-D-glucopyranosyl)formamidoxime (2)
5.1.5. 3-(2′,3′,4′,6′-Tetra-O-benzoyl-β-D-glucopyranosyl)-5-(pyridin-2-yl)-1,2,4-oxadiazole (L-1)
5.1.6. 3-(β-D-Glucopyranosyl)-5-(pyridin-2-yl)-1,2,4-oxadiazole (L-2)
5.1.7. Complex Ru-1
5.1.8. Complex Ru-2
5.1.9. Complex Os-3
5.1.10. Complex Os-4
5.1.11. Complex Os-5
5.1.12. Complex Os-6
5.1.13. Complex Ir-1
5.1.14. Complex Ir-2
5.1.15. Complex Ir-3
5.1.16. Complex Ir-4
5.1.17. Complex Ir-5
5.1.18. Complex Ir-6
5.1.19. Complex Ir-7
5.1.20. Complex Ir-8
5.1.21. Complex Ir-9
5.1.22. Complex Ir-10
5.1.23. Complex Ir-11
5.1.24. Complex Ir-12
5.1.25. Complex Rh-3
5.1.26. Complex Rh-4
5.1.27. Complex Rh-5
5.1.28. Complex Rh-6
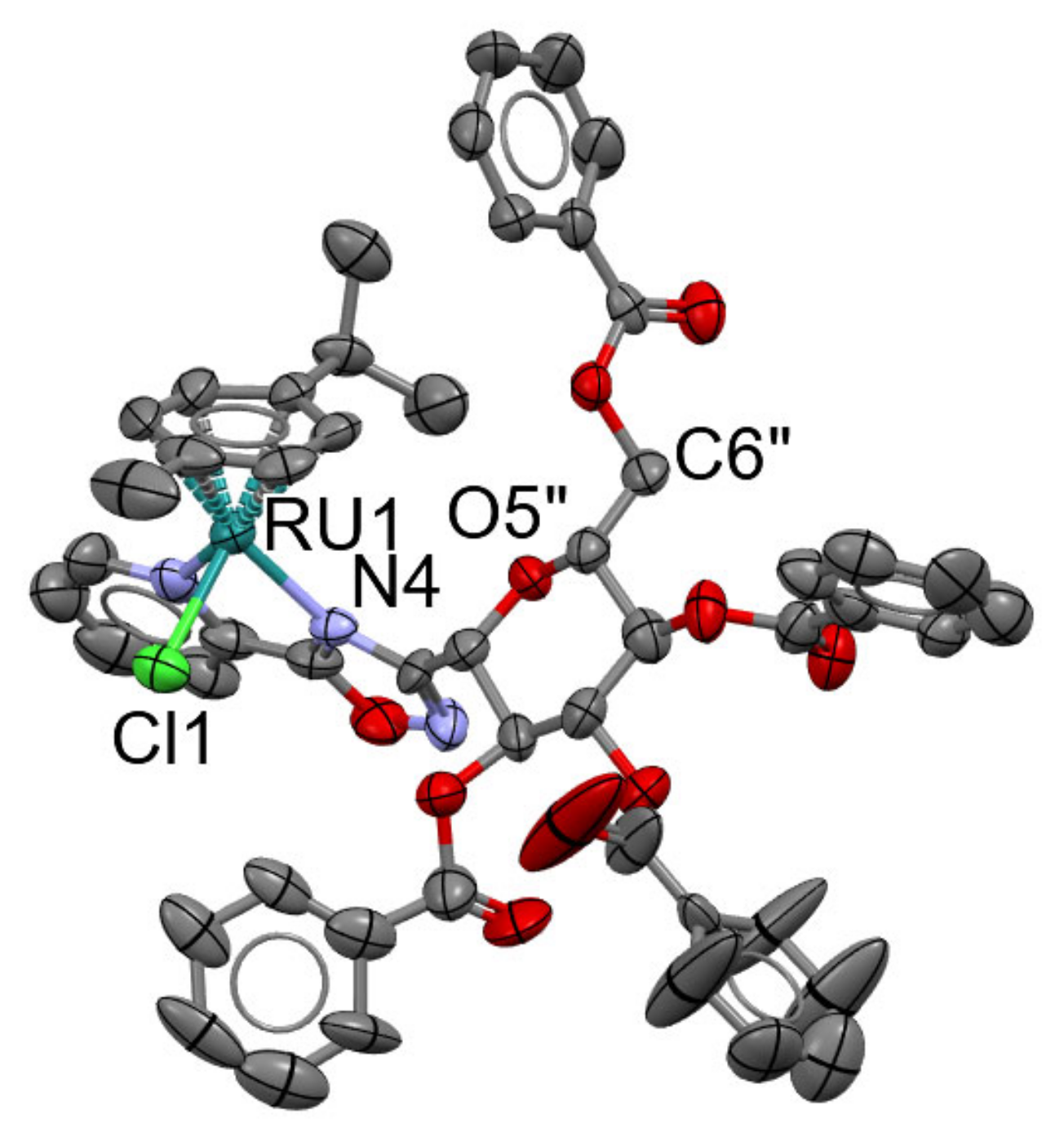
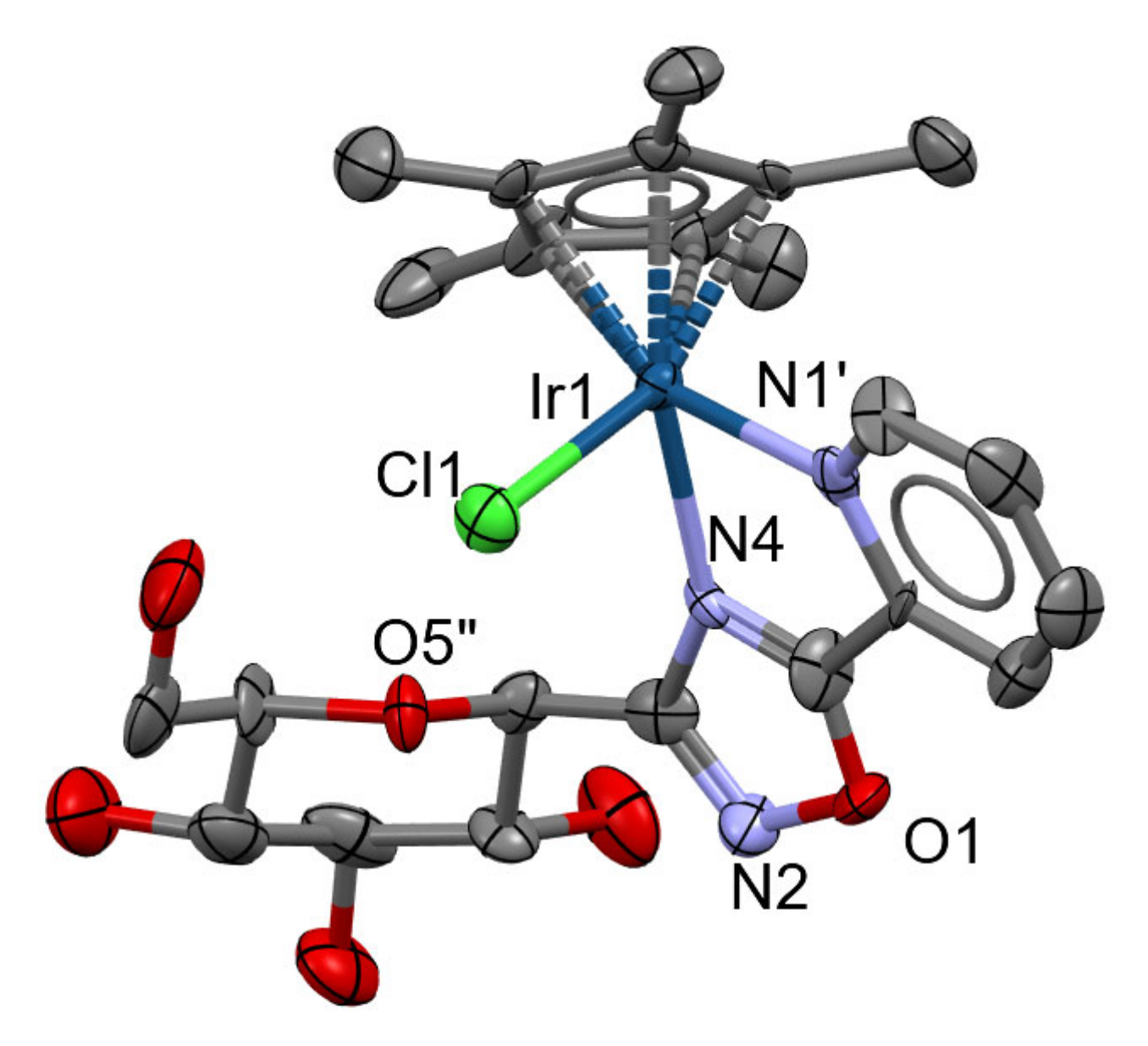
5.2. X-ray Crystallography
5.3. Determination of the Distribution Coefficients (logD)
5.4. Materials
5.5. Cell culture
5.6. Methylthiazolyldiphenyl-Tetrazolium Bromide (MTT) Reduction Assay
5.7. Sulforhodamine B (SRB) Binding Assay
5.8. Cell Death
5.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kenny, R.G.; Marmion, C.J. Toward Multi-Targeted Platinum and Ruthenium Drugs-A New Paradigm in Cancer Drug Treatment Regimens? Chem. Rev. 2019, 119, 1058–1137. [Google Scholar] [CrossRef] [PubMed]
- Konkankit, C.C.; Marker, S.C.; Knopf, K.M.; Wilson, J.J. Anticancer activity of complexes of the third row transition metals, rhenium, osmium, and iridium. Dalton Trans. 2018, 47, 9934–9974. [Google Scholar] [CrossRef]
- Fetoni, A.R.; Paciello, F.; Mezzogori, D.; Rolesi, R.; Eramo, S.L.; Paludetti, G.; Troiani, D. Molecular targets for anticancer redox chemotherapy and cisplatin-induced ototoxicity: The role of curcumin on pSTAT3 and Nrf-2 signalling. Br. J. Cancer 2015, 113, 1434–1444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukherjea, D.; Dhukhwa, A.; Sapra, A.; Bhandari, P.; Woolford, K.; Franke, J.; Ramkumar, V.; Rybak, L. Strategies to reduce the risk of platinum containing antineoplastic drug-induced ototoxicity. Exp. Opin. Drug Metab. Toxicol. 2020, 16, 965–982. [Google Scholar] [CrossRef]
- McMullen, M.; Madariaga, A.; Lheureux, S. New approaches for targeting platinum-resistant ovarian cancer. Semin. Cancer Biol. 2021, 77, 167–181. [Google Scholar] [CrossRef] [PubMed]
- Lund, R.J.; Huhtinen, K.; Salmi, J.; Rantala, J.; Nguyen, E.V.; Moulder, R.; Goodlett, D.R.; Lahesmaa, R.; Carpen, O. DNA methylation and Transcriptome Changes Associated with Cisplatin Resistance in Ovarian Cancer. Sci. Rep. 2017, 7, 1469. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.; Wang, Z.; Sun, Z.; Zhang, L.; Zhang, W.; Xu, Y.; Zhang, J.J. Platinum-Based Combination Therapy: Molecular Rationale, Current Clinical Uses, and Future Perspectives. J. Med. Chem. 2020, 63, 13397–13412. [Google Scholar] [CrossRef] [PubMed]
- Burris, H.A.; Bakewell, S.; Bendell, J.C.; Infante, J.; Jones, S.F.; Spigel, D.R.; Weiss, G.J.; Ramanathan, R.K.; Ogden, A.; Von Hoff, D. Safety and activity of IT-139, a ruthenium-based compound, in patients with advanced solid tumours: A first-in-human, open-label, dose-escalation phase I study with expansion cohort. ESMO Open 2016, 1, e000154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meier-Menches, S.M.; Gerner, C.; Berger, W.; Hartinger, C.G.; Keppler, B.K. Structure–activity relationships for ruthenium and osmium anticancer agents – towards clinical development. Chem. Soc. Rev. 2018, 47, 909–928. [Google Scholar] [CrossRef]
- Zeng, L.; Gupta, P.; Chen, Y.; Wang, E.; Ji, L.; Chao, H.; Chen, Z.-S. The development of anticancer ruthenium(II) complexes: From single molecule compounds to nanomaterials. Chem. Soc. Rev. 2017, 46, 5771–5804. [Google Scholar] [CrossRef] [PubMed]
- Melchart, M.; Sadler, P.J. Ruthenium Arene Anticancer Complexes. In Bioorganometallics; Wiley: Hoboken, NJ, USA, 2005; pp. 39–64. [Google Scholar]
- Štarha, P.; Trávníček, Z. Non-platinum complexes containing releasable biologically active ligands. Coord. Chem. Rev. 2019, 395, 130–145. [Google Scholar] [CrossRef]
- Gichumbi, J.M.; Friedrich, H.B. Half-sandwich complexes of platinum group metals (Ir, Rh, Ru and Os) and some recent biological and catalytic applications. J. Organomet. Chem. 2018, 866, 123–143. [Google Scholar] [CrossRef]
- Hartinger, C.G.; Phillips, A.D.; Nazarov, A.A. Polynuclear ruthenium, osmium and gold complexes. The quest for innovative anticancer chemotherapeutics. Curr. Top. Med. Chem. 2011, 11, 2688–2702. [Google Scholar] [CrossRef]
- Li, Y.; Liu, B.; Shi, H.; Wang, Y.; Sun, Q.; Zhang, Q. Metal complexes against breast cancer stem cells. Dalton Trans. 2021, 50, 14498–14512. [Google Scholar] [CrossRef] [PubMed]
- Hanif, M.; Babak, M.V.; Hartinger, C.G. Development of anticancer agents: Wizardry with osmium. Drug Discov. Today 2014, 19, 1640–1648. [Google Scholar] [CrossRef] [PubMed]
- Nabiyeva, T.; Marschner, C.; Blom, B. Synthesis, structure and anti-cancer activity of osmium complexes bearing π-bound arene substituents and phosphane Co-Ligands: A review. Eur. J. Med. Chem. 2020, 201, 112483. [Google Scholar] [CrossRef]
- Leung, C.H.; Zhong, H.J.; Chan, D.S.H.; Ma, D.L. Bioactive iridium and rhodium complexes as therapeutic agents. Coord. Chem. Rev. 2013, 257, 1764–1776. [Google Scholar] [CrossRef]
- Máliková, K.; Masaryk, L.; Štarha, P. Anticancer Half-Sandwich Rhodium(III) Complexes. Inorganics 2021, 9, 26. [Google Scholar] [CrossRef]
- Liu, Z.; Sadler, P.J. Organoiridium complexes: Anticancer agents and catalysts. Acc. Chem. Res. 2014, 47, 1174–1185. [Google Scholar] [CrossRef]
- Graf, N.; Lippard, S.J. Redox activation of metal-based prodrugs as a strategy for drug delivery. Adv. Drug Deliv. Rev. 2012, 64, 993–1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yadav, A.; Janaratne, T.; Krishnan, A.; Singhal, S.S.; Yadav, S.; Dayoub, A.S.; Hawkins, D.L.; Awasthi, S.; MacDonnell, F.M. Regression of lung cancer by hypoxia-sensitizing ruthenium polypyridyl complexes. Mol. Cancer Ther. 2013, 12, 643–653. [Google Scholar] [CrossRef] [Green Version]
- Mihajlovic, K.; Milosavljevic, I.; Jeremic, J.; Savic, M.; Sretenovic, J.; Srejovic, I.M.; Zivkovic, V.I.; Jovicic, N.; Paunovic, M.; Bolevich, S.; et al. Redox and apoptotic potential of novel ruthenium complexes in the rat blood and heart. Can. J. Physiol. Pharmacol. 2021, 99, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Lai, H.; Xiong, Z.; Chen, B.; Chen, T. Functionalization and cancer-targeting design of ruthenium complexes for precise cancer therapy. Chem. Commun. 2019, 55, 9904–9914. [Google Scholar] [CrossRef] [PubMed]
- Gano, L.; Pinheiro, T.; Matos, A.P.; Tortosa, F.; Jorge, T.F.; Gonçalves, M.S.; Martins, M.; Morais, T.S.; Valente, A.; Tomaz, A.I.; et al. Antitumour and Toxicity Evaluation of a Ru(II)-Cyclopentadienyl Complex in a Prostate Cancer Model by Imaging Tools. Anticancer Agents Med. Chem. 2019, 19, 1262–1275. [Google Scholar] [CrossRef] [PubMed]
- Mello-Andrade, F.; Cardoso, C.G.; Silva, C.R.E.; Chen-Chen, L.; Melo-Reis, P.R.; Lima, A.P.; Oliveira, R.; Ferraz, I.B.M.; Grisolia, C.K.; Almeida, M.A.P.; et al. Acute toxic effects of ruthenium (II)/amino acid/diphosphine complexes on Swiss mice and zebrafish embryos. Biomed. Pharmacother. 2018, 107, 1082–1092. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, A.C. Synthesis, Biological Activity and Medicinal Applications of Ruthenium Complexes Containing Carbohydrate Ligands. Curr. Med. Chem. 2019, 26, 6412–6437. [Google Scholar] [CrossRef]
- Bononi, G.; Iacopini, D.; Cicio, G.; Di Pietro, S.; Granchi, C.; Di Bussolo, V.; Minutolo, F. Glycoconjugated Metal Complexes as Cancer Diagnostic and Therapeutic Agents. ChemMedChem 2020, 16, 30–64. [Google Scholar] [CrossRef]
- Hartinger, C.G.; Nazarov, A.A.; Ashraf, S.M.; Dyson, P.J.; Keppler, B.K. Carbohydrate-Metal Complexes and their Potential as Anticancer Agents. Curr. Med. Chem. 2008, 15, 2574–2591. [Google Scholar] [CrossRef]
- Kacsir, I.; Sipos, A.; Ujlaki, G.; Buglyó, P.; Somsák, L.; Bai, P.; Bokor, É. Ruthenium Half-Sandwich Type Complexes with Bidentate Monosaccharide Ligands Show Antineoplastic Activity in Ovarian Cancer Cell Models through Reactive Oxygen Species Production. Int. J. Mol. Sci. 2021, 22, 10454. [Google Scholar] [CrossRef]
- Bakewell, S.; Conde, I.; Fallah, Y.; McCoy, M.; Jin, L.; Shajahan-Haq, A.N. Inhibition of DNA Repair Pathways and Induction of ROS Are Potential Mechanisms of Action of the Small Molecule Inhibitor BOLD-100 in Breast Cancer. Cancers 2020, 12, 2647. [Google Scholar] [CrossRef]
- Xu, Z.; Kong, D.; He, X.; Guo, L.; Ge, X.; Liu, X.; Zhang, H.; Li, J.; Yang, Y.; Liu, Z. Mitochondria-targeted half-sandwich rutheniumII diimine complexes: Anticancer and antimetastasis via ROS-mediated signalling. Inorg. Chem. Front. 2018, 5, 2100–2105. [Google Scholar] [CrossRef]
- De Camargo, M.S.; De Grandis, R.A.; da Silva, M.M.; da Silva, P.B.; Santoni, M.M.; Eismann, C.E.; Menegario, A.A.; Cominetti, M.R.; Zanelli, C.F.; Pavan, F.R.; et al. Determination of in vitro absorption in Caco-2 monolayers of anticancer Ru(II)-based complexes acting as dual human topoisomerase and PARP inhibitors. Biometals 2019, 32, 89–100. [Google Scholar] [CrossRef]
- Yusoh, N.A.; Ahmad, H.; Gill, M.R. Combining PARP Inhibition with Platinum, Ruthenium or Gold Complexes for Cancer Therapy. ChemMedChem 2020, 15, 2121–2135. [Google Scholar] [CrossRef]
- Benltifa, M.; Vidal, S.; Fenet, B.; Msaddek, M.; Goekjian, P.G.; Praly, J.P.; Brunyánszki, A.; Docsa, T.; Gergely, P. In search of glycogen phosphorylase inhibitors: 5-substituted 3-C-glucopyranosyl-1,2,4-oxadiazoles from β-D-glucopyranosyl cyanides upon cyclization of O-acylamidoxime intermediates. Eur. J. Org. Chem. 2006, 2006, 4242–4256. [Google Scholar] [CrossRef]
- Cecioni, S.; Argintaru, O.A.; Docsa, T.; Gergely, P.; Praly, J.P.; Vidal, S. Probing multivalency for the inhibition of an enzyme: Glycogen phosphorylase as a case study. New J. Chem. 2009, 33, 148–156. [Google Scholar] [CrossRef]
- Godó, A.J.; Bényei, A.C.; Duff, B.; Egan, D.A.; Buglyó, P. Synthesis and X-ray diffraction structures of novel half-sandwich Os(II)- and Ru(II)-hydroxamate complexes. RSC Adv. 2012, 2, 1486–1495. [Google Scholar] [CrossRef] [Green Version]
- Spek, A.L. Single-crystal structure validation with the program PLATON. J. Appl. Crystallogr. 2003, 36, 7–13. [Google Scholar] [CrossRef] [Green Version]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. B Struct. Sci. Crys. Eng. Mater. 2016, 72, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Virág, L.; Salzman, A.L.; Szabó, C. Poly(ADP-ribose) synthetase activation mediates mitochondrial injury during oxidant-induced cell death. J. Immunol. 1998, 161, 3753–3759. [Google Scholar] [PubMed]
- Henslee, E.A.; Torcal Serrano, R.M.; Labeed, F.H.; Jabr, R.I.; Fry, C.H.; Hughes, M.P.; Hoettges, K.F. Accurate quantification of apoptosis progression and toxicity using a dielectrophoretic approach. Analyst 2016, 141, 6408–6415. [Google Scholar] [CrossRef] [Green Version]
- Skehan, P.; Storeng, R.; Scudiero, D.; Monks, A.; McMahon, J.; Vistica, D.; Warren, J.T.; Bokesch, H.; Kenney, S.; Boyd, M.R. New colorimetric cytotoxicity assay for anticancer-drug screening. J. Natl. Cancer Inst. 1990, 82, 1107–1112. [Google Scholar] [CrossRef] [PubMed]
- Bai, P.; Bakondi, É.; Szabó, E.; Gergely, P.; Szabó, C.; Virág, L. Partial protection by poly(ADP-ribose) polymerase inhibitors from nitroxyl-induced cytotoxity in thymocytes. Free Radic. Biol. Med. 2001, 31, 1616–1623. [Google Scholar] [CrossRef]
- Kandioller, W.; Balsano, E.; Meier, S.M.; Jungwirth, U.; Göschl, S.; Roller, A.; Jakupec, M.A.; Berger, W.; Keppler, B.K.; Hartinger, C.G. Organometallic anticancer complexes of lapachol: Metal centre-dependent formation of reactive oxygen species and correlation with cytotoxicity. Chem. Commun. 2013, 49, 3348–3350. [Google Scholar] [CrossRef] [Green Version]
- Romero-Canelón, I.; Mos, M.; Sadler, P.J. Enhancement of Selectivity of an Organometallic Anticancer Agent by Redox Modulation. J. Med. Chem. 2015, 58, 7874–7880. [Google Scholar] [CrossRef] [PubMed]
- Scalcon, V.; Top, S.; Lee, H.Z.; Citta, A.; Folda, A.; Bindoli, A.; Leong, W.K.; Salmain, M.; Vessières, A.; Jaouen, G.; et al. Osmocenyl-tamoxifen derivatives target the thioredoxin system leading to a redox imbalance in Jurkat cells. J. Inorg. Biochem. 2016, 160, 296–304. [Google Scholar] [CrossRef] [Green Version]
- Ramos, R.; Zimbron, J.M.; Thorimbert, S.; Chamoreau, L.M.; Munier, A.; Botuha, C.; Karaiskou, A.; Salmain, M.; Sobczak-Thépot, J. Insights into the antiproliferative mechanism of (C^N)-chelated half-sandwich iridium complexes. Dalton Trans. 2020, 49, 17635–17641. [Google Scholar] [CrossRef] [PubMed]
- Carrasco, A.C.; Rodríguez-Fanjul, V.; Habtemariam, A.; Pizarro, A.M. Structurally Strained Half-Sandwich Iridium(III) Complexes As Highly Potent Anticancer Agents. J. Med. Chem. 2020, 63, 4005–4021. [Google Scholar] [CrossRef] [PubMed]
- Hamala, V.; Martišová, A.; Červenková Šťastná, L.; Karban, J.; Dančo, A.; Šimarek, A.; Lamač, M.; Horáček, M.; Kolářová, T.; Hrstka, R.; et al. Ruthenium tetrazene complexes bearing glucose moieties on their periphery: Synthesis, characterization, and in vitro cytotoxicity. App. Organomet. Chem. 2020, 34, e5896. [Google Scholar] [CrossRef]
- Berger, I.; Hanif, M.; Nazarov, A.A.; Hartinger, C.G.; John, R.O.; Kuznetsov, M.L.; Groessl, M.; Schmitt, F.; Zava, O.; Biba, F.; et al. In Vitro Anticancer Activity and Biologically Relevant Metabolization of Organometallic Ruthenium Complexes with Carbohydrate-Based Ligands. Chem. Eur. J. 2008, 14, 9046–9057. [Google Scholar] [CrossRef]
- Hanif, M.; Meier, S.; Nazarov, A.; Risse, J.; Legin, A.; Casini, A.; Jakupec, M.; Keppler, B.; Hartinger, C. Influence of the π-coordinated arene on the anticancer activity of ruthenium(II) carbohydrate organometallic complexes. Front. Chem. 2013, 1, 27. [Google Scholar] [CrossRef] [Green Version]
- Florindo, P.R.; Pereira, D.M.; Borralho, P.M.; Costa, P.J.; Piedade, M.F.M.; Rodrigues, C.M.P.; Fernandes, A.C. New [(η5-C5H5)Ru(N-N)(PPh3)][PF6] compounds: Colon anticancer activity and GLUT-mediated cellular uptake of carbohydrate-appended complexes. Dalton Trans. 2016, 45, 11926–11930. [Google Scholar] [CrossRef]
- Florindo, P.R.; Pereira, D.M.; Borralho, P.M.; Rodrigues, C.M.P.; Piedade, M.F.M.; Fernandes, A.C. Cyclopentadienyl-Ruthenium(II) and Iron(II) Organometallic Compounds with Carbohydrate Derivative Ligands as Good Colorectal Anticancer Agents. J. Med. Chem. 2015, 58, 4339–4347. [Google Scholar] [CrossRef] [PubMed]
- Florindo, P.; Marques, I.J.; Nunes, C.D.; Fernandes, A.C. Synthesis, characterization and cytotoxicity of cyclopentadienyl ruthenium(II) complexes containing carbohydrate-derived ligands. J. Organomet. Chem. 2014, 760, 240–247. [Google Scholar] [CrossRef]
- Böge, M.; Fowelin, C.; Bednarski, P.; Heck, J. Diaminohexopyranosides as Ligands in Half-Sandwich Ruthenium(II), Rhodium(III), and Iridium(III) Complexes. Organometallics 2015, 34, 1507–1521. [Google Scholar] [CrossRef]
- Greenwood, N.N.; Earnshaw, A. Chemistry of the Elements; Elsevier: Amsterdam, The Netherlands, 1997. [Google Scholar] [CrossRef]
- Bruijnincx, P.C.A.; Sadler, P.J. Controlling Platinum, Ruthenium and Osmium Reactivity for Anticancer Drug Design. Adv. Inorg. Chem. 2009, 61, 1–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gesztelyi, R.; Zsuga, J.; Kemény-Beke, A.; Varga, B.; Juhász, B.; Tosaki, A. The Hill equation and the origin of quantitative pharmacology. Arch. Hist. Exact. Sci. 2012, 66, 427–438. [Google Scholar] [CrossRef]
- Li, G.; Liu, H.; Feng, R.; Kang, T.S.; Wang, W.; Ko, C.N.; Wong, C.Y.; Ye, M.; Ma, D.L.; Wan, J.B.; et al. A bioactive ligand-conjugated iridium(III) metal-based complex as a Keap1-Nrf2 protein-protein interaction inhibitor against acetaminophen-induced acute liver injury. Redox. Biol. 2021, 48, 102129. [Google Scholar] [CrossRef]
- Parveen, S.; Hanif, M.; Leung, E.; Tong, K.K.H.; Yang, A.; Astin, J.; De Zoysa, G.H.; Steel, T.R.; Goodman, D.; Movassaghi, S.; et al. Anticancer organorhodium and -iridium complexes with low toxicity in vivo but high potency in vitro: DNA damage, reactive oxygen species formation, and haemolytic activity. Chem. Commun. 2019, 55, 12016–12019. [Google Scholar] [CrossRef]
- Smolková, K.; Mikó, E.; Kovács, T.; Leguina-Ruzzi, A.; Sipos, A.; Bai, P. NRF2 in regulating cancer metabolism. Antiox. Redox. Signal. 2020, 33, 966–997. [Google Scholar] [CrossRef] [Green Version]
- Kovács, P.; Csonka, T.; Kovács, T.; Sári, Z.; Ujlaki, G.; Sipos, A.; Karányi, Z.; Szeőcs, D.; Hegedűs, C.; Uray, K.; et al. Lithocholic acid, a metabolite of the microbiome, increases oxidative stress in breast cancer. Cancers 2019, 11, 1255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sári, Z.; Mikó, E.; Kovács, T.; Boratkó, A.; Ujlaki, G.; Jankó, L.; Kiss, B.; Uray, K.; Bai, P. Indoxylsulfate, a Metabolite of the Microbiome, Has Cytostatic Effects in Breast Cancer via Activation of AHR and PXR Receptors and Induction of Oxidative Stress. Cancers 2020, 12, 2915. [Google Scholar] [CrossRef]
- Sári, Z.; Mikó, E.; Kovács, T.; Jankó, L.; Csonka, T.; Sebő, E.; Toth, J.; Tóth, D.; Árkosy, P.; Boratkó, A.; et al. Indolepropionic acid, a metabolite of the microbiome, has cytostatic properties in breast cancer by activating AHR and PXR receptors and inducing oxidative stress. Cancers 2020, 12, 2411. [Google Scholar] [CrossRef]
- Sipos, A.; Ujlaki, G.; Mikó, E.; Maka, E.; Szabó, J.; Uray, K.; Krasznai, Z.; Bai, P. The role of the microbiome in ovarian cancer: Mechanistic insights into oncobiosis and to bacterial metabolite signaling. Mol. Med. 2021, 27, 33. [Google Scholar] [CrossRef] [PubMed]
- Kiss, B.; Mikó, E.; Sebő, É.; Toth, J.; Ujlaki, G.; Szabó, J.; Uray, K.; Bai, P.; Árkosy, P. Oncobiosis and Microbial Metabolite Signaling in Pancreatic Adenocarcinoma. Cancers 2020, 12, 1068. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [Green Version]
- Westrip, S.P. publCIF: Software for editing, validating and formatting crystallographic information files. J. Appl. Crystallogr. 2010, 43, 920–925. [Google Scholar] [CrossRef] [Green Version]
- Macrae, C.F.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Shields, G.P.; Taylor, R.; Towler, M.; van De Streek, J. Mercury: Visualization and analysis of crystal structures. J. Appl. Crystallogr. 2006, 39, 453–457. [Google Scholar] [CrossRef] [Green Version]
- Parsons, S.; Flack, H.D.; Wagner, T. Use of intensity quotients and differences in absolute structure refinement. Acta Crystallogr. B Struct. Sci. Crys. Eng. Mater. 2013, 69, 249–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozsup, M.; Dömötör, O.; Nagy, S.; Farkas, E.; Enyedy, É.A.; Buglyó, P. Synthesis, characterization and albumin binding capabilities of quinizarin containing ternary cobalt(III) complexes. J. Inorg. Biochem. 2020, 204, 110963. [Google Scholar] [CrossRef] [PubMed]
- Bakondi, E.; Gönczi, M.; Szabó, É.; Bai, P.; Pacher, P.; Gergely, P.; Kovács, L.; Hunyadi, J.; Szabó, C.; Csernoch, L.; et al. Role of intracellular calcium mobilization and cell-density-dependent signaling in oxidative-stress-induced cytotoxicity in HaCaT keratinocytes. J. Invest. Dermatol. 2003, 121, 88–95. [Google Scholar] [CrossRef] [Green Version]
- Márton, J.; Fodor, T.; Nagy, L.; Vida, A.; Kis, G.; Brunyánszki, A.; Antal, M.; Lüscher, B.; Bai, P. PARP10 (ARTD10) modulates mitochondrial function. PLoS ONE 2018, 13, e0187789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Box, G.E.P.; Cox, D.R. An analysis of transformations. J. R. Stat. Soc. B 1964, 26, 211–234. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||||
|---|---|---|---|---|---|---|
| Heterocyclic Ligand | R1 | R2 | R3 | Product | Yield (%) | Diastereomeric Ratio |
| L-3 | OBz | H | CH2OBz | Os-3 | 71 | 7:6 |
| L-4 | H | OBz | CH2OBz | Os-4 | 78 | 3:2 |
| L-5 | OBz | H | H | Os-5 | 84 | 7:5 |
| L-6 | - | - | - | Os-6 | 85 | 1:1 |
 | |||||||
|---|---|---|---|---|---|---|---|
| Heterocyclic Ligand | R | R1 | R2 | R3 | Product | Yield (%) | Diastereomeric Ratio |
| L-1 | Bz | - | - | - | Ir-1 | 70 | 6:1 |
| L-2 | H | - | - | - | Ir-2 | 86 | 1:1 |
| L-3 | Bz | OBz | H | CH2OBz | Ir-3 | 80 | 5:2 |
| L-4 | Bz | H | OBz | CH2OBz | Ir-4 | 77 | 2:1 |
| L-5 | Bz | OBz | H | H | Ir-5 | 72 | 3:1 |
| L-6 | Bz | - | - | - | Ir-6 | 84 | 2:1 |
| L-7 | Ac | H | OAc | CH2OAc | Ir-7 | 81 | 5:2 |
| L-8 | H | OH | H | CH2OH | Ir-8 | 79 | 1:1 |
| L-9 | H | H | OH | CH2OH | Ir-9 | 61 | 1:1 |
| L-10 | H | OH | H | H | Ir-10 | 74 | 1:1 |
| L-11 | Ac | - | - | - | Ir-11 | 81 | 6:5 |
| L-12 | H | - | - | - | Ir-12 | 56 | 1:1 |
 | ||||||
|---|---|---|---|---|---|---|
| Heterocyclic Ligand | R1 | R2 | R3 | Product | Yield (%) | Diastereomeric Ratio |
| L-3 | OBz | H | CH2OBz | Rh-3 | 88 | 3:1 |
| L-4 | H | OBz | CH2OBz | Rh-4 | 85 | 9:2 |
| L-5 | OBz | H | H | Rh-5 | 87 | 5:2 |
| L-6 | - | - | - | Rh-6 | 91 | 2:1 |
| Compound | Maximum Inhibition (%) | Compound | Maximum Inhibition (%) | ||||
|---|---|---|---|---|---|---|---|
| A2780 | ID8 | Fibroblast | A2780 | ID8 | Fibroblast | ||
| L-1 | ND | Ir-7 | ND | ||||
| L-2 | ND | Ir-8 | ND | ||||
| Ru-1 | ND | Ir-9 | ND | ||||
| Ru-2 | ND | Ir-10 | ND | ||||
| Os-3 | 37.42 | 35.24 | ND | Ir-11 | ND | ||
| Os-4 | 26.67 | 22.91 | ND | Ir-12 | ND | ||
| Os-5 | 31.38 | 59.51 | ND | Rh-3 | ND | ND | ND |
| Os-6 | 21.38 | 64.44 | ND | Rh-4 | ND | ND | ND |
| Ir-1 | ND | Rh-5 | ND | 21.87 | ND | ||
| Ir-2 | ND | Rh-6 | ND | 28.28 | ND | ||
| Ir-3 | ND | 26.11 | ND | Ir-dimer | ND | ND | ND |
| Ir-4 | ND | ND | 31.19 | Rh-dimer | ND | ND | ND |
| Ir-5 | ND | ND | ND | Os-dimer | ND | ND | ND |
| Ir-6 | 43.07 | 62.71 | ND | ||||
| A2780 | ID8 | Capan2 | Saos | L428 | Fibroblast | LogD | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Max. Inh. (%) | IC50 (µM) | Hill (µM) | Max. Inh. (%) | IC50 (µM) | Hill (µM) | Max. Inh. (%) | IC50 (µM) | Hill (µM) | Max. Inh. (%) | IC50 (µM) | Hill (µM) | Max. Inh. (%) | IC50 (µM) | Hill (µM) | Max. Inh. (%) | IC50 (µM) | Hill (µM) | ||
| L-1 | ND | ND | ND | ||||||||||||||||
| L-2 | ND | ND | ND | ||||||||||||||||
| Ru-1 | ND | ND | ND | 2.79 | |||||||||||||||
| Ru-2 | ND | ND | ND | −0.96 | |||||||||||||||
| Os-3 | 100 | 2.52 | 1.82 | 100 | 5.94 | 2.39 | 100 | 8.80 | 2.30 | 65 | ND | ND | 100 | ND | ND | ND | ND | ND | 3.32 |
| Os-4 | 100 | 3.23 | 2.52 | 100 | 6.95 | 3.69 | 100 | 6.20 | 1.85 | 100 | 8.78 | ND | 100 | ND | ND | ND | ND | ND | 2.87 |
| Os-5 | 100 | 2.81 | 1.85 | 100 | 1.96 | 2.90 | 100 | 8.89 | 2.26 | 100 | 8.52 | ND | 100 | ND | ND | ND | ND | ND | 2.36 |
| Os-6 | 100 | 0.73 | 3.01 | 100 | 1.96 | 3.14 | 100 | 1.71 | 1.33 | 100 | 3.60 | ND | 100 | ND | ND | ND | ND | ND | 2.33 |
| Ir-1 | 35.13 | ND | ND | 2.87 | |||||||||||||||
| Ir-2 | 36.38 | ND | ND | −1.15 | |||||||||||||||
| Ir-3 | 69.25 | ND | ND | 67.93 | ND | ND | 27.62 | ND | ND | ND | ND | ND | 1.46 | ||||||
| Ir-4 | 32.45 | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | 1.46 | ||||||
| Ir-5 | 45.45 | ND | ND | 53.15 | ND | ND | ND | ND | ND | ND | ND | ND | 1.80 | ||||||
| Ir-6 | 100 | 1.64 | 2.08 | 100 | 0.98 | 2.46 | 100 | 4.15 | 1.58 | 100 | 2.71 | ND | 100 | ND | ND | ND | ND | ND | 2.36 |
| Ir-7 | 26.91 | ND | ND | −0.75 | |||||||||||||||
| Ir-8 | ND | ND | ND | −1.60 | |||||||||||||||
| Ir-9 | 21.29 | ND | ND | −1.47 | |||||||||||||||
| Ir-10 | 23.12 | ND | ND | −1.56 | |||||||||||||||
| Ir-11 | 20.26 | ND | ND | −1.31 | |||||||||||||||
| Ir-12 | 19.11 | ND | ND | −2.08 | |||||||||||||||
| Rh-3 | 39.86 | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | 1.60 | ||||||
| Rh-4 | 35.30 | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | 1.64 | ||||||
| Rh-5 | 51.28 | ND | ND | 75.25 | ND | ND | ND | ND | ND | ND | ND | ND | 1.8 | ||||||
| Rh-6 | 59 | 25.28 | 0.71 | 43.70 | ND | ND | ND | ND | ND | ND | ND | ND | 1.85 | ||||||
| Ir-dimer | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | |
| Rh-dimer | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | |
| Os-dimer | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | |
| Ru-3 * | 100 | 6.19 | 2.62 | 100 | 5.66 | 3.04 | 100 | 3.27 | 1.81 | ND | ND | ND | 2.41 | ||||||
| Ru-4 * | 100 | 4.27 | 3.5 | 100 | 7.94 | 3.31 | 100 | 3.82 | 1.66 | ND | ND | ND | 2.44 | ||||||
| Ru-5 * | 100 | 8.54 | 3.74 | 100 | 6.76 | 3.59 | 100 | 4.97 | 1.66 | 80 | 24.63 | 3.52 | 2.04 | ||||||
| Ru-6 * | 100 | 0.87 | 1.95 | 100 | 2.15 | 2.5 | 100 | 2.18 | 2.43 | ND | ND | ND | 2.85 | ||||||
| Ru-1 | Ir-2 | |
|---|---|---|
| Crystal data | ||
| Chemical formula | 2(C51H45ClN3O10Ru)·2(F6P)·5(CHCl3) | C23H30ClIrN3O6·F6P |
| Mr | 2879.61 | 817.12 |
| Crystal system, space group | Monoclinic, P21 | Orthorhombic, P212121 |
| Temperature (K) | 150 | 295 |
| a, b, c (Å) | 14.531 (3), 24.879 (4), 16.855 (4) | 9.3075 (8), 12.1090 (8), 24.6823 (18) |
| α,β,γ (°) | 90, 93.610 (5), 90 | 90, 90, 90 |
| V (Å3) | 6081 (2) | 2781.8 (4) |
| Z | 2 | 4 |
| Radiation type | Mo Kα | |
| µ (mm−1) | 0.73 | 5.04 |
| Crystal size (mm) | 0.19 × 0.18 × 0.05 | 0.51 × 0.10 × 0.08 |
| Data collection | ||
| Diffractometer | Bruker D8 VENTURE | |
| Absorption correction | Multi-scan SADABS2016/2—Bruker AXS area detector scaling and absorption correction | |
| Tmin, Tmax | 0.89, 0.97 | 0.53, 0.70 |
| No. of measured, independent and observed [I > 2σ(I)] reflections | 71,476, 23,083, 12,753 | 20076, 5675, 4789 |
| Rint | 0.115 | 0.111 |
| (sin θ/λ)max (Å−1) | 0.612 | 0.626 |
| Refinement | ||
| R[F2 > 2σ (F2)], wR(F2), S | 0.090, 0.175, 1.32 | 0.059, 0.104, 1.07 |
| No. of reflections | 23083 | 5675 |
| No. of parameters | 1424 | 387 |
| No. of restraints | 10 | 34 |
| H-atom treatment | H-atom parameters constrained | H atoms treated by a mixture of independent and constrained refinement |
| w = 1/[σ2(Fo2) + (0.0123P)2 + 11.6544P] where P = (Fo2 + 2Fc2)/3 | w = 1/[σ2(Fo2) + (0.0285P)2] where P = (Fo2 + 2Fc2)/3 | |
| Δ〉max, Δ〉min (e Å−3) | 1.55, −0.85 | 1.81, −3.07 |
| Absolute structure | Flack x determined using 3978 quotients [(I+) − (I−)]/[(I+) + (I−)] [70] | Flack x determined using 1573 quotients [(I+) − (I−)]/[(I+) + (I−)] [70] |
| Absolute structure parameter | −0.04 (2) | 0.017 (11) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kacsir, I.; Sipos, A.; Bényei, A.; Janka, E.; Buglyó, P.; Somsák, L.; Bai, P.; Bokor, É. Reactive Oxygen Species Production Is Responsible for Antineoplastic Activity of Osmium, Ruthenium, Iridium and Rhodium Half-Sandwich Type Complexes with Bidentate Glycosyl Heterocyclic Ligands in Various Cancer Cell Models. Int. J. Mol. Sci. 2022, 23, 813. https://doi.org/10.3390/ijms23020813
Kacsir I, Sipos A, Bényei A, Janka E, Buglyó P, Somsák L, Bai P, Bokor É. Reactive Oxygen Species Production Is Responsible for Antineoplastic Activity of Osmium, Ruthenium, Iridium and Rhodium Half-Sandwich Type Complexes with Bidentate Glycosyl Heterocyclic Ligands in Various Cancer Cell Models. International Journal of Molecular Sciences. 2022; 23(2):813. https://doi.org/10.3390/ijms23020813
Chicago/Turabian StyleKacsir, István, Adrienn Sipos, Attila Bényei, Eszter Janka, Péter Buglyó, László Somsák, Péter Bai, and Éva Bokor. 2022. "Reactive Oxygen Species Production Is Responsible for Antineoplastic Activity of Osmium, Ruthenium, Iridium and Rhodium Half-Sandwich Type Complexes with Bidentate Glycosyl Heterocyclic Ligands in Various Cancer Cell Models" International Journal of Molecular Sciences 23, no. 2: 813. https://doi.org/10.3390/ijms23020813