
AGC1 Deficiency: Pathology and Molecular and Cellular Mechanisms of the Disease

 , and
, and
Abstract
:1. Introduction
2. AGC1/Slc25a12/Aralar-MAS Pathway in Brain Metabolism
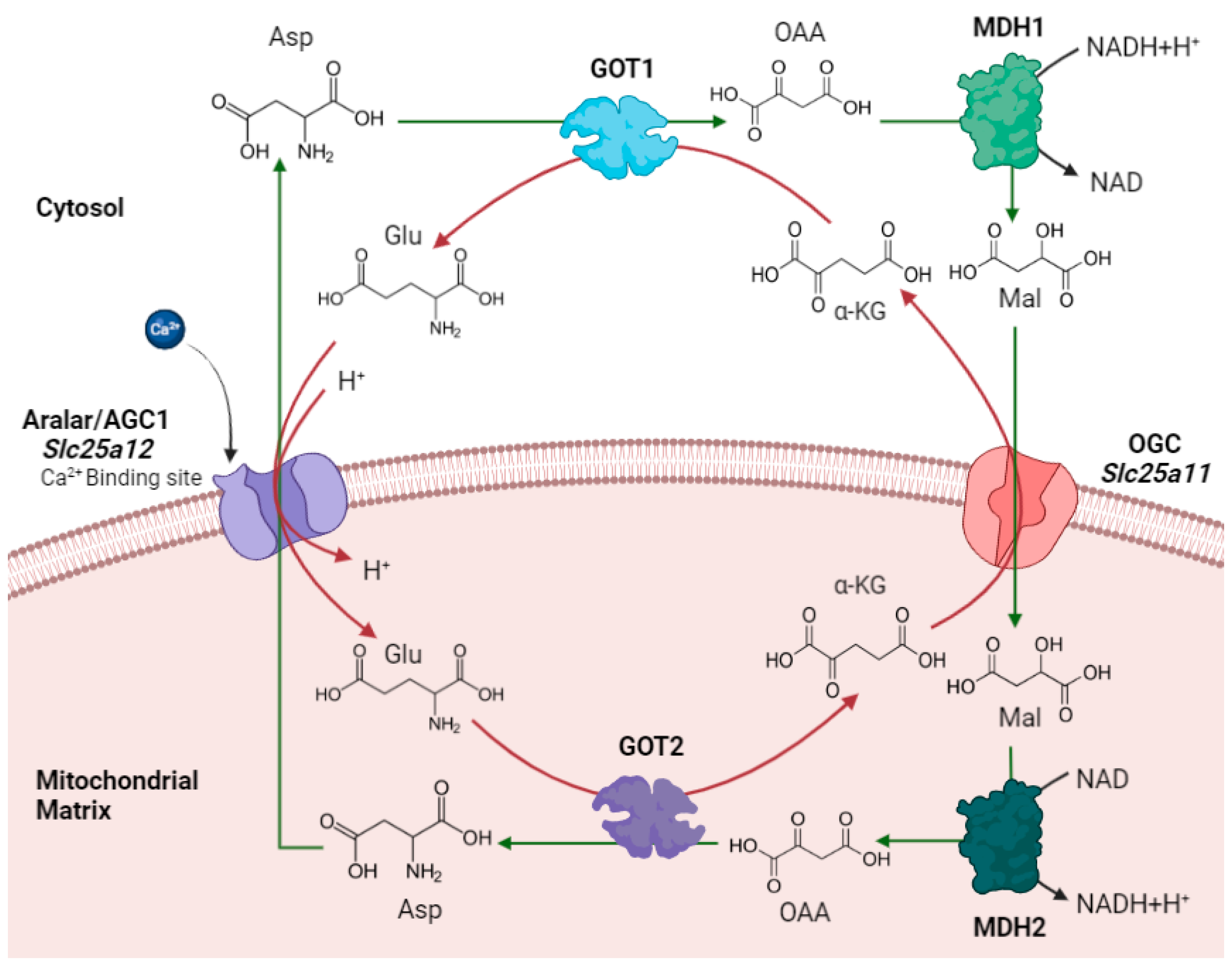
2.1. MAS as the Main NADH Shuttle in the Brain
2.2. AGC1/ARALAR/SLC25a12, the Regulatory Component of MAS in Brain: Structure, and Cellular Expression
2.3. Use of Experimental Models of AGC1 Deficiency Revealed New Functions of ARALAR-MAS
2.3.1. Experimental Models of Mice with AGC1 Deficiency (Aralar-KO Mice)
2.3.2. New Aralar Functions Discovered in Aralar-KO Mice
3. AGC1/Aralar- and MAS-Deficiency in Humans
3.1. Mutations in AGC1 Related to Human AGC1 Deficiency
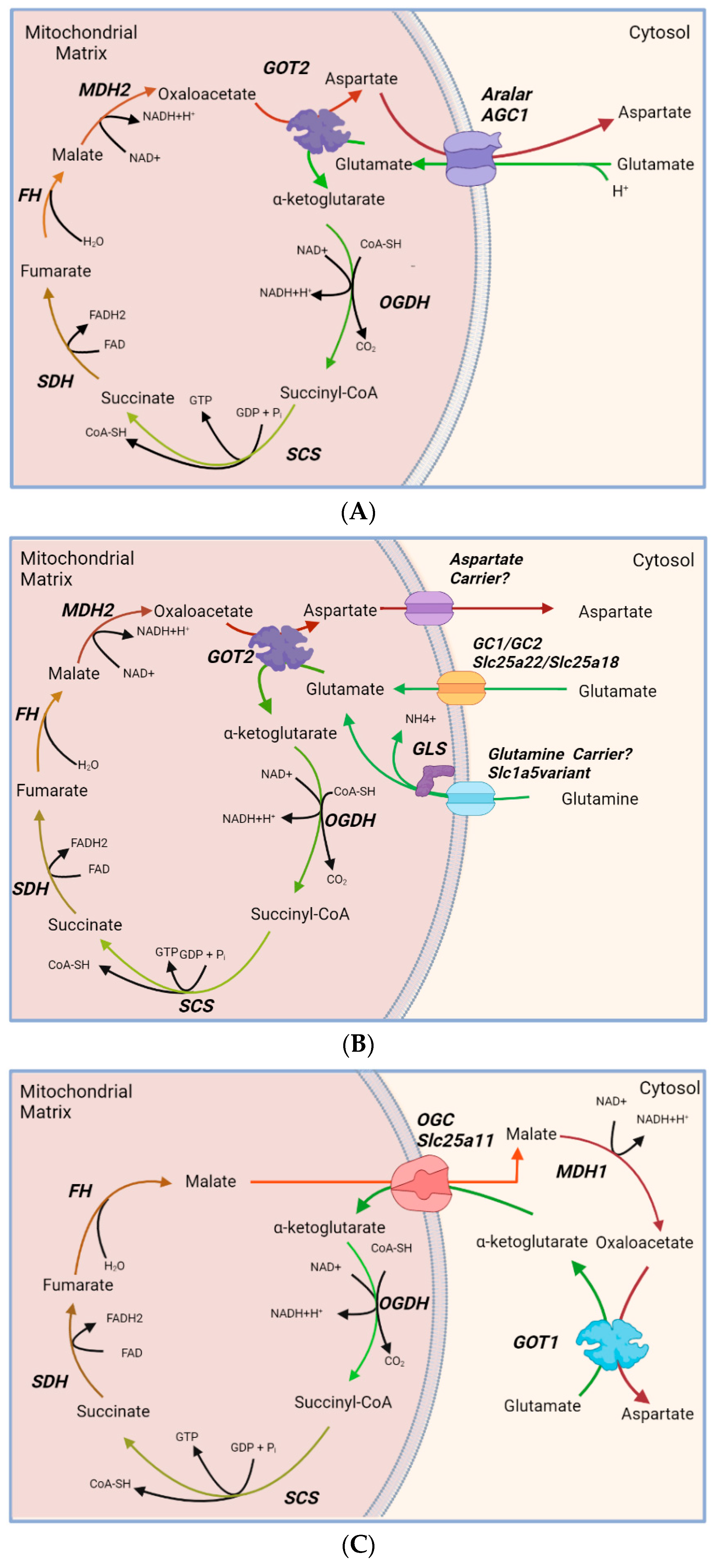
3.2. Other Inborn MAS Deficiencies in Humans
4. AGC1 Deficiency: Pathophysiology
4.1. Epilepsy
4.2. Postnatal Hypomyelination
4.3. Failure in Astroglial Glu and Gln Synthesis
4.4. Deficits in the Nigrostriatal DAergic System
5. Treatment of Patients and Murine Models of AGC1/Aralar Deficiency
5.1. Treatment of AGC1-Deficient Patients with Ketogenic Diet (KD)
5.2. Treatment of Agc1/Aralar- KO Mice with KD or with the KB, β-Hydroxybutyrate (βOHB)
6. Conclusions
Funding
Conflicts of Interest
References
- Dawson, A.G. Oxidation of cytosolic NADH formed during aerobic metabolism in mammalian cells. Trends Biochem. Sci. 1979, 4, 171–176. [Google Scholar] [CrossRef]
- Del Arco, A.; Satrústegui, J. Molecular cloning of Aralar, a new member of the mitochondrial carrier superfamily that binds calcium and is present in human muscle and brain. J. Biol. Chem. 1998, 273, 23327–23334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmieri, L.; Pardo, B.; Lasorsa, F.M.; del Arco, A.; Kobayashi, K.; Iijima, M.; Runswick, M.J.; Walker, J.E.; Saheki, T.; Satrústegui, J.; et al. Citrin and aralar1 are Ca2+-stimulated aspartate/glutamate transporters in mitochondria. EMBO J. 2001, 20, 5060–5069. [Google Scholar] [CrossRef]
- Jalil, M.A.; Begum, L.; Contreras, L.; Pardo, B.; Iijima, M.; Li, M.X.; Ramos, M.; Mármol, P.; Horiuchi, M.; Shimotsu, K.; et al. Reduced N-Acetylaspartate levels in mice lacking aralar, a brain- and muscle-type mitochondrial aspartate-glutamate carrier. J. Biol. Chem. 2005, 280, 31333–31339. [Google Scholar] [CrossRef] [Green Version]
- Del Arco, A.; Morcillo, J.; Martínez-Morales, J.R.; Galián, C.; Martos, V.; Bovolenta, P.; Satrústegui, J. Expression of the aspartate/glutamate mitochondrial carriers aralar1 and citrin development and in adult rat tissues. Eur. J. Biochem. 2002, 269, 3313–3320. [Google Scholar] [CrossRef]
- Ramos, M.; Del Arco, A.; Pardo, B.; Martínez-Serrano, A.; Martínez-Morales, J.R.; Kobayashi, K.; Yasuda, T.; Bogónez, E.; Bovolenta, P.; Saheki, T.; et al. Developmental changes in the Ca2+-regulated mitochondrial aspartate-glutamate carrier aralar1 in brain and prominent expression in the spinal cord. Dev. Brain Res. 2003, 143, 33–46. [Google Scholar] [CrossRef]
- Pardo, B.; Rodrigues, T.B.; Contreras, L.; Garzón, M.; Llorente-Folch, I.; Kobayashi, K.; Saheki, T.; Cerdán, S.; Satrústegui, J. Brain glutamine synthesis requires neuronal-born aspartate as amino donor for glial glutamate formation. J. Cereb. Blood Flow Metab. 2011, 31, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, K.; Sinasac, D.S.; Iijima, M.; Boright, A.P.; Begum, L.; Lee, J.R.; Yasuda, T.; Ikeda, S.; Hirano, R.; Terazono, H.; et al. The gene mutated in adult-onset type II citrullinaemia encodes a putative mitochondrial carrier protein. Nat. Genet. 1999, 22, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Del Arco, A.; Agudo, M.; Satrústegui, J. Characterization of a second member of the subfamily of calcium-binding mitochondrial carriers expressed in human non-excitable tissues. Biochem. J. 2000, 345, 725–732. [Google Scholar] [CrossRef]
- Wibom, R.; Lasorsa, F.M.; Töhönen, V.; Barbaro, M.; Sterky, F.H.; Kucinski, T.; Naess, K.; Jonsson, M.; Pierri, C.L.; Palmieri, F.; et al. AGC1 deficiency associated with global cerebral hypomyelination. N. Engl. J. Med. 2009, 361, 489–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falk, M.J.; Li, D.; Gai, X.; McCormick, E.; Place, E.; Lasorsa, F.M.; Otieno, F.G.; Hou, C.; Kim, C.E.; Abdel-Magid, N. AGC1 Deficiency Causes Infantile Epilepsy, Abnormal Myelination, and Reduced N-Acetylaspartate. JIMD Reports 2014, 4, 77–85. [Google Scholar]
- Parnes, M.; Robak, L.; Shulman, J.M.; Stocco, A.; Jankovic, J. Dystonia-spasticity in a patient with a novel SLC25A12 mutation. Mov. Disord. 2015, 30 (Suppl. 1), 1234. [Google Scholar]
- Pronicka, E.; Piekutowska-Abramczuk, D.; Ciara, E.; Trubicka, J.; Rokicki, D.; Karkucinska-Wieckowska, A.; Pajdowska, M.; Jurkiewicz, E.; Halat, P.; Kosinska, J.; et al. New perspective in diagnostics of mitochondrial disorders: Two years´experience with whole-eexome sequencing at a national paediatric centre. J. Transl. Med. 2016, 14, 174. [Google Scholar] [CrossRef] [Green Version]
- Kavanaugh, B.C.; Warren, E.B.; Baytas, O.; Schmidt, M.; Merck, D.; Buch, K.; Liu, J.S.; Phornphutkul, C.; Caruso, P.; Morrow, E.M. Longitudinal MRI findings in patient with SLC25A12 pathogenic variants inform disease progression and classification. Am. J. Med. Genet. 2019, 179, 2284–2291. [Google Scholar] [CrossRef] [PubMed]
- Nashabat, M.; Al Qahtani, X.S.; Almakdob, S.; Altwaijri, W.; Ba-Armah, D.M.; Hundallah, K.; Al Hashem, A.; Al Tala, S.; Maddirevula, S.; Alkuraya, F.S.; et al. The landscape of early infantile epileptic encephalopathy in a consanguineous population. Seizure Europ. J. Epilepsy 2019, 69, 154–172. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, B.; Sen, K.; Kaur, S.; Pappas, K. Expanding Phenotypic Spectrum of Cerebral Aspartate-Glutamate Carrier Isoform 1 (AGC1) Deficiency. Neuropediatrics 2020, 51, 160–163. [Google Scholar] [CrossRef]
- Saleh, M.; Helmi, M.; Yacop, B. A novel nonsense gene variant responsible for early infantile epileptic encephalopathy type 39: Case report. Pak. J. Biol. Sci. 2020, 23, 973–976. [Google Scholar] [CrossRef]
- Kose, M.; Isik, E.; Aykut, A.; Durmaz, A.; Kose, E.; Ersoy, M.; Diniz, G.; Adebali, O.; Ünalp, A.; Yilmaz, Ü.; et al. The utility of next-generation sequencing technologies in diagnosis of Mendelian mitochondrial diseases and refelctions on clinical spectrum. J. Pediatr. Endocrinol. Metab. 2021, 34, 417–430. [Google Scholar] [CrossRef]
- Gómez-Galán, M.; Makarova, J.; Llorente-Folch, I.; Saheki, T.; Pardo, B.; Satrústegui, J.; Herreras, O. Altered postnatal development of cortico-hippocampal neuronal electric activity in mice deficient for the mitochondrial aspartate-glutamate transporter. J. Cereb. Blood Flow Metab. 2012, 32, 306–317. [Google Scholar] [CrossRef] [Green Version]
- Llorente-Folch, I.; Sahún, I.; Contreras, L.; Casarejos, M.J.; Grau, J.M.; Saheki, T.; Mena, M.A.; Satrústegui, J.; Dierssen, M.; Pardo, B. AGC1-malate aspartate shuttle activity is critical for dopamine handling in the nigrostriatal pathway. J. Neurochem. 2013, 124, 347–362. [Google Scholar] [CrossRef]
- Ramos, M.; Pardo, B.; Llorente-Folch, I.; Saheki, T.; del Arco, A.; Satrústegui, J. Deficiency of the mitochondrial transporter of aspartate/glutamate aralar/AGC1 causes hypomyelination and neuronal defects unrelated to myelin deficits in mouse brain. J. Neurosci. Res. 2011, 89, 2008–2017. [Google Scholar] [CrossRef] [PubMed]
- Berkich, D.A.; Ola, M.S.; Cole, J.; Sweatt, A.J.; Hutson, S.M.; LaNoue, K.F. Mitochondrial transport proteins of the brain. J. Neurosci. Res. 2007, 85, 3367–3377. [Google Scholar] [CrossRef]
- Xu, J.; Mashiomo, T.; Südhof, T.C. Synaptotagmin-1, -2, and -9: Ca(2+) sensors for fast release that specify distinct presynaptic properties in subsets of neurons. Neuron. 2007, 24, 567–581. [Google Scholar] [CrossRef] [Green Version]
- Satrústegui, J.; Pardo, B.; del Arco, A. Mitochondrial transporters as novel targets for intracellular calcium signaling. Physiol. Rev. 2007, 87, 29–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardo, B.; Contreras, L.; Serrano, A.; Ramos, M.; Kobayashi, K.; Iijima, M.; Saheki, T.; Satrústegui, J. Essential role of aralar in the transduction of small Ca2+ signals to neuronal mitochondria. J. Biol. Chem. 2006, 281, 1039–1047. [Google Scholar] [CrossRef] [Green Version]
- Llorente-Folch, I.; Rueda, C.B.; Pérez-Liébana, I.; Satrústegui, J.; Pardo, B. L-Lactate-Mediated Neuroprotection against Glutamate-Induced Excitotoxicity Requires ARALAR/AGC1. J. Neurosci. 2016, 36, 4443–4456. [Google Scholar] [CrossRef] [Green Version]
- Llorente-Folch, I.; Rueda, C.B.; Amigo, I.; del Arco, A.; Saheki, T.; Pardo, B.; Satrústegui, J. Calcium-regulation of mitochondrial respiration maintains ATP homeostasis and requires ARALAR/AGC1-malate aspartate shuttle in intact cortical neurons. J. Neurosci. 2013, 33, 13957–13971. [Google Scholar] [CrossRef] [PubMed]
- Dahlin, M.; Martin, D.A.; Hedlund, Z.; Jonsson, M.; Von Döbeln, U.; Wedell, A. The ketogenic diet compensates for AGC1 deficiency and improves myelination. Epilepsia 2015, 56, e176–e181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez-Liébana, I.; Casarejos, M.J.; Alcaide, A.; Herrada-Soler, A.; Llorente-Folch, I.; Contreras, L.; Satrústegui, J.; Pardo, B. βOHB protective pathways in Aralar-KO neurons and brain: An alternative to ketogenic diet. J. Neurosci. 2020, 40, 9293–9305. [Google Scholar] [CrossRef] [PubMed]
- Luongo, T.S.; Eller, J.M.; Lu, M.-J.; Niere, M.; Raith, F.; Perry, C.; Bornstein, M.R.; Oliphint, P.; Wang, L.; McReynolds, M.R.; et al. SLC25A51 is a mammalian mitochondrial NAD+ transporter. Nature 2020, 588, 174–179. [Google Scholar] [CrossRef] [PubMed]
- Lehninger, A.L. Phosphorylation coupled to oxidation of dihydrophosphopyridine nucleotide. J. Biol. Chem. 1951, 190, 345–359. [Google Scholar] [CrossRef]
- Purvis, J.L.; Lowenstein, J.M. The relation between intra- and extramitochondrial pyridine nucleotides. J. Biol. Chem. 1961, 236, 2794–2803. [Google Scholar] [CrossRef]
- Borst, P. Interrelations between Cytoplasmic and Mitochondrial Diphosphopyridine Nucleotide in Ehrlich Ascites Tumour Cells; Pergamon Press: Oxford, UK, 1963; pp. 233–247. [Google Scholar]
- Liu, S.; Song, F.; Guodong, W.; Cao, Y.; Li, L.; Li, X.; Yang, J.; Li, N.; Shan, Y.; Cao, Y.; et al. Glycerol-3-phosphate biosynthesis regenerates cytosolic NAD + to alleviate mitochondrial disease. Cell Metab. 2021, 33, 1974–1987. [Google Scholar] [CrossRef]
- Eto, K.; Tsubamoto, Y.; Terauchi, Y.; Sugiyama, T.; Kishimoto, T.; Takahashi, N.; Yamauchi, N.; Kubota, N.; Murayama, S.; Aizawa, T.; et al. Role of NADH shuttle system in glucose-induced activation of mitochondrial metabolism and insulin secretion. Science 1999, 283, 981–985. [Google Scholar] [CrossRef] [PubMed]
- Pardo, B.; Contreras, L. Redox shuttles in the brain. In Advances in Neurobiology. Neuronal Metabolism In Vivo; Choi, I.-Y., Gruetter, R., Eds.; Springer: Boston, MA, USA, 2012; Volume 4, pp. 841–883. ISBN 978-1-4614-1787-3. [Google Scholar]
- Cahoy, J.D.; Emery, B.; Kaushal, A.; Foo, L.C.; Zamanian, J.L.; Christopherson, K.S.; Xing, Y.; Lubischer, J.L.; Krieg, P.A.; Krupenko, S.A.; et al. A transcriptome database for astrocytes, neurons, and oligodendrocytes: A new resource for understanding brain development and function. J. Neurosci. 2008, 28, 264–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juaristi, I.; García-Martín, M.L.; Rodrigues, T.B.; Satrústegui, J.; Llorente-Folch, I.; Pardo, B. ARALAR/AGC1 deficiency, a neurodevelopmental disorder with severe impairment of neuronal mitochondrial respiration, does not produce a primary increase in brain lactate. J. Neurochem. 2017, 142, 132–139. [Google Scholar] [CrossRef]
- LaNoue, K.F.; Tischler, M.E. Electrogenic characteristics of the mitochondrial glutamate-aspartate antiporter. J. Biol. Chem. 1974, 249, 7522–7528. [Google Scholar] [CrossRef]
- Sanz, R.; del Arco, A.; Ayuso, C.; Ramos, C.; Satrústegui, J. Assignement of the calcium-binding mitochondrial carrier Aralar1 gene (SLC25A12) to human chromosome band 2q31 by in situ hybridization. Cytogenet Cell Genet 2000, 89, 143–144. [Google Scholar] [CrossRef] [PubMed]
- Sinasac, D.S.; Crackower, M.A.; Lee, J.R.; Kobayashi, K.; Saheki, T.; Scherer, S.W.; Tsui, L.C. Genomic structure of the adult-onset type II citrullinemia gene, SLC25A13, and cloning and expression of its mouse homologue. Genomics 1999, 62, 289–292. [Google Scholar] [CrossRef] [PubMed]
- LaNoue, K.F.; Schoolwerth, A.C. Metabolite transport in mitochondria. Annu. Rev. Biochem. 1979, 48, 871–922. [Google Scholar] [CrossRef]
- Saheki, T.; Kobayashi, K. Mitochondrial aspartate glutamate carrier (citrin) deficiency as the cause of adult-onset type II citrullinemia (CTLN2) and idiopathic neonatal hepatitis (NICCD). J. Hum. Genet. 2002, 47, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.Z.; Guo, L.; Yang, Y.L.; Han, L.S.; Kobayashi, K.; Saheki, T. Failure to thrive and dyslipidemia caused by citrin deficiency: A novel clinical phenotype. Chin. J. Contemp. Pediatrics 2009, 11, 328–332. [Google Scholar]
- Saheki, T.; Moriyama, M.; Funahashi, A.; Kuroda, E. AGC2 (Citrin) deficiency- From Recognition of the Disease till Construction of Therapeutic Procedures. Biomolecules 2020, 10, 1100. [Google Scholar] [CrossRef] [PubMed]
- Del Arco, A.; Satrústegui, J. Identification of a novel human subfamioy of mitochondrial carriers with calcium-binding domains. J. Biol. Chem. 2004, 279, 24701–24713. [Google Scholar] [CrossRef] [Green Version]
- Fiermonte, G.; De Leonardis, F.; Todisco, S.; Palmieri, L.; Lasorsa, F.M.; Palmieri, F. Identification of the mitochondrial ATP-Mg/Pi transporter: Bacterial expression, reconstitution, functional characterization, and tissue distribution. J. Biol. Chem. 2004, 279, 30722–30730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Contreras, L.; Satrústegui, J. Calcium signaling in brain mitochondria: Interplay of malate aspartate NADH shuttle and calcium uniporter/mitochondrial dehydrogenase pathways. J. Biol. Chem. 2009, 284, 7091–7099. [Google Scholar] [CrossRef] [Green Version]
- Harborne, S.P.D.; Ruprecht, J.J.; Kunji, E.R.S. Calcium-induced conformational changes in the regulatory domain of the human mitochondrial ATP-Mg/Pi carrier. Biochim. Biophys. Acta 2015, 1847, 1245–1253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pebay-Peyroula, E.; Dahout-Gonzalez, C.; Kahn, R.; Trézéguet, V.; Lauquin, G.J.; Brandolin, G. Structure of mitochondrial ADP/ATP carrier in complex with carboxyatractyloside. Nature 2003, 426, 39–44. [Google Scholar] [CrossRef]
- Ruprecht, J.J.; King, M.S.; Zögg, T.; Aleksandrova, A.A.; Pardon, E.; Crichton, P.G.; Steyaert, J.; Kunji, E.R.S. The Molecular Mechanism of Transport by the Mitochondrial ADP/ATP Carrier. Cell 2019, 176, 435–447. [Google Scholar] [CrossRef] [Green Version]
- Ruprecht, J.J.; Kunji, E.R.S. The SLC25 mitochondrial carrier family: Structure and mechanism. Trends Biochem. Sci. 2020, 45, 244–258. [Google Scholar] [CrossRef] [Green Version]
- Ruprecht, J.J.; Kunji, E.R.S. Structural Mechanism of Transport of Mitochondrial Carriers. Annu. Rev. Biochem. 2021, 90, 535–558. [Google Scholar] [CrossRef]
- Kunji, E.R.S.; Robinson, A.J. The conserved substrate binding site of mitochondrial carriers. Biochim. Biophys. Acta 2006, 1757, 1237–1248. [Google Scholar] [CrossRef] [Green Version]
- Thangaratnarajah, C.; Ruprecht, J.J.; Kunji, E.R.S. Calcium-induced conformational changes of the regulatory domain of human mitochondrial aspartate/glutamate carriers. Nature Commun. 2014, 5, 5491. [Google Scholar] [CrossRef] [Green Version]
- Contreras, L.; Urbieta, A.; Kobayashi, K.; Saheki, T.; Satrústegui, J. Low Levels of Citrin (SLC25A13) Expression in Adult Mouse Brain Restricted to Neuronal Clusters. J. Neurosci. Res. 2010, 88, 1009–1016. [Google Scholar] [CrossRef] [PubMed]
- Lovatt, D.; Sonnewald, U.; Waagepetersen, H.S.; Schousboe, A.; He, W.; Lin, J.H.; Han, X.; Takano, T.; Wang, S.; Sim, F.J.; et al. The transcriptome and metabolic gene signature of protoplasmic astrocytes in the adult murine cortex. J. Neurosci. 2007, 27, 12255–12266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Hertz, L.; Peng, L. Aralar mRNA and protein levels in neurons and astrocytes freshly isolated from young and adult mouse brain and in maturing cultured astrocytes. Neurochem. Int. 2012, 61, 1325–1332. [Google Scholar] [CrossRef]
- The Tabula Muris Consortium. Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature 2018, 562, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, T.; Ramoz, N.; Barreto, M.; Gazdoiu, M.; Takahashi, N.; Gerner, M.; Dorr, N.; Gama Sosa, M.A.; De Gasperi, R.; Perez, G.; et al. Slc25a12 disruption alters myelination and neurofilaments: A model for hypomyelination syndrome and childhood neurodevelopment disorders. Biol. Psychiatry 2010, 67, 887–894. [Google Scholar] [CrossRef] [PubMed]
- Petralla, S.; Peña-Altamira, L.E.; Poeta, E.; Massenzio, F.; Virgili, M.; Barile, S.N.; Sbano, L.; Profilo, E.; Corricelli, M.; Danese, A.; et al. Deficiency of mitochondrial Aspartate-glutamate carrier 1 leads to oligodendrocyte precursor cell proliferation defects both in vitro and in vivo. Int. J. Mol. Sci. 2019, 20, 4486. [Google Scholar] [CrossRef] [Green Version]
- Llorente-Folch, I.; Rueda, C.B.; Pardo, B.; Szabadkai, G.; Duchen, M.R.; Satrustegui, J. The regulation of neuronal mitochondrial metabolism by calcium. J. Physiol. 2015, 593, 3447–3462. [Google Scholar] [CrossRef] [Green Version]
- Rueda, C.B.; Llorente-Folch, I.; Amigo, I.; Contreras, L.; González-Sánchez, P.; Martínez-Valero, P.; Juaristi, I.; Pardo, B.; del Arco, A.; Satrústegui, J. Ca(2+) regulation of mitochondrial function in neurons. Biochim. Biophys. Acta 2014, 1837, 1617–1624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rueda, C.B.; Llorente-Folch, I.; Traba, J.; Amigo, I.; González-Sánchez, P.; Contreras, L.; Juaristi, I.; Martínez-Valero, P.; Pardo, B.; Del Arco, A.; et al. Glutamate excitotoxicity and Ca2+-regulation of respiration: Role of the Ca2+ activated mitochondrial transporters (CaMCs). Biochim. Biophys. Acta 2016, 1857, 1158–1166. [Google Scholar] [CrossRef]
- Babetto, E.; Wong, K.M.; Beirowski, B. A glycolytic shift in Schwann cells supports injured axons. Nat. Neurosci. 2020, 23, 1215–1228. [Google Scholar] [CrossRef] [PubMed]
- Solano Fonseca, R.; Metang, P.; Egge, N.; Liu, Y.; Zuurbier, K.R.; Sivaprakasam, K.; Shirazi, S.; Chuah, A.; Arneaud, S.L.; Konopka, G.; et al. Glycolytic preconditioning in astrocytes mitigates trauma-induced neurodegeneration. Elife 2021, 10, e69438. [Google Scholar] [CrossRef]
- Lhomme, T.; Clasadonte, J.; Imbernon, M.; Fernandois, D.; Sauve, F.; Caron, E.; da Silva Lima, N.; Heras, V.; Martinez-Corral, I.; Mueller-Fielitz, H.; et al. Tanycytic networks mediate energy balance by feeding lactate to glucose-insensitive POMC neurons. J. Clin. Investig. 2021, 131, e140521. [Google Scholar] [CrossRef]
- Chen, W.W.; Freinkman, E.; Wang, T.; Birsoy, K.; Sabatini, D.M. Absolute quantification of matrix metabolites reveals the dynamics of mitochondrial metabolism. Cell 2016, 166, 1324–1337. [Google Scholar] [CrossRef]
- Urenjak, J.; Williams, S.R.; Gadian, D.G.; Noble, M. Specific expression of N-acetylaspartate in neurons, oligodendrocyte-type-2 astrocyte progenitors, and immature oligodendrocytes in vitro. J. Neurochem. 1992, 59, 55–61. [Google Scholar] [CrossRef] [PubMed]
- McNair, L.F.; Andersen, J.V.; Aldana, B.I.; Hohnholt, M.C.; Nissen, J.D.; Sun, Y.; Fischer, K.D.; Sonnewald, U.; Nyberg, N.; Webster, S.C.; et al. Deletion of Neuronal GLT-1 in Mice Reveals Its Role in Synaptic Glutamate Homeostasis and Mitochondrial Function. J. Neurosci. 2019, 39, 4847–4863. [Google Scholar] [CrossRef] [Green Version]
- McNair, L.F.; Andersen, J.V.; Nissen, J.D.; Sun, Y.; Fischer, K.D.; Hodgson, N.W.; Du, M.; Aoki, C.J.; Waagepetersen, H.S.; Rosenberg, P.A.; et al. Conditional Knockout of GLT-1 in Neurons Leads to Alterations in Aspartate Homeostasis and Synaptic Mitochondrial Metabolism in Striatum and Hippocampus. Neurochem. Res. 2020, 45, 1420–1437. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.V.; Markussen, K.A.; Jakobsen, E.; Schousboe, A.; Waagepetersen, H.S.; Rosenberg, P.A.; Aldana, B.I. Glutamate metabolism and recycling at the excitatory synapse in health and neurodegeneration. Neuropharmacology 2021, 196, 108719. [Google Scholar] [CrossRef]
- Ralphe, J.C.; Segar, J.L.; Schutte, B.C.; Scholz, T.D. Localization and function of the brain excitatory amino acid transporter type 1 in cardiac mitochondria. J. Mol. Cell. Cardiol. 2004, 37, 33–41. [Google Scholar] [CrossRef]
- Piccirillo, S.; Magi, S.; Castaldo, P.; Preziuso, A.; Lariccia, V.; Amoroso, S. NCX and EAAT transporters in ischemia: At the crossroad between glutamate metabolism and cell survival. Cell Calcium 2020, 86, 102160. [Google Scholar] [CrossRef]
- Contreras, L.; Gomez-Puertas, P.; Iijima, M.; Kobayashi, K.; Saheki, T.; Satrústegui, J. Ca2+ activation kinetics of the two aspartate-glutamate mitochondrial carriers, aralar and citrin: Role in the heart malate-aspartate NADH shuttle. J. Biol. Chem. 2007, 282, 7098–7106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailis, W.; Shyer, J.A.; Zhao, J.; Canaveras, J.C.G.; Al Khazal, F.J.; Qu, R.; Steach, H.R.; Bielecki, P.; Khan, O.; Jackson, R.; et al. Distinct modes of mitochondrial metabolism uncouple T cell differentiation and function. Nature 2019, 571, 403–407. [Google Scholar] [CrossRef]
- Juaristi, I.; Llorente-Folch, I.; Satrústegui, J.; del Arco, A. Extracellular ATP and glutamate drive pyruvate production and energy demand to regulate mitochondrial respiration in astrocytes. Glia 2019, 67, 759–774. [Google Scholar] [CrossRef] [PubMed]
- Juaristi, I.; Contreras, L.; González-Sánchez, P.; Pérez-Liébana, I.; González-Moreno, L.; Pardo, B.; Del Arco, A.; Satrústegui, J. The Response to Stimulation in Neurons and Astrocytes. Neurochem. Res. 2019, 44, 2385–2391. [Google Scholar] [CrossRef] [PubMed]
- Yu, A.C.; Drejer, J.; Hertz, L.; Schousboe, A. Pyruvate carboxylase activity in primary cultures of astrocytes and neurons. J. Neurochem. 1983, 41, 1484–1487. [Google Scholar] [CrossRef] [PubMed]
- Voss, C.M.; Arildsen, L.; Nissen, J.D.; Waagepetersen, H.S.; Schousboe, A.; Maechler, P.; Ott, P.; Vilstrup, H.; Walls, A.B. Glutamate Dehydrogenase Is Important for Ammonia Fixation and Amino Acid Homeostasis in Brain During Hyperammonemia. Front. Neurosci. 2021, 15, 646291. [Google Scholar] [CrossRef]
- Skytt, D.M.; Klawonn, A.M.; Stridh, M.H.; Pajęcka, K.; Patruss, Y.; Quintana-Cabrera, R.; Bolaños, J.P.; Schousboe, A.; Waagepetersen, H.S. siRNA knock down of glutamate dehydrogenase in astrocytes affects glutamate metabolism leading to extensive accumulation of the neuroactive amino acids glutamate and aspartate. Neurochem. Int. 2012, 61, 490–497. [Google Scholar] [CrossRef] [PubMed]
- Hertz, L.; Rothman, D.L. Glutamine-Glutamate Cycle Flux Is Similar in Cultured Astrocytes and Brain and Both Glutamate Production and Oxidation Are Mainly Catalyzed by Aspartate Aminotransferase. Biology 2017, 6, 17. [Google Scholar] [CrossRef] [Green Version]
- Lindsay, K.J.; Du, J.; Sloat, S.R.; Contreras, L.; Linton, J.D.; Turner, S.J.; Sadilek, M.; Satrústegui, J.; Hurley, J.B. Pyruvate kinase and aspartate-glutamate carrier distributions reveal key metabolic links between neurons and glia in retina. Proc. Natl. Acad. Sci. USA 2014, 111, 15579–15584. [Google Scholar] [CrossRef] [Green Version]
- Contreras, L. Role of AGC1/aralar in the metabolic synergies between neuron and glia. Neurochem. Int. 2015, 88, 38–46. [Google Scholar] [CrossRef] [Green Version]
- McKenna, M.C.; Tildon, J.T.; Stevenson, J.H.; Boatright, R.; Huang, S. Regulation of energy metabolism in synaptic terminals and cultured rat brain astrocytes: Differences revealed using aminooxyacetate. Dev. Neurosci. 1993, 15, 320–329. [Google Scholar] [CrossRef]
- Nissen, J.D.; Pajecka, K.; Stridh, M.H.; Skytt, D.M.; Waagepetersen, H.S. Dysfunctional TCA-cycle metabolism in glutamate dehydrogenase deficient astrocytes. Glia 2015, 63, 2313–2326. [Google Scholar] [CrossRef]
- Hohnholt, M.C.; Andersen, V.H.; Andersen, J.V.; Christensen, S.K.; Karaca, M.; Maechler, P.; Waagepetersen, H.S. Glutamate dehydrogenase is essential to sustain neuronal oxidative energy metabolism during stimulation. J. Cereb. Blood Flow Metab. 2018, 38, 1754–1768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yudkoff, M.; Nelson, D.; Daikhin, Y.; Erecińska, M. Tricarboxylic acid cycle in rat brain synaptosomes. Fluxes and interactions with aspartate aminotransferase and malate/aspartate shuttle. J. Biol. Chem. 1994, 269, 27414–27420. [Google Scholar] [CrossRef]
- Sutherland, G.R.; Tyson, R.L.; Auer, R.N. Truncation of the Krebs cycle during hypoglycemic coma. Med. Chem. 2008, 4, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Cleghorn, W.M.; Contreras, L.; Lindsay, K.; Rountree, A.M.; Chertov, A.O.; Turner, S.J.; Sahaboglu, A.; Linton, J.; Sadilek, M.; et al. Inhibition of mitochondrial pyruvate transport by zaprinast causes massive accumulation of aspartate at the expense of glutamate in the retina. J. Biol. Chem. 2013, 288, 36129–36140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olszewski, K.; Barsotti, A.; Feng, X.-J.; Momcilovic, M.; Liu, K.G.; Kim, J.-I.; Morris, K.; Lamarque, C.; Gaffney, J.; Yu, X.; et al. Inhibition of glucose transport synergizes with chemical or genetic disruption of mitochondrial metabolism and suppresses TCA cycle-deficient tumors. Cell Chem. Biol. 2022, 29, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Robinson, A.J.; Kunji, E.R.S. Mitochondrial carriers in the cytoplasmic state have a common substrate binding site. Proc. Natl. Acad. Sci. USA 2006, 103, 2617–2622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pierri, C.L.; Palmieri, F.; De Grassi, A. Single-nucleotide evolution quantifies the importance of each site along the structure of mitochondrial carriers. Cell. Mol. Life Sci. 2014, 71, 349–364. [Google Scholar] [CrossRef]
- Van der Knaap, M.S.; Bugiani, M. Leukodystrophies: A proposed classification system based on pathological changes and pathogenetic mechanisms. Acta Neuropathol. 2017, 134, 351–382. [Google Scholar] [CrossRef] [Green Version]
- Almeida, A.; Bolaños, J.P.; Moncada, S. E3 ubiquitin ligase APC/C-Cdh1 accounts for the Warburg effect by linking glycolysis to cell proliferation. Proc. Natl. Acad. Sci. USA 2010, 107, 738–741. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Chen, K.; Sloan, S.A.; Bennett, M.L.; Scholze, A.R.; O’Keeffe, S.; Phatnani, H.P.; Guarnieri, P.; Caneda, C.; Ruderisch, N.; et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 2014, 34, 11929–11947. [Google Scholar] [CrossRef]
- Ait-El-Mkadem, S.; Dayem-Quere, M.; Gusic, M.; Chaussenot, A.; Bannwarth, S.; Francois, B.; Genin, E.C.; Fragaki, K.; Volker-Touw, C.L.M.; Vasnier, C.; et al. Mutations in MDH2, encoding a Krebs cycle enzyme, cause early-onset severe encephalopathy. Am. J. Hum. Genet. 2017, 100, 151–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broeks, M.H.; Shamseldin, H.E.; Alhashem, A.; Hashem, M.; Abdulwahab, F.; Alshedi, T.; Alobaid, I.; Zwartkruis, F.; Westland, D.; Fuchs, S.; et al. MDH1 deficiency is a metabolic disorder of the malate-aspartate shuttle associated with early onset severe encephalopathy. Hum. Genet. 2019, 138, 1247–1257. [Google Scholar] [CrossRef] [PubMed]
- Van Karnebeek, C.D.M.; Ramos, R.J.; Wen, X.-Y.; Tarailo-Graovac, M.; Gleeson, J.G.; Skrypnyk, C.; Brand-Arzamendi, K.; Rousseau, J.; Campeau, P.M.; Wang, Y.; et al. Bi-allelic GOT2 mutations cause a treatable malate-aspartate shuttle-related encephalopathy. Am. J. Hum. Genet. 2019, 105, 1–15. [Google Scholar] [CrossRef] [Green Version]
- McKenna, M.C.; Waagepetersen, H.S.; Schousboe, A.; Sonnewald, U. Neuronal and astrocytic shuttle mechanisms for cytosolic-mitochondrial transfer of reducing equivalents: Current evidence and pharmacological tools. Biochem. Pharmacol. 2006, 71, 399–407. [Google Scholar] [CrossRef] [PubMed]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Pascual, J.M.; Van Heertum, R.L.; Wang, D.; Engelstad, K.; De Vivo, D.C. Imaging the metabolic footprint of Glut1 deficiency on the brain. Ann Neurol. 2002, 52, 458–464. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Prasad, A.N. Inborn Errors of Metabolism and Epilepsy: Current Understanding, Diagnosis, and Treatment Approaches. Int. J. Mol. Sci. 2017, 18, 1384. [Google Scholar] [CrossRef] [Green Version]
- De la Rossa, A.; Laporte, M.H.; Astori, S.; Marissal, T.; Montessuit, S.; Sheshadri, P.; Ramos-Fernández, E.; Mendez, P.; Khani, A.; Quairiaux, H.; et al. Paradoxical neuronal hyperexcitability in a mouse model of mitochondrial pyruvate 1 import deficiency. bioRxiv 2020. [Google Scholar] [CrossRef]
- Bliss, T.M.; Miranda, I.; Cheng, E.; Minami, M.; Pellerin, L.; Magistretti, P.; Sapolsky, R.M. Dual-gene, dual-cell type therapy against an ecitotoxic insult by bolstering neuroenergetics. J. Neurosci. 2004, 24, 6202–6208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blüml, S. In vivo quantitation of cerebral metabolite concentrations using natural abundance 13C MRS at 1.5 T. J. Magn. Reson. 1999, 136, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Nave, K.-A. Myelination and the trophic support of long axons. Nat. Rev. Neurosci. 2010, 11, 275–283. [Google Scholar] [CrossRef]
- Snaidero, J.; Simons, M. Myelination at a glance. J. Cell Sci. 2014, 127 Pt 14, 2999–3004. [Google Scholar] [CrossRef] [Green Version]
- Moffett, J.R.; Namboodiri, M.A.; Cangro, C.B.; Neale, J.H. Immunohistochemical localization of N-acetylaspartate in rat brain. Neuroreport 1991, 2, 131–134. [Google Scholar] [CrossRef] [Green Version]
- Moffett, J.R.; Namboodiri, M.A. Differential distribution of N-acetylaspartylglutamate and N-acetylaspartate immunoreactivities in rat forebrain. J. Neurocytol. 1995, 24, 409–433. [Google Scholar] [CrossRef]
- Bhakoo, K.K.; Pearce, D. In vitro expression of N-acetyl-aspartate by oligodendrocytes: Implications for proton magnetic resonance spectroscopy signal in vivo. J. Neurochem. 2000, 74, 254–262. [Google Scholar] [CrossRef]
- Maier, H.; Wang-Eckhardt, L.; Hartmann, D.; Gieselmann, V.; Eckhardt, M. N-acetylaspartate synthase deficiency corrects the myelin phenotype in a Canavan disease mouse model but does not affect survival time. J. Neurosci. 2015, 35, 14501–14516. [Google Scholar] [CrossRef] [Green Version]
- Guo, F.; Bannerman, P.; Mills, K.E.; Miers, L.; Xu, J.; Burns, T.; Li, S.; Freeman, E.; McDonough, J.A.; Pleasure, D. Ablating N-acetylaspartate prevents leukodystrophy in a Canavan disease model. Ann. Neurol. 2015, 77, 884–888. [Google Scholar] [CrossRef] [PubMed]
- Sohn, J.; Bannerman, P.; Guo, F.; Burns, T.; Miers, L.; Croteau, C.; Singhal, N.K.; McDonough, J.A.; Pleasure, D. Suppresing N-acetyl-L-aspartate synthesis prevents loss of neurons in a murine model of Canavan leukodystrophy. J. Neurosci. 2017, 37, 413–421. [Google Scholar] [CrossRef] [Green Version]
- Martin, E.; Capone, A.; Schneider, J.; Hennig, J.; Thiel, T. Absence of N-acetylaspartate in the human brain: Impact on neurospectroscopy? Ann. Neurol. 2001, 49, 518–521. [Google Scholar] [CrossRef]
- Fünfschilling, U.; Supplie, L.M.; Mahad, D.; Boretius, S.; Saab, A.S.; Edgar, J.; Brinkmann, B.G.; Kassmann, C.M.; Tzvetanova, I.D.; Möbius, W.; et al. Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature 2012, 485, 517–522. [Google Scholar] [CrossRef] [Green Version]
- Bak, L.K.; Schousboe, A.; Waagepetersen, H.S. The glutamate/GABA-glutamine cycle: Aspects of transport, neurotransmitter homeostasis and ammonia transfer. J. Neurochem. 2006, 98, 641–653. [Google Scholar] [CrossRef]
- Rothman, D.L.; Behar, K.L.; Hyder, F.; Shulman, R.G. In vivo NMR studies of the glutamate neurotransmitter flux and neuroenergetics: Implications for brain function. Annu. Rev. Physiol. 2003, 65, 401–427. [Google Scholar] [CrossRef] [PubMed]
- Hertz, L.; Kala, G. Energy metabolism in brain cells: Effects of elevated ammonia concentrations. Metab. Brain Dis. 2007, 22, 199–218. [Google Scholar] [CrossRef]
- Shank, R.P.; Bennett, G.S.; Freytag, S.O.; Campbell, G.L. Pyruvate carboxylase: An astrocyte-specific enzyme implicated in the replenishment of amino acid neurotransmitter pools. Brain Res. 1985, 329, 364–367. [Google Scholar] [CrossRef]
- Eid, T.; Williamson, A.; Lee, T.-S.W.; Petroff, O.A.; de Lanerolle, N.C. Glutamate and astrocytes--key players in human mesial temporal lobe epilepsy? Epilepsia 2008, 49, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Sepkuty, J.P.; Cohen, A.S.; Eccles, C.; Rafiq, A.; Behar, K.; Ganel, R.; Coulter, D.A.; Rothstein, J.D. A neuronal glutamate transporter contributes to neurotransmitter GABA synthesis and epilepsy. J. Neurosci. 2002, 22, 6372–6379. [Google Scholar] [CrossRef]
- Liang, S.L.; Carlson, G.C.; Coulter, D.A. Dynamic regulation of synaptic GABA release by the glutamate-glutamine cycle in hippocampal area CA1. J. Neurosci. 2006, 26, 8537–8548. [Google Scholar] [CrossRef]
- Wetherington, J.; Serrano, G.; Dingledine, R. Astrocytes in the epileptic brain. Neuron 2008, 58, 168–178. [Google Scholar] [CrossRef] [Green Version]
- Dawson, T.D.; Dawson, V.L. Molecular pathways of neurodegeneration in Parkinson´s disease. Science 2003, 302, 819–822. [Google Scholar] [CrossRef] [PubMed]
- Zampese, E.; Surmeier, D.J. Calcium, Bioenergetics, and Parkinson´s disease. Cells 2020, 9, 2045. [Google Scholar] [CrossRef] [PubMed]
- González-Rodríguez, P.; Zampese, E.; Stout, K.A.; Guzmán, J.N.; Ilijic, E.; Yang, B.; Tkatch, T.; Stavarache, M.A.; Wokosin, D.L.; Gao, L.; et al. Disruption of mitochondrial complex I induces progressive parkinsonism. Nature 2021, 599, 650–656. [Google Scholar] [CrossRef]
- Katsu-Jiménez, Y.; Alves, R.M.P.; Giménez-Cassina, A. Food for thought: Impact of metabolism on neuronal excitability. Exp. Cell Res. 2017, 360, 41–46. [Google Scholar] [CrossRef]
- Klepper, J.; Engelbrecht, V.; Scheffer, H.; van der Knaap, M.S.; Fiedler, A. GLUT1 deficiency with delayed myelination responding to ketogenic diet. Pediatr. Neurol. 2007, 37, 130–133. [Google Scholar] [CrossRef] [PubMed]
- Kossoff, E.H.; Zupec-Kania, B.A.; Rho, J.M. Ketogenic diets: An update for child neurologists. J. Child Neurol. 2009, 24, 979–988. [Google Scholar] [CrossRef]
- Villeneuve, N.; Pinton, F.; Bahi-Buisson, N.; Dulac, O.; Chiron, C.; Nabbout, R. The ketogenic diet improves recently worsened focal epilepsy. Dev. Med. Child Neurol. 2009, 51, 276–281. [Google Scholar] [CrossRef]
- Alter, A.S.; Engelstad, K.; Hinton, V.J.; Montes, J.; Pearson, T.S.; Akman, C.I.; De Vivo, D.C. Long-term clinical course of Glut1 deficiency syndrome. J. Child Neurol. 2015, 30, 160–169. [Google Scholar] [CrossRef]
- Kessler, S.K.; Neal, E.G.; Camfield, C.S.; Kossoff, E.H. Dietary therapies for epilepsy: Future research. Epilepsy Behav. 2011, 22, 17–22. [Google Scholar] [CrossRef] [Green Version]
- Vanderperre, B.; Herzig, S.; Krznar, P.; Hörl, M.; Ammar, Z.; Montessuit, S.; Pierredon, S.; Zamboni, N.; Martinou, J.-C. Embryonic lethality of mitochondrial pyruvate carrier 1 deficient mouse can be rescued by a ketogenic diet. PLoS Genet. 2016, 12, e1006056. [Google Scholar] [CrossRef]
- Maalouf, M.; Rho, J.M.; Mattson, M.P. The neuroprotective properties of calorie restriction, the ketogenic diet, and ketone bodies. Brain Res. Rev. 2009, 59, 293–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lutas, A.; Yellen, G. The ketogenic diet: Metabolic influences on brain excitability and epilepsy. Trends Neurosci. 2013, 36, 32–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.Y.; Simeone, K.A.; Simeone, T.A.; Pandya, J.D.; Wilke, J.C.W.; Ahn, Y.; Geddes, J.W.; Sullivan, P.G.; Rho, J.M. Ketone bodies mediate anti-seizure effects through mitochondrial permeability transition. Ann. Neurol. 2015, 78, 77–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kashiwaya, Y.; Takeshima, T.; Mori, N.; Nakashima, K.; Clarke, K.; Veech, R.L. D-beta- hydroxybutyrate protects neurons in models of Alzheimer´s and Parkinson´s disease. Proc. Natl. Acad. Sci. USA 2000, 97, 5440–5444. [Google Scholar] [CrossRef] [Green Version]
- Tieu, K.; Perier, C.; Caspersen, C.; Teismann, P.; Wu, D.C.; Yan, S.D.; Naini, A.; Vila, M.; Jackson-Lewis, V.; Ramasamy, R.; et al. D-beta-hydroxybutyrate rescues mitochondrial respiration and mitigates features of Parkinson’s disease. J. Clin. Investig. 2003, 112, 892–901. [Google Scholar] [CrossRef] [Green Version]
- Imamura, K.; Takeshima, T.; Kashiwaya, Y.; Nakaso, K.; Nakashima, K. D-β-hydroxybutyrate protects dopaminergic SH-SY5Y cells in a rotenone model of Parkinson’s disease. J. Neurosci. Res. 2006, 84, 1376–1384. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.K.; Kuwabara, H.; Zhou, Y.; Adams, R.J.; Brasic, J.R.; McGlothan, J.L.; Verina, T.; Burton, N.C.; Alexander, M.; Kumar, A.; et al. VMAT2 and dopamine neuron loss in a primate model of Parkinson’s disease. J. Neurochem. 2008, 105, 78–90. [Google Scholar] [CrossRef] [Green Version]
- Randle, P.J.; Garland, P.B.; Hales, C.N.; Newsholme, E.A. The glucose fatty acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet 1963, 1, 785–789. [Google Scholar] [CrossRef]
- Thevenet, J.; De Marchi, U.; Domingo, J.S.; Christinat, N.; Bultot, L.; Lefebvre, G.; Sakamoto, K.; Descombes, P.; Masoodi, M.; Wiederkerhr, A. Medium-chain fatty acids inhibit mitochondrial metabolism in astrocytes promoting astrocyte-neuron lactate and ketone body shuttle systems. FASEB J. 2016, 30, 1913–1926. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| AGC1-Deficiency | Main Traits | Molecular | MRI | MRS | Biochemistry | Treatment |
|---|---|---|---|---|---|---|
| Wibom et al. (2009) | Delayed psychomotor development, seizures, hypotonia, spasticity and hyperreflexia | AGC1 (p.Gln590Arg) | Cerebral Hypomyelination, Reduced supratentorial cerebral volume, enlarged ventricles and subarachnoid space, reduced putamen | NAA ↓↓ Myoinositol ↑ Choline = Lactate = Lipids = | Lactateplasma ↑↑ LactateCSF ↑ Aminoacidsplasma = | AED KD (Dahlin et al. 2015) |
| Falk et al. (2014) | Profound developmental delay, congenital hypotonia, refractory epilepsy, multiple dysmorphic features. | AGC1 (p.Arg353Gln) | Global Hypomyelination, Enlarged subarachnoid spaces, ventricles and sulci Cerebral volume loss, Diffuse brain atrophy | NAA ↓↓ Choline ↑ Myoinositol ↑ Lactate (parenchyma and CSF) ↑ | Lactateplasma = LactateCSF = Aminoacidsplasma = AminoacidsCSF = | AED |
| Parnes et al. (2015) | Delayed psychomotor development, epilepsy, hypotonia, spasticity and hyperreflexia, non-verbal | AGC1 (p.K100fs) (p.I72T) | Hypomyelination, brain atrophy | NAA ↓↓ | NA | AED |
| Pronicka et al. (2016) | NA | AGC1 (p.Asn445Lys) | NA | NA | NA | AED |
| Pfeiffer et al. (2019) | Intractable epilepsy, psychomotor delay, cerebral atrophy, severe hypotonia | AGC1 (p.Thr444Ile) | Unaffected myelination Cortical brain atrophy Ventricles not mentioned | NAA ↓↓ Choline ↑ Myoinositol ↑ Lactate peak not resolved | Acylcarnitine = Lactateplasma ↑ LactateUrineOrganicAcids ↑ Aminoacidsplasma = | KD |
| Kavanaugh et al. (2019) | Global developmental delay, optic neuropathy and visual impairment, spasticity and cerebral palsy, epilepsy without status epilepticus, non-verbal | AGC1 (p.A432V) (c.1447-2_1447-1delAG) | Diffuse white matter volume loss Increased ventricular volume and thinned corpus callosum Enlarged subarachnoid spaces and sulci | NAA ↓↓ Lactate ↑ (Hypothesized to be a seizure sequel) | NA | AED |
| Nashabat et al. (2019) | Refractory epilepsy, optic neuropathy and visual impairment, no hypotonia no microcephaly | AGC1 (p.Thr462Met) | Unremarkable | NA | NA | AED |
| Saleh et al. (2020) | Global developmental delay, epilepsy, no speech, hypertonia | AGC1 (p.Arg134 *) | Thin Corpus Callosum Diffuse brain atrophy Enlarged ventricles | NA | NA | NA |
| Kose et al. (2021) | NA | AGC1 (p.Arg42Pro) | NA | NA | NA | NA |
| Other MAS Defects | Main Tracts | Molecular | MRI | MRS | Biochemistry | Treatment |
| Broeks MH et al. (2019) | Global developmental delay, Infantile Epileptic Encephalopathy, progressive microcephaly, dysmorphic facies, axial hypotonia/hypertonia of extremities | MDH1 (p.Ala138Val) | Partial agenesis of corpus callosum, enlarged ventricles, mild hypoplasia of inferior vermis and pons | NA | Lactateplasma = Aminoacidsplasma = Acylcarnitine = Organic Acids = Dried-Blood Spot: Glutamate ↑ G-3-P ↑ | AED |
| Ait-El-MKadem S et al. (2017) | Generalized hypotonia, psychomotor delay and refractory epilepsy, muscle atrophy and dyskinesia | MDH2 (p.Pro133Leu) (p.Pro207Leu) (p.Gly199Alafs*10) (p.Gly37Arg) | Global brain atrophy, Corpus callosum atrophy, delayed myelination; cortical, frontal and parietal atrophy | Lactate ↑ | Lactateplasma ↑ LactateCSF ↑ | AED, KD |
| Karnebeek et al. (2019) | Progressive microcephaly, hypotonia, myoclonic epilepsy, profound intellectual disability, spasticity, frequent infections, non-verbal | GOT2 (p.Leu209del) (p.Arg337Gly) (p.Arg262Gly) (p.Gly33Val) | Mild cerebral atrophy, thinned corpus callosum, hypoplastic vermis (only in 1 case: multicistic encephalomalacia and asymmetric dilated lateral ventricles | NA | Aminoacidsplasma = Lactateplasma ↑ Ammoniaplasma ↑ Organic Acids and Acylcarnitine = | Pyridoxine and serine (2 out of 4 patients) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pardo, B.; Herrada-Soler, E.; Satrústegui, J.; Contreras, L.; del Arco, A. AGC1 Deficiency: Pathology and Molecular and Cellular Mechanisms of the Disease. Int. J. Mol. Sci. 2022, 23, 528. https://doi.org/10.3390/ijms23010528
Pardo B, Herrada-Soler E, Satrústegui J, Contreras L, del Arco A. AGC1 Deficiency: Pathology and Molecular and Cellular Mechanisms of the Disease. International Journal of Molecular Sciences. 2022; 23(1):528. https://doi.org/10.3390/ijms23010528
Chicago/Turabian StylePardo, Beatriz, Eduardo Herrada-Soler, Jorgina Satrústegui, Laura Contreras, and Araceli del Arco. 2022. "AGC1 Deficiency: Pathology and Molecular and Cellular Mechanisms of the Disease" International Journal of Molecular Sciences 23, no. 1: 528. https://doi.org/10.3390/ijms23010528