
Novel Mechanism by a Bis-Pyridinium Fullerene Derivative to Induce Apoptosis by Enhancing the MEK-ERK Pathway in a Reactive Oxygen Species-Independent Manner in BCR-ABL-Positive Chronic Myeloid Leukemia-Derived K562 Cells

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
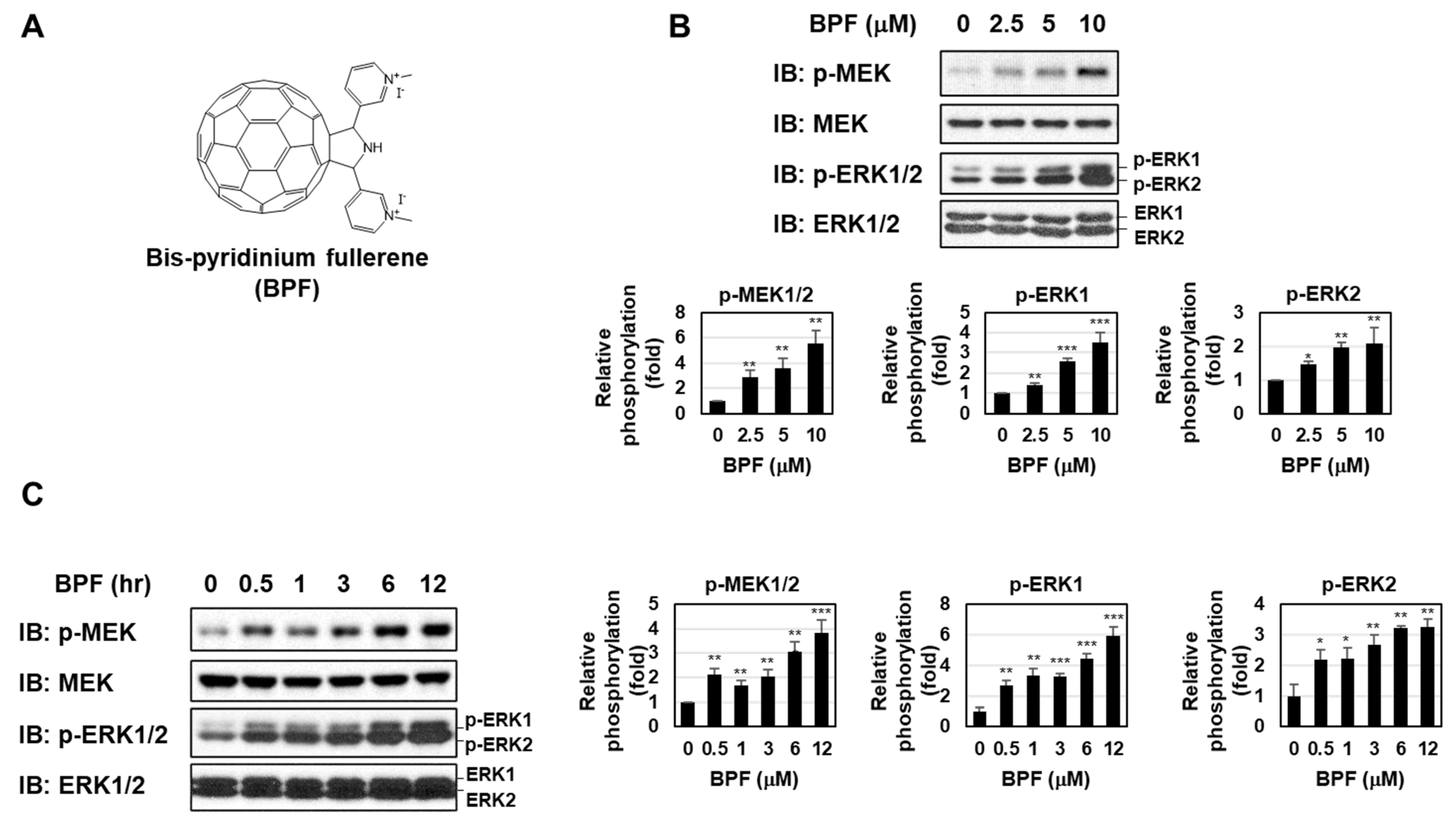
2.1. BPF Enhanced the Activation of the MEK-ERK Pathway in K562 Cells
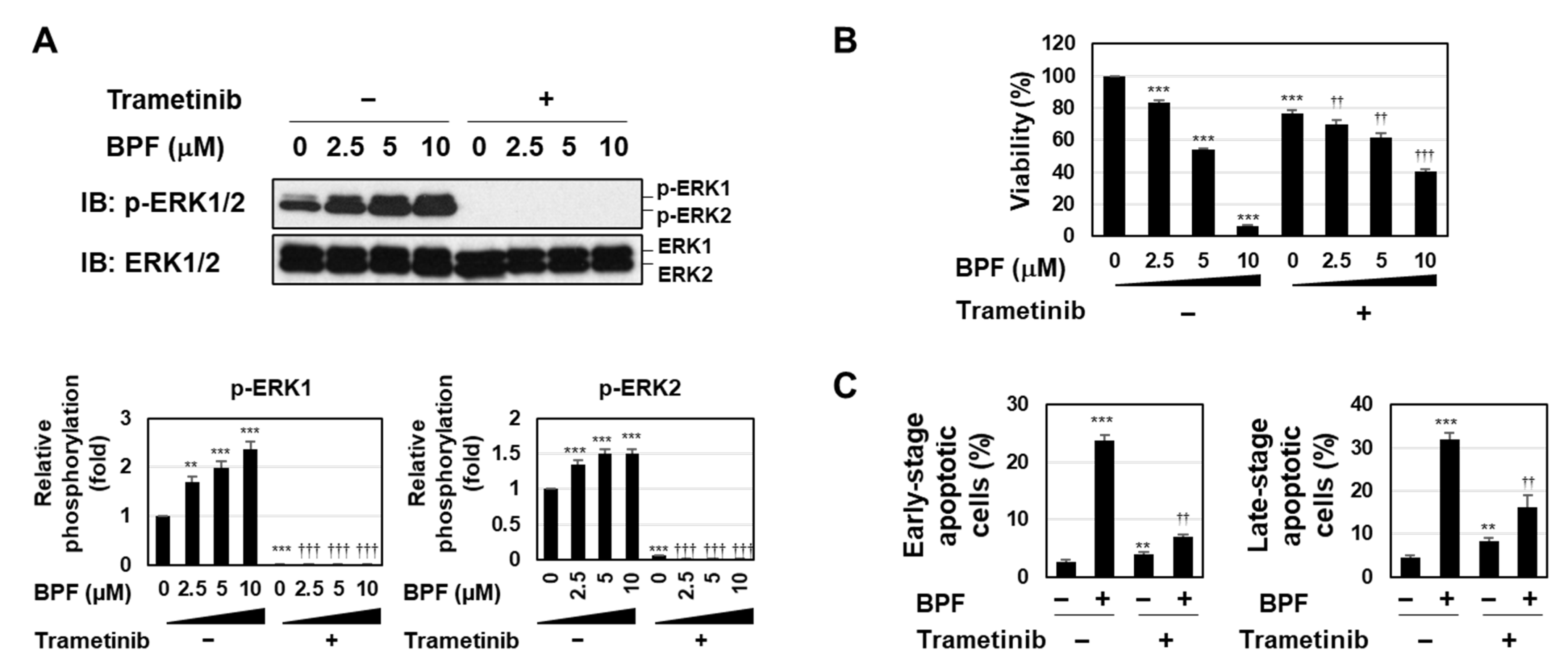
2.2. BPF Induced Apoptosis by Enhancing the Activation of the MEK-ERK Pathway in K562 Cells
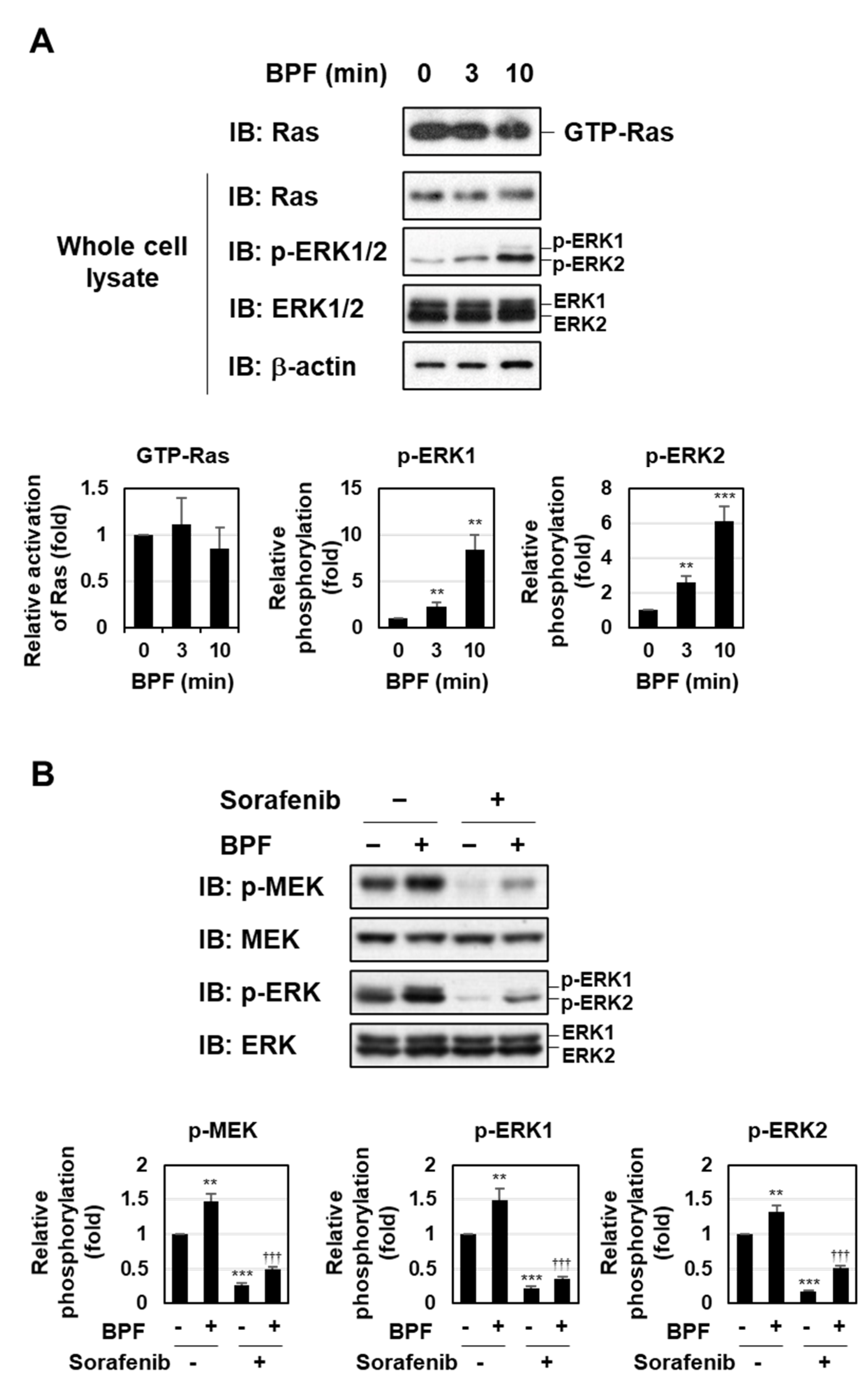
2.3. BPF Induced the Activation of the MEK-ERK Pathway Independently of ROS Generation in K562 Cells
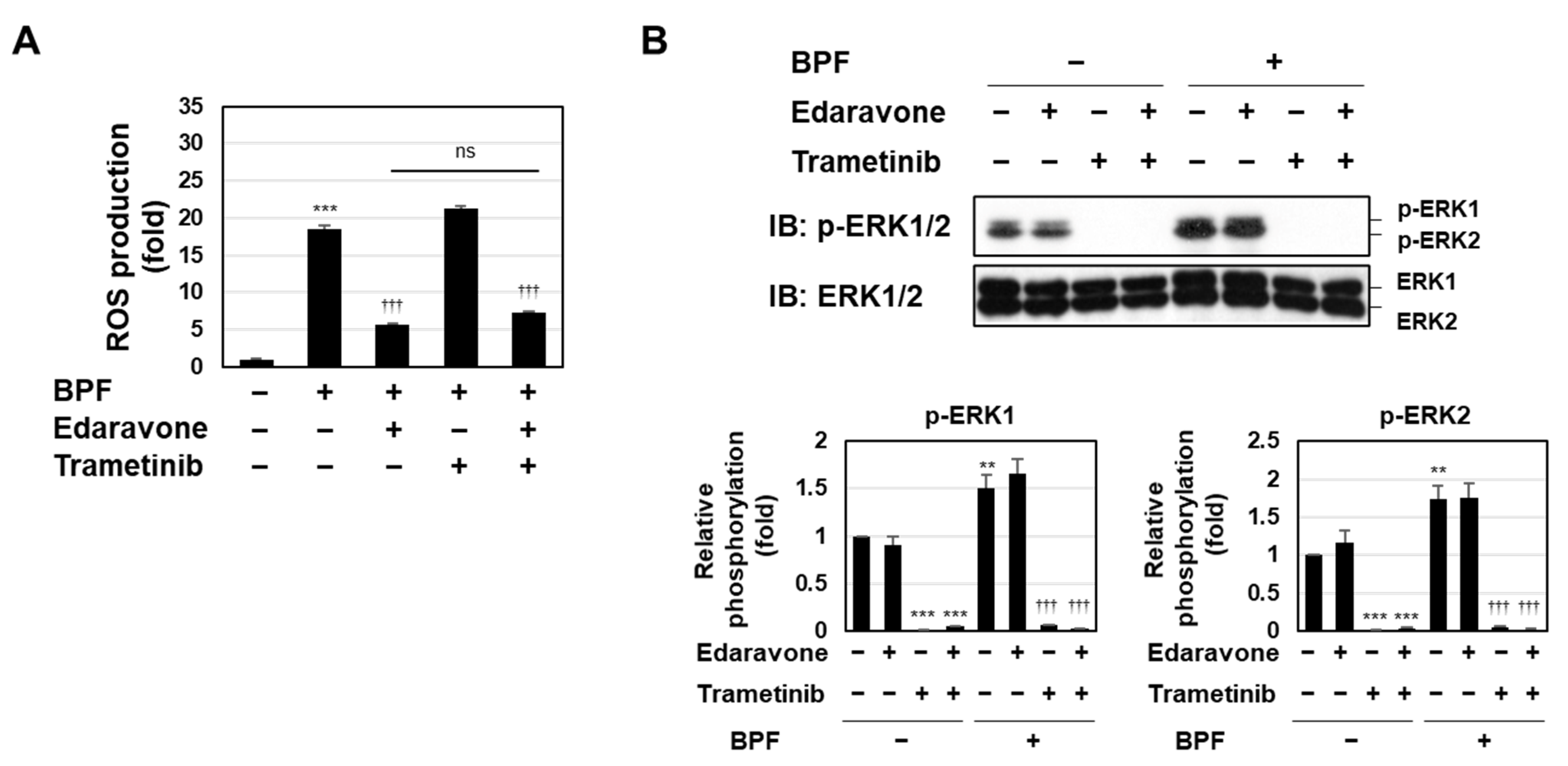
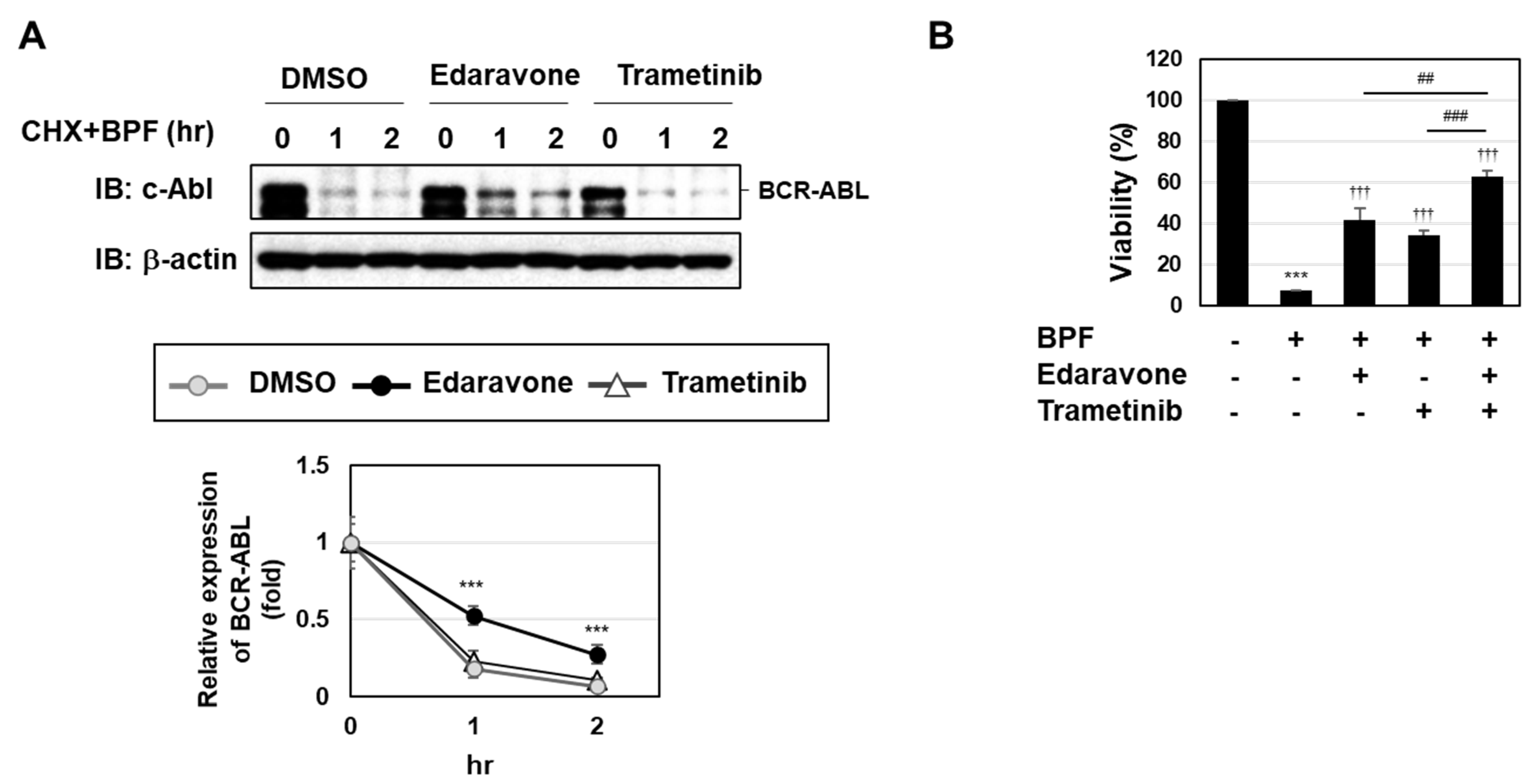
2.4. BPF Induced Cell Death through Two Pathways, ROS Generation and the Activation of the MEK-ERK Pathway, in K562 Cells
2.5. Enforced Expression of the BRAFV600E Mutant Enhanced the Sensitivity of K562 Cells to BPF
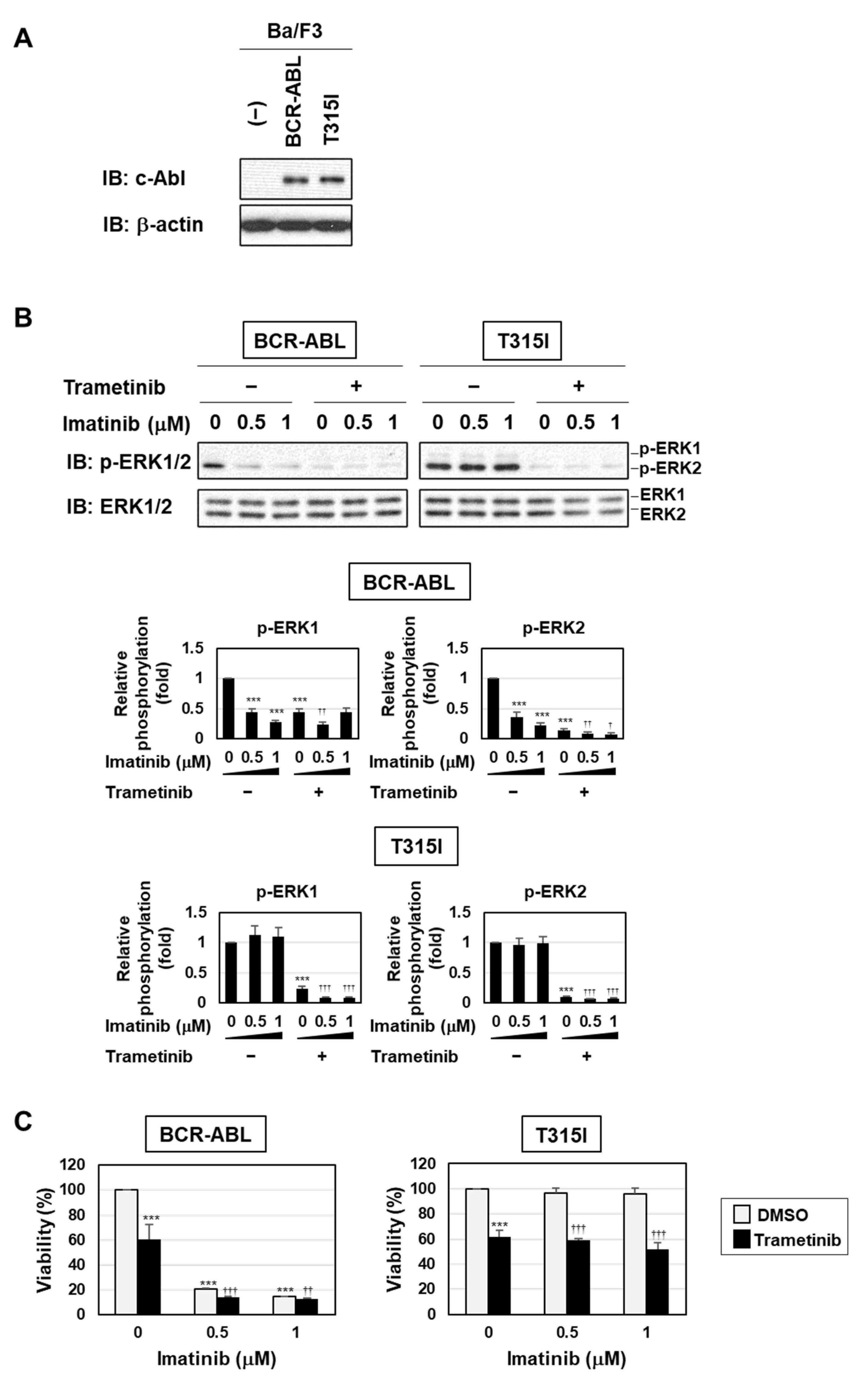
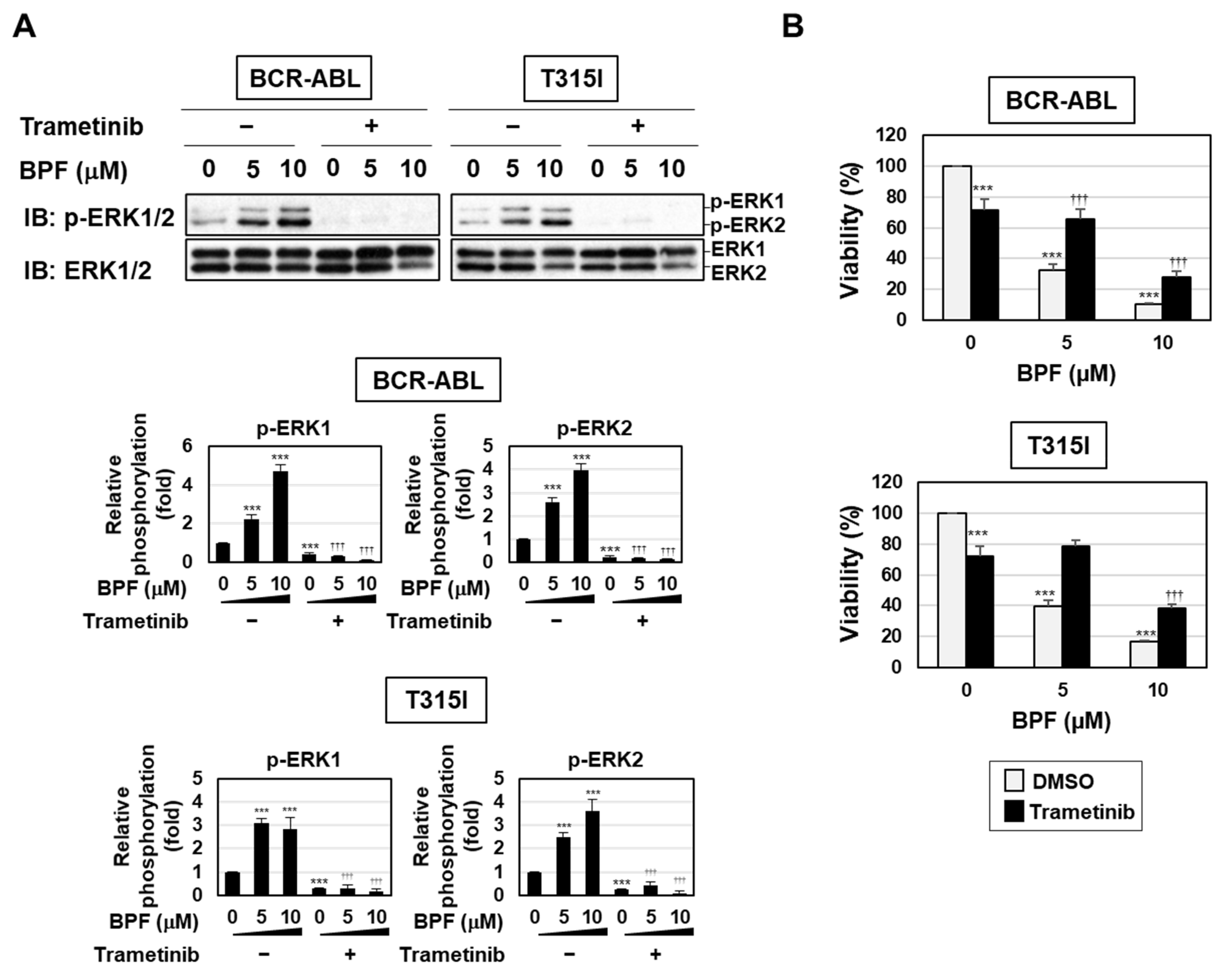
2.6. BPF Overcame Gatekeeper Mutation-Induced Resistance to BCR-ABL Inhibitors
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Retrovirus Infection
4.2. Measurement of Cell Viability
4.3. Annexin V/PI Assay
4.4. Measurement of Intracellular ROS
4.5. Immunoblotting
4.6. Measurement of Ras Activity
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Abl | Abelson |
| ALL | acute lymphocytic leukemia |
| Bcr | breakpoint cluster region |
| BPF | bis-pyridinium fullerene derivative |
| CHX | cycloheximide |
| CML | chronic myeloid leukemia |
| DCFH-DA | 2′, 7′-dichlorodihydrofluorescein diacetate |
| ERK | extracellular signal-regulated kinase |
| FBS | fetal bovine serum |
| MEK | mitogen-activated protein kinase/extracellular signal-regulated kinase kinase |
| PBS | phosphate-buffered saline |
| Ph | Philadelphia chromosome |
| RBD | Ras-binding domain |
| ROS | reactive oxygen species |
| SOS | Son of Sevenless |
| STAT5 | signal transducers and activator of transcription 5 |
| WST | water-soluble tetrazolium |
References
- Pinilla-Ibarz, J.; Bello, C. Modern approaches to treating chronic myelogenous leukemia. Curr. Oncol. Rep. 2008, 10, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Melo, J.V. BCR-ABL gene variants. Baillieres Clin. Haematol. 1997, 10, 203–222. [Google Scholar] [CrossRef]
- Kang, Z.J.; Liu, Y.F.; Xu, L.Z.; Long, Z.J.; Huang, D.; Yang, Y.; Liu, B.; Feng, J.X.; Pan, Y.J.; Yan, J.S.; et al. The Philadelphia chromosome in leukemogenesis. Chin. J. Cancer 2016, 27, 35–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hazlehurst, L.A.; Bewry, N.N.; Nair, R.R.; Pinilla-Ibarz, J. Signaling networks associated with BCR-ABL-dependent transformation. Cancer Control. 2009, 16, 100–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shuai, K.; Halpern, J.; ten Hoeve, J.; Rao, X.; Sawyers, C.L. Constitutive activation of STAT5 by the BCR-ABL oncogene in chronic myelogenous leukemia. Oncogene 1996, 13, 247–254. [Google Scholar] [PubMed]
- Nieborowska-Skorska, M.; Wasik, M.A.; Slupianek, A.; Salomoni, P.; Kitamura, T.; Calabretta, B.; Skorski, T. Signal transducer and activator of transcription (STAT)5 activation by BCR/ABL is dependent on intact Src homology (SH)3 and SH2 domains of BCR/ABL and is required for leukemogenesis. J. Exp. Med. 1999, 189, 1229–1242. [Google Scholar] [CrossRef]
- Puil, L.; Liu, J.; Gish, G.; Mbamalu, G.; Bowtell, D.; Pelicci, P.G.; Arlinghaus, R.; Pawson, T. Bcr-Abl oncoproteins bind directly to activators of the Ras signalling pathway. EMBO J. 1994, 13, 764–773. [Google Scholar] [CrossRef]
- Chu, S.; Li, L.; Singh, H.; Bhatia, R. BCR-tyrosine 177 plays an essential role in RAS and Akt activation and in human hematopoietic progenitor transformation in chronic myelogenous leukemia. Cancer Res. 2007, 67, 7045–7053. [Google Scholar] [CrossRef] [Green Version]
- Goga, A.; McLaughlin, J.; Afar, D.E.; Saffran, D.C.; Witte, O.N. Alternative signals to RAS for hematopoietic transformation by the BCR-ABL oncogene. Cell 1995, 82, 981–988. [Google Scholar] [CrossRef] [Green Version]
- O’Hare, T.; Deininger, M.W.; Eide, C.A.; Clackson, T.; Druker, B.J. Targeting the BCR-ABL signaling pathway in therapy-resistant Philadelphia chromosome-positive leukemia. Clin. Cancer Res. 2011, 17, 212–221. [Google Scholar] [CrossRef] [Green Version]
- Kantarjian, H.; Shah, N.P.; Hochhaus, A.; Cortes, J.; Shah, S.; Ayala, M.; Moiraghi, B.; Shen, Z.; Mayer, J.; Pasquini, R.; et al. Dasatinib versus imatinib in newly diagnosed chronic-phase chronic myeloid leukemia. N. Engl. J. Med. 2010, 362, 2260–2270. [Google Scholar] [CrossRef]
- Saglio, G.; Kim, D.W.; Issaragrisil, S.; le Coutre, P.; Etienne, G.; Lobo, C.; Pasquini, R.; Clark, R.E.; Hochhaus, A.; ENESTnd Investigators; et al. Nilotinib versus imatinib for newly diagnosed chronic myeloid leukemia. N. Engl. J. Med. 2010, 362, 2251–2259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortes, J.; Jabbour, E.; Kantarjian, H.; Yin, C.C.; Shan, J.; O’Brien, S.; Garcia-Manero, G.; Giles, F.; Breeden, M.; Reeves, N.; et al. Dynamics of BCR-ABL kinase domain mutations in chronic myeloid leukemia after sequential treatment with multiple tyrosine kinase inhibitors. Blood 2007, 110, 4005–4011. [Google Scholar] [CrossRef] [Green Version]
- Gorre, M.E.; Mohammed, M.; Ellwood, K.; Hsu, N.; Paquette, R.; Rao, P.N.; Sawyers, C.L. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science 2001, 293, 876–880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azam, M.; Latek, R.R.; Daley, G.Q. Mechanisms of autoinhibition and STI-571/imatinib resistance revealed by mutagenesis of BCR-ABL. Cell 2003, 112, 831–843. [Google Scholar] [CrossRef] [Green Version]
- Nishizawa, C.; Hashimoto, N.; Yokoo, S.; Funakoshi-Tago, M.; Kasahara, T.; Takahashi, K.; Nakamura, S.; Mashino, T. Pyrrolidinium-type fullerene derivative-induced apoptosis by the generation of reactive oxygen species in HL-60 cells. Free Radic. Res. 2009, 43, 1240–1247. [Google Scholar] [CrossRef]
- Funakoshi-Tago, M.; Nagata, T.; Tago, K.; Tsukada, M.; Tanaka, K.; Nakamura, S.; Mashino, T.; Kasahara, T. Fullerene derivative prevents cellular transformation induced by JAK2 V617F mutant through inhibiting c-Jun N-terminal kinase pathway. Cell. Signal. 2012, 24, 2024–2034. [Google Scholar] [CrossRef]
- Funakoshi-Tago, M.; Tsukada, M.; Watanabe, T.; Mameda, Y.; Tago, K.; Ohe, T.; Nakamura, S.; Mashino, T.; Kasahara, T. Effect of chemical modification on the ability of pyrrolidinium fullerene to induce apoptosis of cells transformed by JAK2 V617F mutant. Int. Immunopharmacol. 2014, 20, 258–263. [Google Scholar] [CrossRef] [PubMed]
- Yasuno, T.; Ohe, T.; Ikeda, H.; Takahashi, K.; Nakamura, S.; Mashino, T. Synthesis and antitumor activity of novel pyridinium fullerene derivatives. Int. J. Nanomed. 2019, 14, 6325–6337. [Google Scholar] [CrossRef] [Green Version]
- Sumi, K.; Tago, K.; Nakazawa, Y.; Takahashi, K.; Ohe, T.; Mashino, T.; Funakoshi-Tago, M. A bis-pyridinium fullerene derivative induces apoptosis through the generation of ROS in BCR-ABL-positive leukemia cells. Eur. J. Pharmacol. 2021, 916, 174714. [Google Scholar] [CrossRef]
- Lowinger, T.B.; Riedl, B.; Dumas, J.; Smith, R.A. Design and discovery of small molecules targeting raf-1 kinase. Curr. Pharm. Des. 2002, 8, 2269–2278. [Google Scholar] [CrossRef] [PubMed]
- Gilmartin, A.G.; Bleam, M.R.; Groy, A.; Moss, K.G.; Minthorn, E.A.; Kulkarni, S.G.; Rominger, C.M.; Erskine, S.; Fisher, K.E.; Yang, J.; et al. GSK1120212 (JTP-74057) is an inhibitor of MEK activity and activation with favorable pharmacokinetic properties for sustained in vivo pathway inhibition. Clin. Cancer. Res. 2011, 17, 989–1000. [Google Scholar] [CrossRef] [Green Version]
- Tabrizchi, R. Edaravone Mitsubishi-Tokyo. Curr. Opin. Investig. Drugs 2000, 1, 347–354. [Google Scholar] [PubMed]
- Holderfield, M.; Deuker, M.M.; McCormick, F.; McMahon, M. Targeting RAF kinases for cancer therapy: BRAF-mutated melanoma and beyond. Nat. Rev. Cancer 2014, 14, 455–467. [Google Scholar] [CrossRef] [Green Version]
- Foley, S.; Crowley, C.; Smaihi, M.; Bonfils, C.; Erlanger, B.F.; Seta, P.; Larroque, C. Cellular localisation of a water-soluble fullerene derivative. Biochem. Biophys. Res. Commun. 2002, 294, 116–119. [Google Scholar] [CrossRef]
- Chirico, F.; Fumelli, C.; Marconi, A.; Tinari, A.; Straface, E.; Malorni, W.; Pellicciari, R.; Pincelli, C. Carboxyfullerenes localize within mitochondria and prevent the UVB-induced intrinsic apoptotic pathway. Exp. Dermatol. 2007, 16, 429–436. [Google Scholar] [CrossRef]
- Yamakoshi, Y.; Sueyoshi, S.; Fukuhara, K.; Miyata, N.; Masumizu, T.; Kohno, M. •OH and O2•− generation in aqueous C60 and C70 solutions by photoirradiation: An EPR study. J. Am. Chem. Soc. 1998, 120, 12363–12364. [Google Scholar] [CrossRef]
- Zhang, B.; Bian, W.; Pal, A.; He, Y. Macrophage apoptosis induced by aqueous C60 aggregates changing the mitochondrial membrane potential. Environ. Toxicol. Pharmacol. 2015, 39, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Harhaji, L.; Isakovic, A.; Raicevic, N.; Markovic, Z.; Todorovic-Markovic, B.; Nikolic, N.; Vranjes-Djuric, S.; Markovic, I.; Trajkovic, V. Multiple mechanisms underlying the anticancer action of nanocrystalline fullerene. Eur. J. Pharmacol. 2007, 568, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Miao, Y.; Chen, L.; Xu, J.; Wang, X.; Zhao, H.; Shen, Y.; Hu, Y.; Bian, Y.; Shen, Y.; et al. The role of low levels of fullerene C60 nanocrystals on enhanced learning and memory of rats through persistent CaMKII activation. Biomaterials 2014, 35, 9269–9279. [Google Scholar] [CrossRef]
- Ueda, Y.; Hirai, S.; Osada, S.; Suzuki, A.; Mizuno, K.; Ohno, S. Protein kinase C activates the MEK-ERK pathway in a manner independent of RAS and dependent on Raf. J. Biol. Chem. 1996, 271, 23512–23519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Zhang, T.; Chang, X.; Wang, K.; Lee, M.H.; Ma, W.Y.; Liu, K.; Dong, Z. Targeting Opsin4/melanopsin with a novel small molecule suppresses PKC/RAF/MEK/ERK signaling and inhibits lung adenocarcinoma progression. Mol. Cancer Res. 2020, 18, 1028–1038. [Google Scholar] [CrossRef]
- Sugiura, R.; Satoh, R.; Takasaki, T. ERK: A double-edged sword in cancer. ERK-dependent apoptosis as a potential therapeutic Strategy for cancer. Cells 2021, 10, 2509. [Google Scholar] [CrossRef]
- Qi, M.; Elion, E.A. MAP kinase pathways. J. Cell Sci. 2005, 118, 3569–3572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chambard, J.C.; Lefloch, R.; Pouysségur, J.; Lenormand, P. ERK implication in cell cycle regulation. Biochim. Biophys. Acta 2007, 1773, 1299–1310. [Google Scholar] [CrossRef]
- Cagnol, S.; Chambard, J.C. ERK and cell death: Mechanisms of ERK-induced cell death—Apoptosis, autophagy and senescence. FEBS J. 2010, 277, 2–21. [Google Scholar] [CrossRef]
- Yao, Y.; Li, W.; Wu, J.; Germann, U.A.; Su, M.S.; Kuida, K.; Boucher, D.M. Extracellular signal-regulated kinase 2 is necessary for mesoderm differentiation. Proc. Natl. Acad. Sci. USA 2003, 100, 12759–12764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatano, N.; Mori, Y.; Oh-hora, M.; Kosugi, A.; Fujikawa, T.; Nakai, N.; Niwa, H.; Miyazaki, J.; Hamaoka, T.; Ogata, M. Essential role for ERK2 mitogen-activated protein kinase in placental development. Genes Cells 2003, 8, 847–856. [Google Scholar] [CrossRef]
- Satoh, Y.; Endo, S.; Ikeda, T.; Yamada, K.; Ito, M.; Kuroki, M.; Hiramoto, T.; Imamura, O.; Kobayashi, Y.; Watanabe, Y.; et al. Extracellular signal-regulated kinase 2 (ERK2) knockdown mice show deficits in long-term memory; ERK2 has a specific function in learning and memory. J. Neurosci. 2007, 27, 10765–10776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satoh, Y.; Endo, S.; Nakata, T.; Kobayashi, Y.; Yamada, K.; Ikeda, T.; Takeuchi, A.; Hiramoto, T.; Watanabe, Y.; Kazama, T. ERK2 contributes to the control of social behaviors in mice. J. Neurosci. 2011, 31, 11953–11967. [Google Scholar]
- Uchihara, Y.; Tago, K.; Taguchi, H.; Narukawa, Y.; Kiuchi, F.; Tamura, H.; Funakoshi-Tago, M. Taxodione induces apoptosis in BCR-ABL-positive cells through ROS generation. Biochem. Pharmacol. 2018, 154, 357–372. [Google Scholar] [CrossRef] [PubMed]
- Funakoshi-Tago, M.; Tago, K.; Li, C.; Hokimoto, S.; Tamura, H. Coffee decoction enhances tamoxifen proapoptotic activity on MCF-7 cells. Sci. Rep. 2020, 10, 19588. [Google Scholar] [CrossRef] [PubMed]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sumi, K.; Tago, K.; Nakazawa, Y.; Takahashi, K.; Ohe, T.; Mashino, T.; Funakoshi-Tago, M. Novel Mechanism by a Bis-Pyridinium Fullerene Derivative to Induce Apoptosis by Enhancing the MEK-ERK Pathway in a Reactive Oxygen Species-Independent Manner in BCR-ABL-Positive Chronic Myeloid Leukemia-Derived K562 Cells. Int. J. Mol. Sci. 2022, 23, 749. https://doi.org/10.3390/ijms23020749
Sumi K, Tago K, Nakazawa Y, Takahashi K, Ohe T, Mashino T, Funakoshi-Tago M. Novel Mechanism by a Bis-Pyridinium Fullerene Derivative to Induce Apoptosis by Enhancing the MEK-ERK Pathway in a Reactive Oxygen Species-Independent Manner in BCR-ABL-Positive Chronic Myeloid Leukemia-Derived K562 Cells. International Journal of Molecular Sciences. 2022; 23(2):749. https://doi.org/10.3390/ijms23020749
Chicago/Turabian StyleSumi, Kazuya, Kenji Tago, Yosuke Nakazawa, Kyoko Takahashi, Tomoyuki Ohe, Tadahiko Mashino, and Megumi Funakoshi-Tago. 2022. "Novel Mechanism by a Bis-Pyridinium Fullerene Derivative to Induce Apoptosis by Enhancing the MEK-ERK Pathway in a Reactive Oxygen Species-Independent Manner in BCR-ABL-Positive Chronic Myeloid Leukemia-Derived K562 Cells" International Journal of Molecular Sciences 23, no. 2: 749. https://doi.org/10.3390/ijms23020749