
Global Analysis of Transcriptome and Translatome Revealed That Coordinated WNT and FGF Regulate the Carapacial Ridge Development of Chinese Soft-Shell Turtle

 ,
,
Abstract
:1. Introduction
2. Results
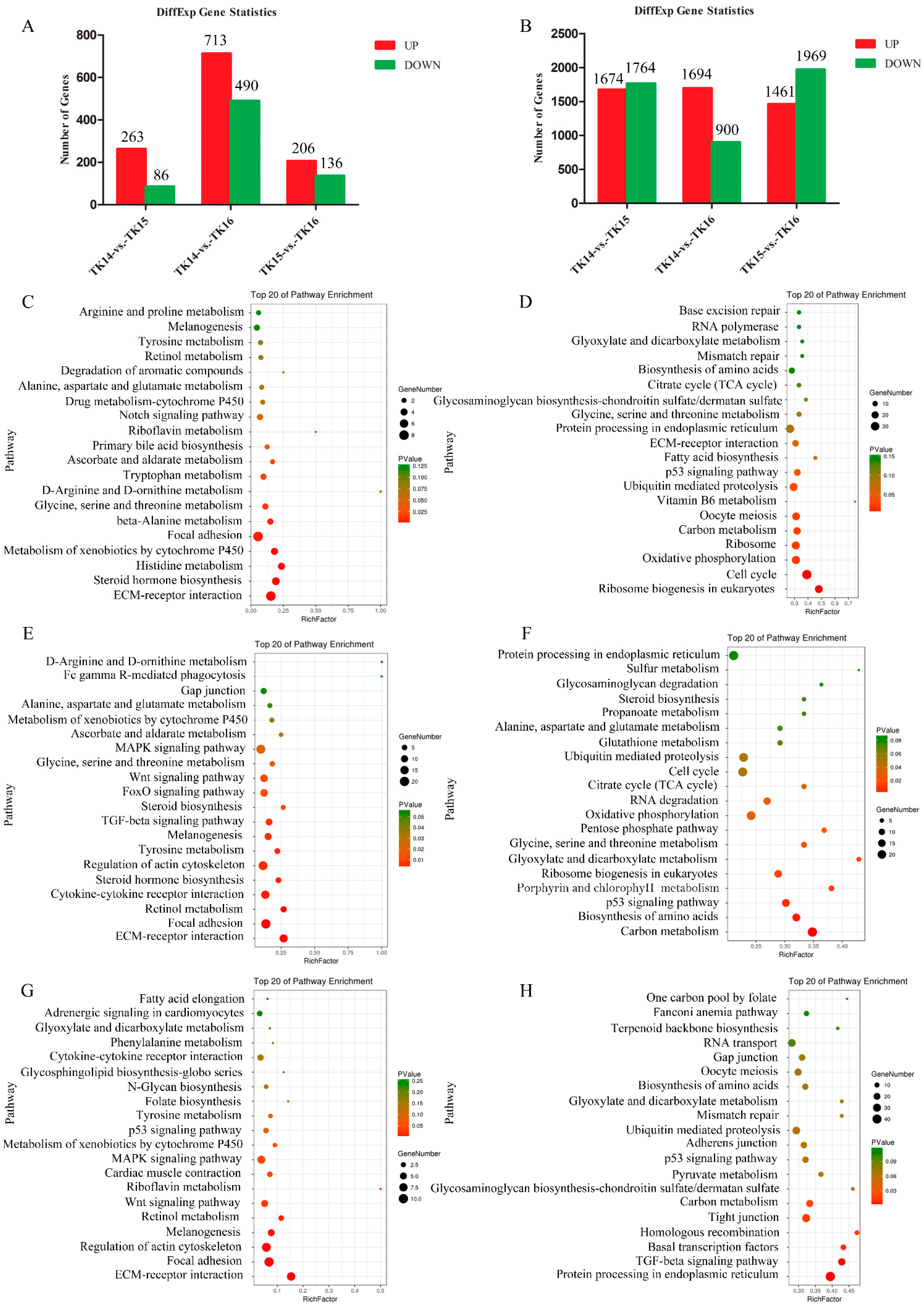
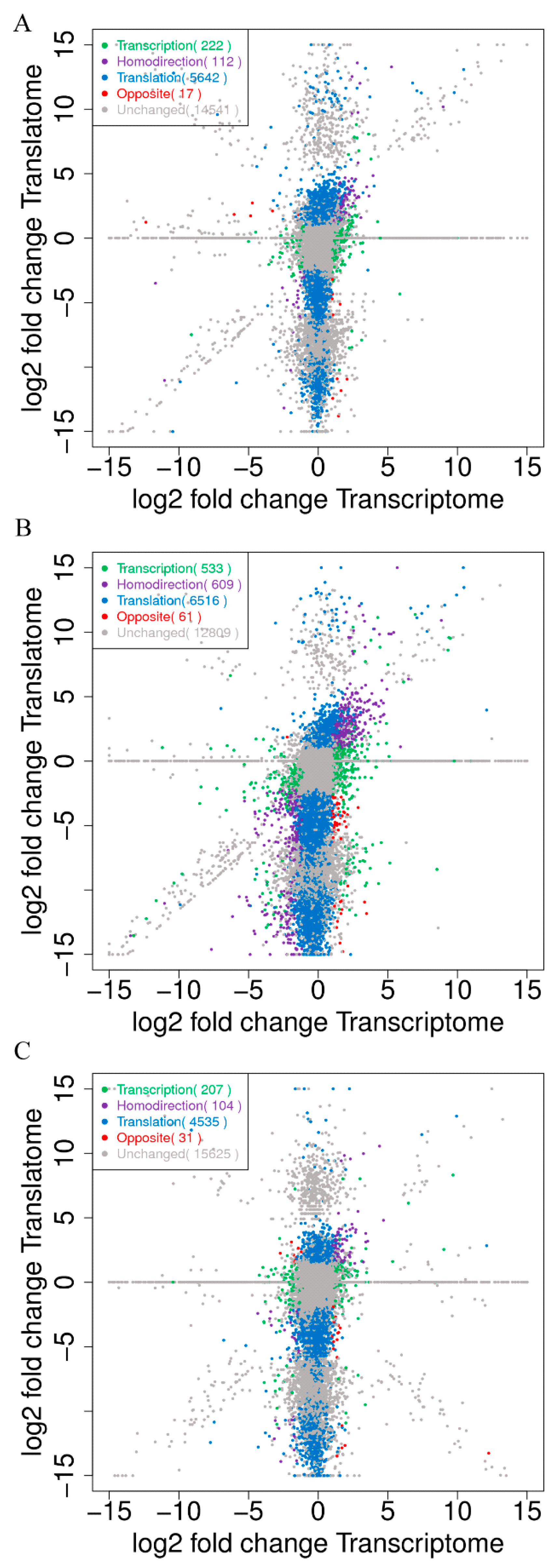
2.1. Transcription and Translation Gene Profiles during CR Development
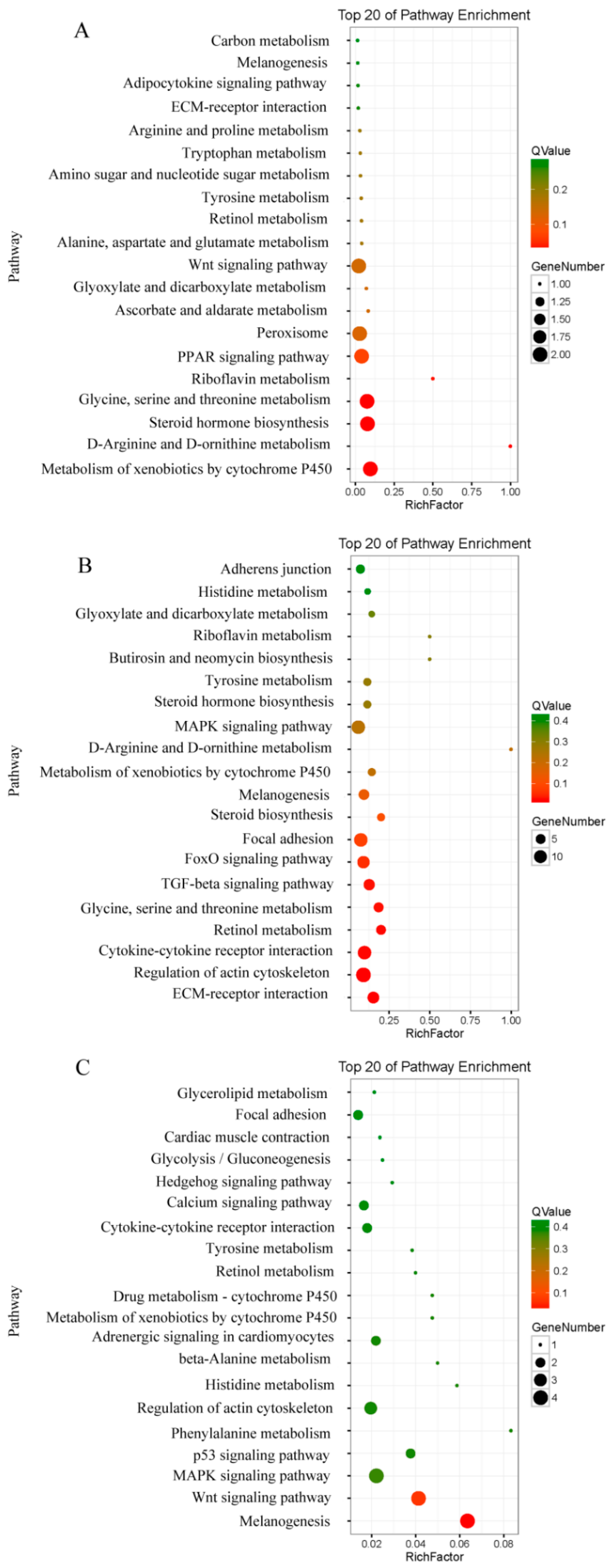
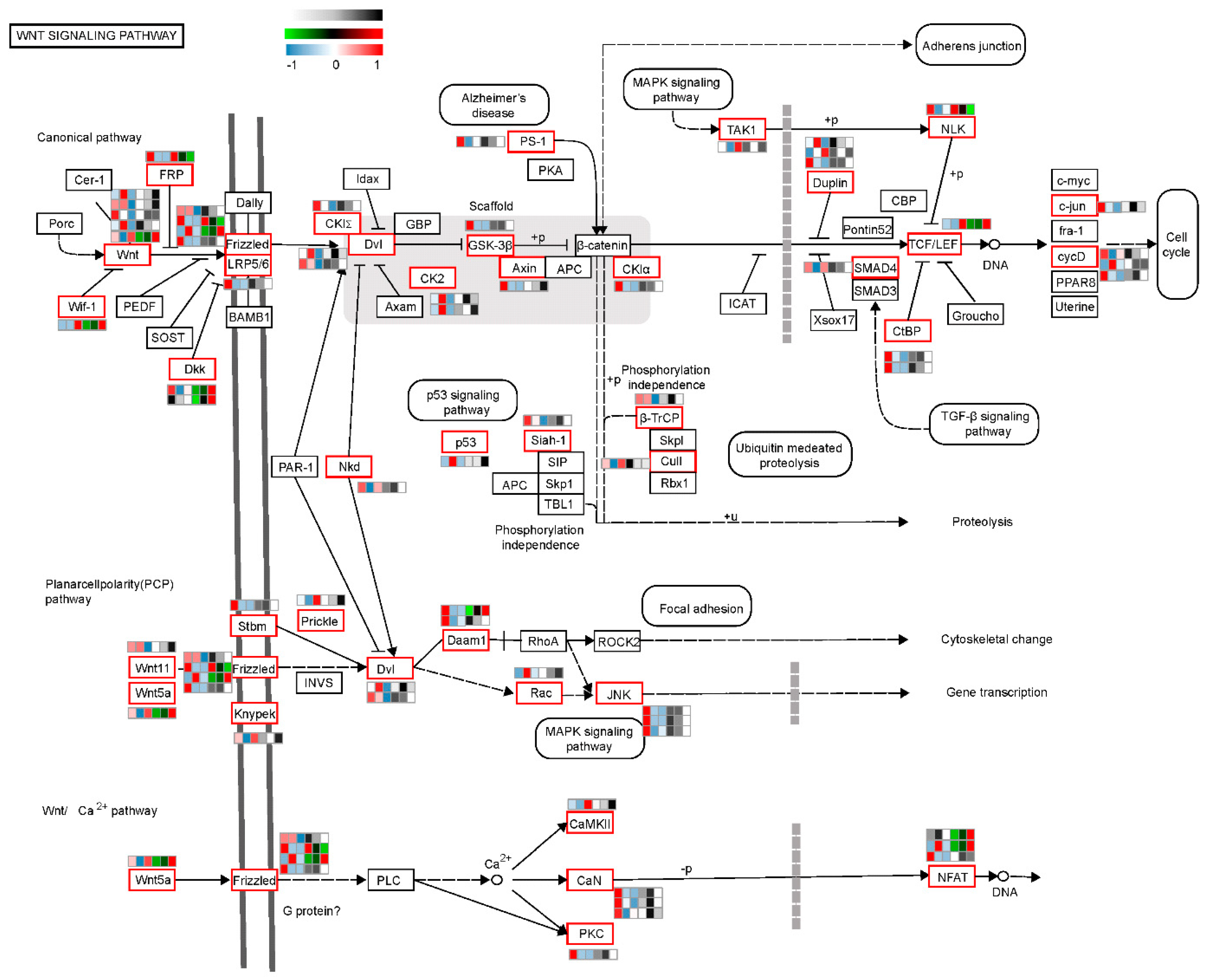
2.2. Dynamic Changes in Signaling Pathways Involved in CR Development
2.3. The Wnt Signaling Pathway Might Have a Key Role in Regulating CR Development through a Correlation Analysis of Transcriptomics and Translatomics
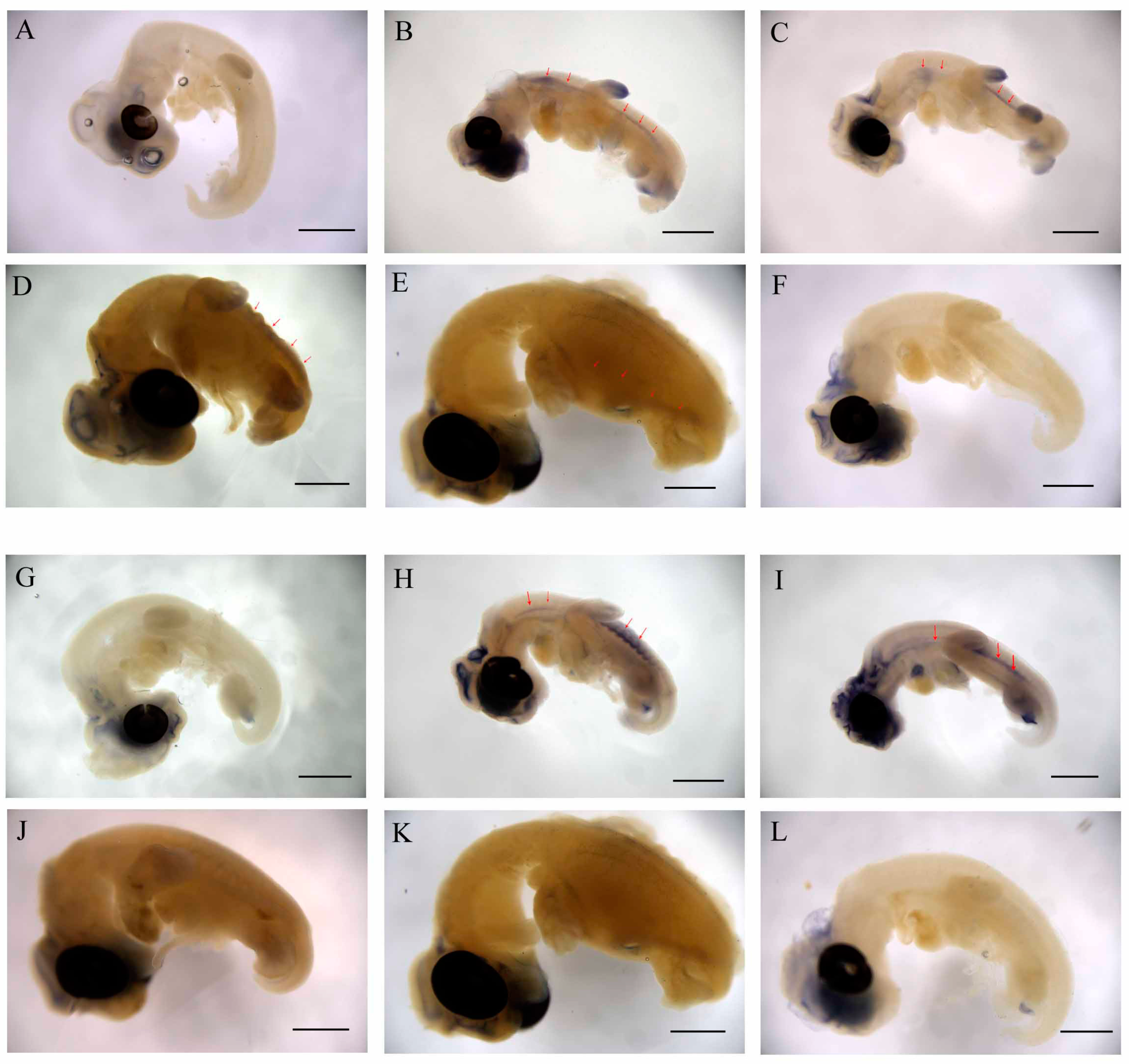
2.4. WNT and FGF Are Specifically Expressed in CR
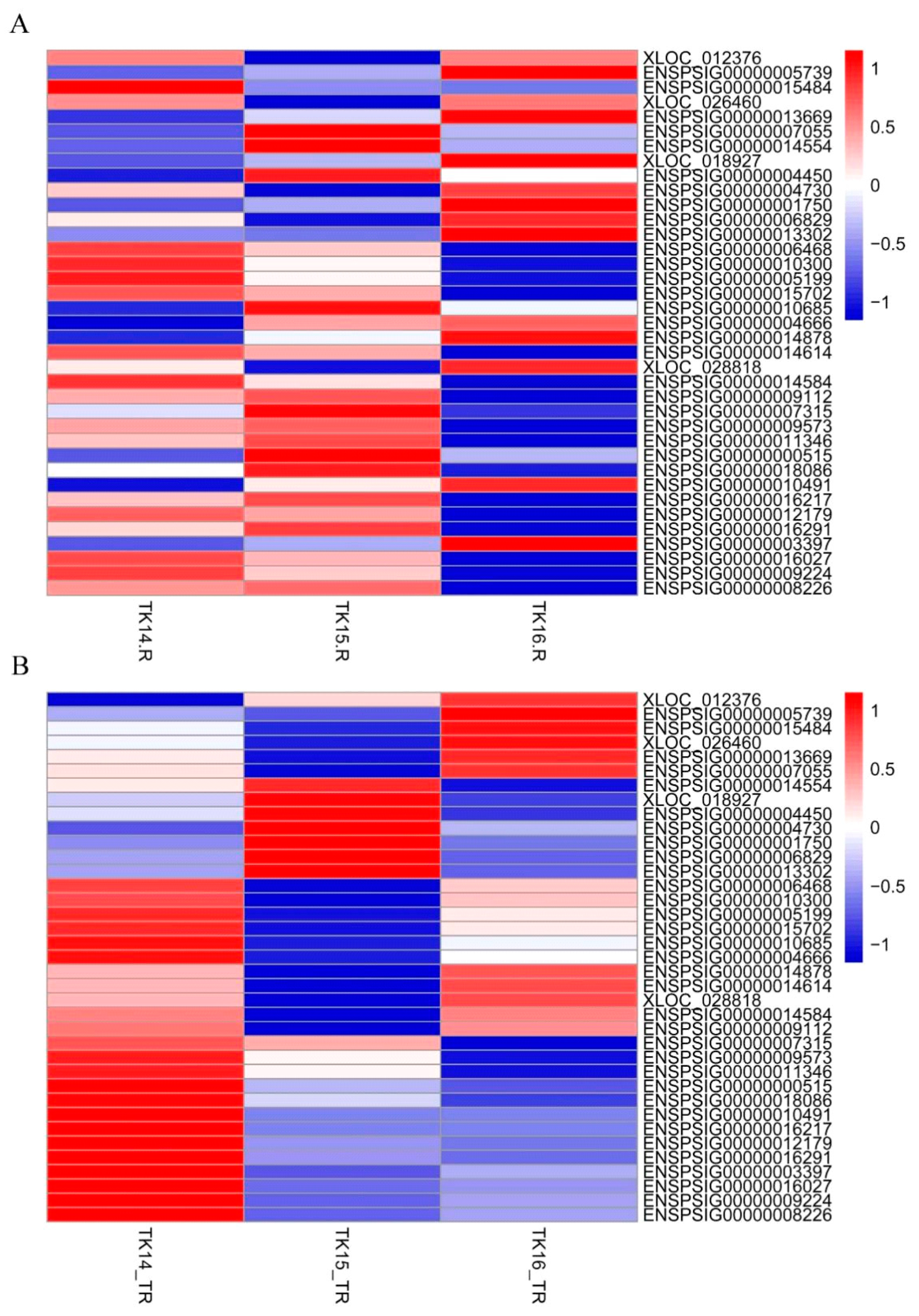
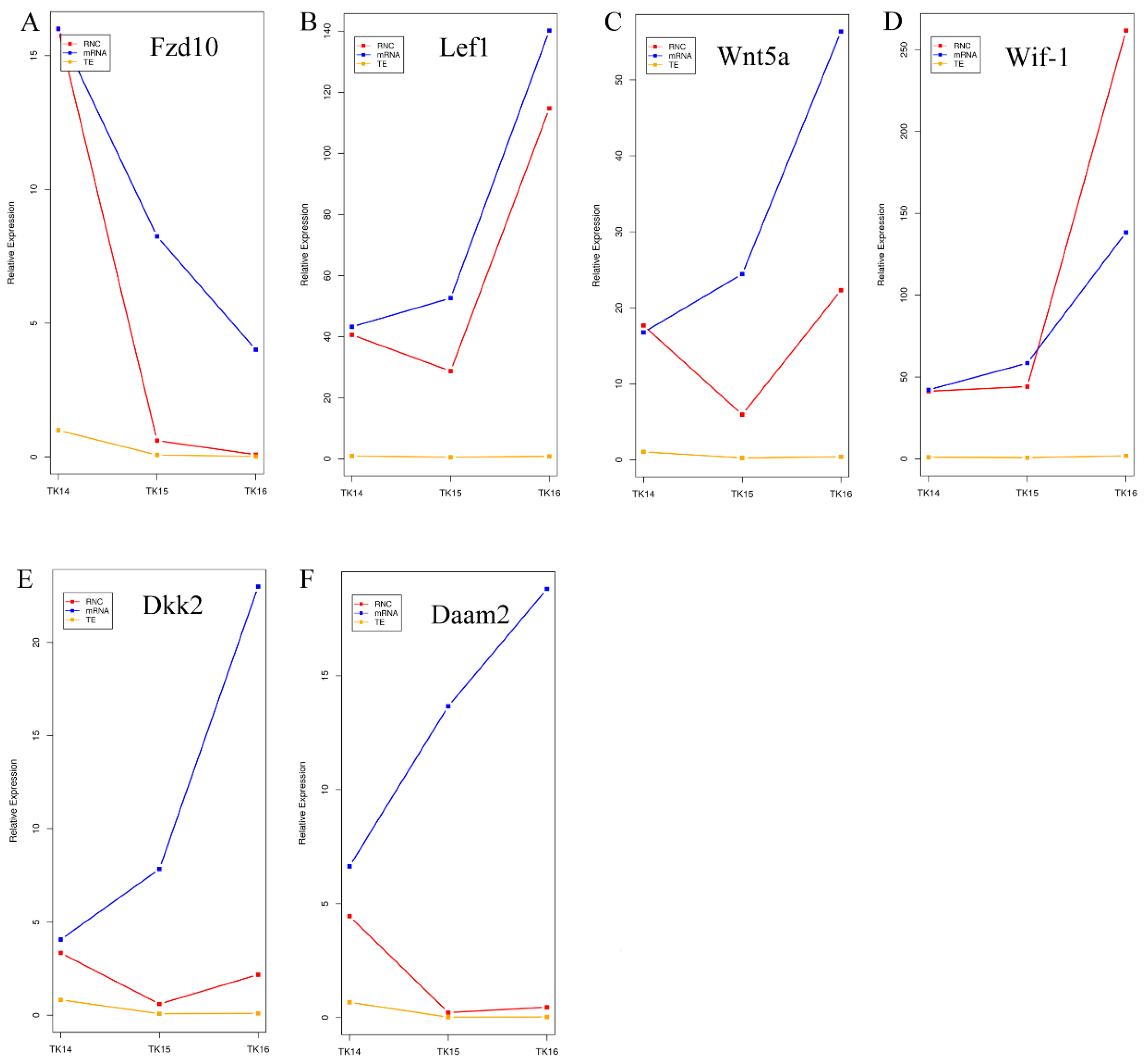
2.5. Wnts Genes Involved in CR Development Are Not Completely Dependent on mRNA Abundance
2.6. The PCP Signaling Pathway Was Activated during CR Development
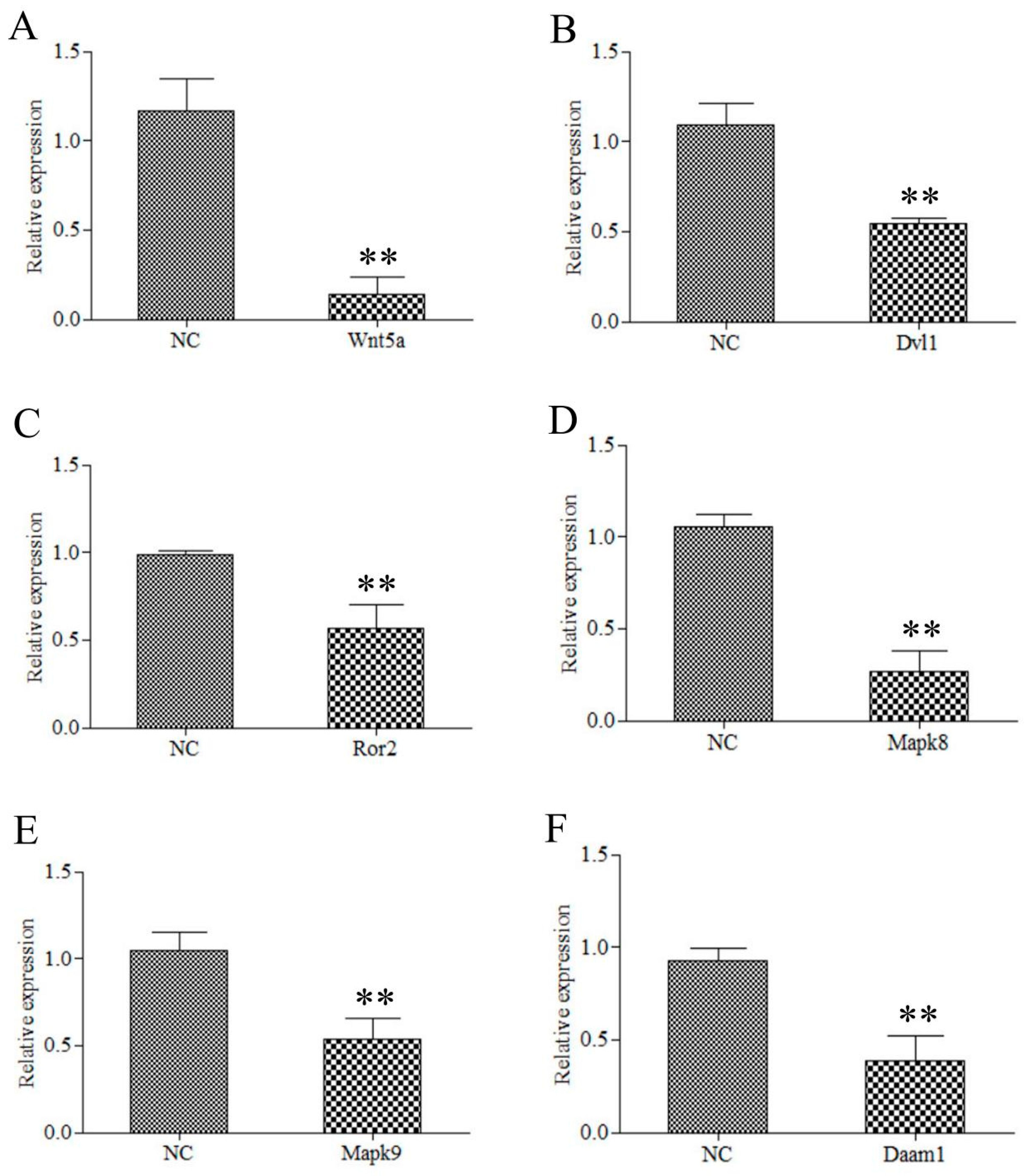
2.7. Wnt5a Participate in the CR Development through the JNK Pathway
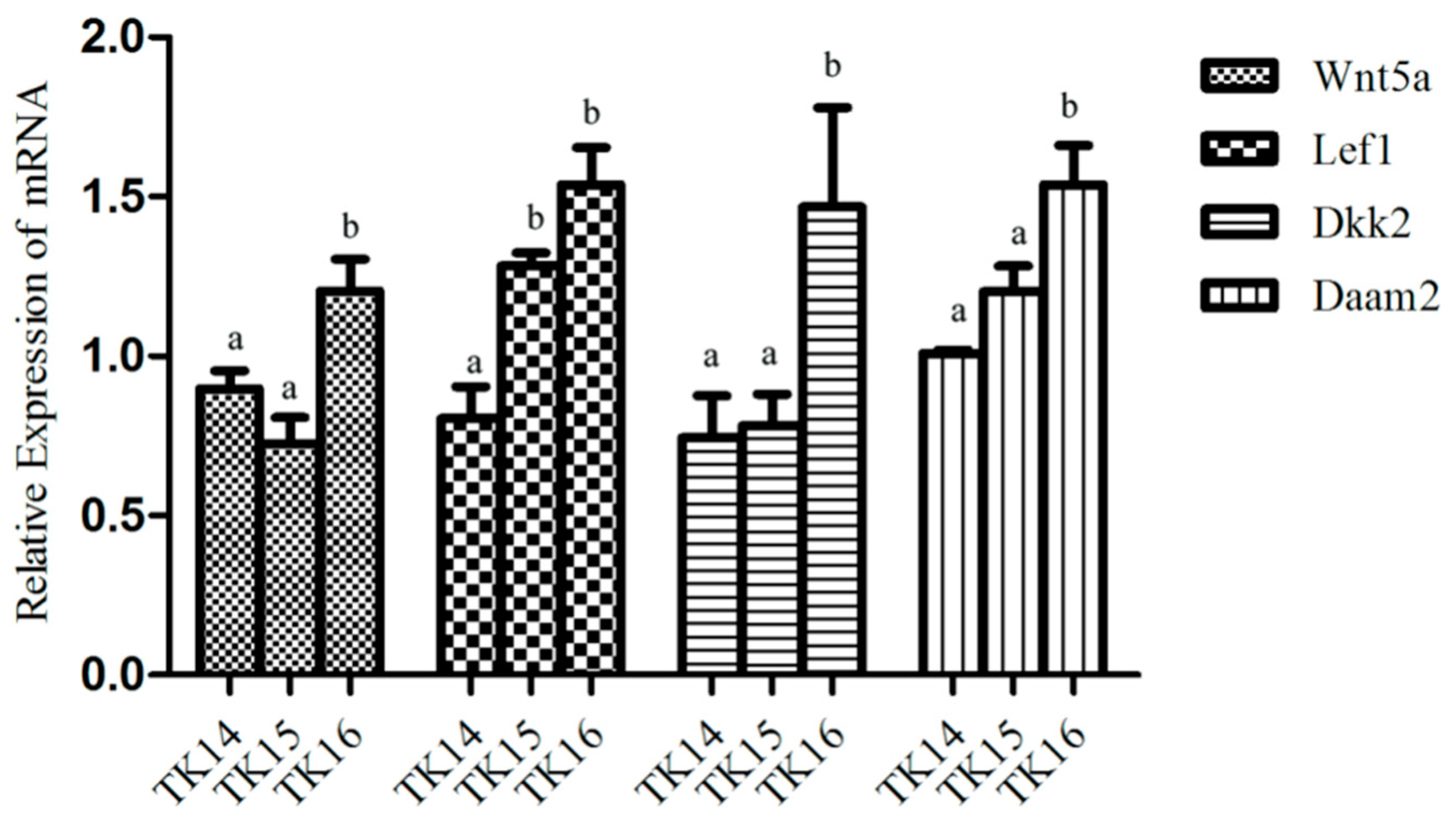
2.8. The Expression of Wnts during the CR Development by qRT-PCR
3. Discussion
4. Materials and Methods
4.1. Embryo Culture and Tissue Collection
4.2. Total RNA, RNC-mRNA, Library Construction, and Sequencing
4.3. Transcriptome and Translatome Assembly
4.4. Quantification of Gene Abundance
4.5. Differentially Transcribed Genes (DTRGs) and Differentially Translated Genes (DTLGs)
4.6. Pathway Enrichment Analysis
4.7. Whole Mount In Situ Hybridization (WISH)
4.8. In Vitro Culture of Carapacial Ridge Tissues
4.9. RNAi Interference
4.10. Quantitative RT-PCR
4.11. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CR | Carapace ridge |
| KEGG | Kyoto encyclopedia of Genes and Genomes |
| RNC-mRNA | ribosome nascent-chain complex-mRNA |
| WNT | Wingless-type |
| PCP | planar cell polarity |
| FGF | fibroblast growth factor |
| JNK | c-Jun N-terminal kinase |
References
- Sanchez-Villagra, M.R.; Muller, H.; Sheil, C.A.; Scheyer, T.M.; Nagashima, H.; Kuratani, S. Skeletal development in the Chinese soft-shelled turtle Pelodiscus sinensis (Testudines: Trionychidae). J. Morphol. 2009, 270, 1381–1399. [Google Scholar] [CrossRef] [PubMed]
- Nagashima, H.; Uchida, K.; Yamamoto, K.; Kuraku, S.; Usuda, R.; Kuratani, S. Turtle-chicken chimera: An experimental approach to understanding evolutionary innovation in the turtle. Dev. Dynam. 2005, 232, 149–161. [Google Scholar] [CrossRef]
- Nagashima, H.; Kuraku, S.; Uchida, K.; Ohya, Y.; Narita, Y.; Kuratani, S. On the carapacial ridge in turtle embryos: Its developmental origin, function and the chelonian body plan. Development 2007, 134, 2219–2226. [Google Scholar] [CrossRef] [Green Version]
- Moustakas-Verho, J.E.; Cebra-Thomas, J.; Gilbert, S.F. Patterning of the turtle shell. Curr. Opin. Genet. Dev. 2017, 45, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Kuraku, S.; Usuda, R.; Kuratani, S. Comprehensive survey of carapacial ridge-specific genes in turtle implies co-option of some regulatory genes in carapace evolution. Evol. Dev. 2005, 7, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Nagashima, H.; Shibata, M.; Taniguchi, M.; Ueno, S.; Kamezaki, N.; Sato, N. Comparative study of the shell development of hard- and soft-shelled turtles. J. Anat. 2014, 225, 60–70. [Google Scholar] [CrossRef]
- Nagashima, H.; Kuraku, S.; Uchida, K.; Kawashima-Ohya, Y.; Narita, Y.; Kuratani, S. Body plan of turtles: An anatomical, developmental and evolutionary perspective. Anat. Sci. Int. 2012, 87, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Hirasawa, T.; Nagashima, H.; Kuratani, S. The endoskeletal origin of the turtle carapace. Nat. Commun. 2013, 4, 2107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bever, G.S.; Lyson, T.R.; Field, D.J.; Bhullar, B.-A.S. Evolutionary origin of the turtle skull. Nature 2015, 525, 239–242. [Google Scholar] [CrossRef]
- Carroll, C. Vertebrate Paleontology and Evolution; W H Freeman and Co: New York, NY, USA, 1988. [Google Scholar]
- Lyson, T.R.; Bever, G.S.; Bhullar, B.A.; Joyce, W.G.; Gauthier, J.A. Transitional fossils and the origin of turtles. Biol. Lett. 2010, 6, 830–833. [Google Scholar] [CrossRef] [Green Version]
- Schoch, R.R.; Sues, H.D. A Middle Triassic stem-turtle and the evolution of the turtle body plan. Nature 2015, 523, 584–587. [Google Scholar] [CrossRef]
- Li, C.; Wu, X.C.; Rieppel, O.; Wang, L.T.; Zhao, L.J. An ancestral turtle from the Late Triassic of southwestern China. Nature 2008, 456, 497–501. [Google Scholar] [CrossRef] [PubMed]
- Lyson, T.R.; Bever, G.S.; Scheyer, T.M.; Hsiang, A.Y.; Gauthier, J.A. Evolutionary origin of the turtle shell. Curr. Biol. 2013, 23, 1113–1119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuratani, S. Acquisition of the turtle shell: Changes in developmental program behind the evolutionary novelty. Mech. Dev. 2017, 145, S12. [Google Scholar] [CrossRef]
- Li, C.; Fraser, N.C.; Rieppel, O.; Wu, X.-C. A Triassic stem turtle with an edentulous beak. Nature 2018, 560, 476. [Google Scholar] [CrossRef] [PubMed]
- Burke, A.C. The Development and Evolution of the Turtle Body Plan, Inferring Intrinsic Aspects of the Evolutionary Process from Experimental Embryology. Am. Zool. 1991, 31, 616–627. [Google Scholar] [CrossRef] [Green Version]
- Brenner, S.; Johnson, M.; Bridgham, J.; Golda, G.; Lloyd, D.H.; Johnson, D.; Luo, S.; McCurdy, S.; Foy, M.; Ewan, M.; et al. Gene expression analysis by massively parallel signature sequencing (MPSS) on microbead arrays. Nat. Biotechnol. 2000, 18, 630–634. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Pascual-Anaya, J.; Zadissa, A.; Li, W.; Niimura, Y.; Huang, Z.; Li, C.; White, S.; Xiong, Z.; Fang, D.; et al. The draft genomes of soft-shell turtle and green sea turtle yield insights into the development and evolution of the turtle-specific body plan. Nat. Genet. 2013, 45, 701–706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pascual-Anaya, J.; Hirasawa, T.; Sato, I.; Kuraku, S.; Kuratani, S. Comparative analysis of pleurodiran and cryptodiran turtle embryos depicts the molecular ground pattern of the turtle carapacial ridge. Int. J. Dev. Biol. 2014, 58, 743–750. [Google Scholar] [CrossRef] [Green Version]
- Cebra-Thomas, J.; Tan, F.; Sistla, S.; Estes, E.; Bender, G.; Kim, C.; Riccio, P.; Gilbert, S. How the turtle forms its shell: A paracrine hypothesis of carapace formation. J. Exp. Zool. Part B Mol. Dev. Evol. 2005, 304, 558–569. [Google Scholar] [CrossRef]
- Gao, B.; Ajima, R.; Yang, W.; Li, C.; Song, H.; Anderson, M.; Liu, R.; Lewandoski, M.; Yamaguchi, T.; Yang, Y. Coordinated directional outgrowth and pattern formation by integration of Wnt5a and Fgf signaling in planar cell polarity. Development 2018, 145, dev163824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiese, K.; Nusse, R.; van Amerongen, R. Wnt signalling: Conquering complexity. Development 2018, 145, dev165902. [Google Scholar] [CrossRef] [Green Version]
- Gao, B.; Yang, Y. Planar cell polarity in vertebrate limb morphogenesis. Curr. Opin. Genet. Dev. 2013, 23, 438–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boutros, M.; Paricio, N.; Strutt, D.; Mlodzik, M. Dishevelled activates JNK and discriminates between JNK pathways in planar polarity and wingless signaling. Cell 1998, 94, 109–118. [Google Scholar] [CrossRef] [Green Version]
- Oishi, I.; Suzuki, H.; Onishi, N.; Takada, R.; Kani, S.; Ohkawara, B.; Koshida, I.; Suzuki, K.; Yamada, G.; Schwabe, G.; et al. The receptor tyrosine kinase Ror2 is involved in non-canonical Wnt5a/JNK signalling pathway. Genes Cells 2003, 8, 645–654. [Google Scholar] [CrossRef]
- Yamanaka, H.; Moriguchi, T.; Masuyama, N.; Kusakabe, M.; Hanafusa, H.; Takada, R.; Takada, S.; Nishida, E. JNK functions in the non-canonical Wnt pathway to regulate convergent extension movements in vertebrates. EMBO Rep. 2002, 3, 69–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gros, J.; Hu, J.; Vinegoni, C.; Feruglio, P.; Weissleder, R.; Tabin, C. WNT5A/JNK and FGF/MAPK pathways regulate the cellular events shaping the vertebrate limb bud. Curr. Biol. 2010, 20, 1993–2002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwanhäusser, B.; Busse, D.; Li, N.; Dittmar, G.; Schuchhardt, J.; Wolf, J.; Chen, W.; Selbach, M. Global quantification of mammalian gene expression control. Nature 2011, 473, 337–342. [Google Scholar] [CrossRef] [Green Version]
- Gebauer, F.; Hentze, M.W. Molecular mechanisms of translational control. Nat. Rev. Mol. Cell Biol. 2004, 5, 827–835. [Google Scholar] [CrossRef]
- Sonenberg, N.; Hinnebusch, A. Regulation of translation initiation in eukaryotes: Mechanisms and biological targets. Cell 2009, 136, 731–745. [Google Scholar] [CrossRef] [Green Version]
- Ingolia, N.; Ghaemmaghami, S.; Newman, J.; Weissman, J. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 2009, 324, 218–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cordero, G.A. The turtle’s shell. Curr. Biol. 2017, 27, R168–R169. [Google Scholar] [CrossRef] [PubMed]
- Baarsma, H.; Königshoff, M.; Gosens, R. The WNT signaling pathway from ligand secretion to gene transcription: Molecular mechanisms and pharmacological targets. Pharmacol. Ther. 2013, 138, 66–83. [Google Scholar] [CrossRef] [PubMed]
- Du, S.; Purcell, S.; Christian, J.; McGrew, L.; Moon, R. Identification of distinct classes and functional domains of Wnts through expression of wild-type and chimeric proteins in Xenopus embryos. Mol. Cell Biol. 1995, 15, 2625–2634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roszko, I.; Sawada, A.; Solnica-Krezel, L. Regulation of convergence and extension movements during vertebrate gastrulation by the Wnt/PCP pathway. Semin. Cell. Dev. Biol. 2009, 20, 986–997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsieh, J.; Kodjabachian, L.; Rebbert, M.; Rattner, A.; Smallwood, P.; Samos, C.; Nusse, R.; Dawid, I.; Nathans, J. A new secreted protein that binds to Wnt proteins and inhibits their activities. Nature 1999, 398, 431–436. [Google Scholar] [CrossRef]
- Liepinsh, E.; Bányai, L.; Patthy, L.; Otting, G. NMR structure of the WIF domain of the human Wnt-inhibitory factor-1. J. Mol. Biol. 2006, 357, 942–950. [Google Scholar] [CrossRef] [PubMed]
- Glinka, A.; Wu, W.; Delius, H.; Monaghan, A.; Blumenstock, C.; Niehrs, C. Dickkopf-1 is a member of a new family of secreted proteins and functions in head induction. Nature 1998, 391, 357–362. [Google Scholar] [CrossRef]
- Gordon, M.; Nusse, R. Wnt signaling: Multiple pathways, multiple receptors, and multiple transcription factors. J. Biol. Chem. 2006, 281, 22429–22433. [Google Scholar] [CrossRef] [Green Version]
- Andre, P.; Song, H.; Kim, W.; Kispert, A.; Yang, Y. Wnt5a and Wnt11 regulate mammalian anterior-posterior axis elongation. Development 2015, 142, 1516–1527. [Google Scholar] [CrossRef] [Green Version]
- Curtin, J.; Quint, E.; Tsipouri, V.; Arkell, R.; Cattanach, B.; Copp, A.; Henderson, D.; Spurr, N.; Stanier, P.; Fisher, E.; et al. Mutation of Celsr1 disrupts planar polarity of inner ear hair cells and causes severe neural tube defects in the mouse. Curr. Biol. 2003, 13, 1129–1133. [Google Scholar] [CrossRef]
- Song, H.; Hu, J.; Chen, W.; Elliott, G.; Andre, P.; Gao, B.; Yang, Y. Planar cell polarity breaks bilateral symmetry by controlling ciliary positioning. Nature 2010, 466, 378–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Mlodzik, M. Wnt-Frizzled/planar cell polarity signaling: Cellular orientation by facing the wind (Wnt). Annu. Rev. Cell Dev. Biol. 2015, 31, 623–646. [Google Scholar] [CrossRef] [Green Version]
- Gao, B.; Song, H.; Bishop, K.; Elliot, G.; Garrett, L.; English, M.; Andre, P.; Robinson, J.; Sood, R.; Minami, Y.; et al. Wnt signaling gradients establish planar cell polarity by inducing Vangl2 phosphorylation through Ror2. Dev. Cell 2011, 20, 163–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, H.; Susman, M.; Bikoff, J.; Ryu, Y.; Jonas, A.; Hu, L.; Kuruvilla, R.; Greenberg, M. Wnt5a-Ror-Dishevelled signaling constitutes a core developmental pathway that controls tissue morphogenesis. Proc. Natl. Acad. Sci. USA 2012, 109, 4044–4051. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, T.; Bradley, A.; McMahon, A.; Jones, S. A Wnt5a pathway underlies outgrowth of multiple structures in the vertebrate embryo. Development 1999, 126, 1211–1223. [Google Scholar] [CrossRef]
- Minden, A.; Lin, A.; Claret, F.; Abo, A.; Karin, M. Selective activation of the JNK signaling cascade and c-Jun transcriptional activity by the small GTPases Rac and Cdc42Hs. Cell 1995, 81, 1147–1157. [Google Scholar] [CrossRef] [Green Version]
- Musti, A.; Treier, M.; Bohmann, D. Reduced ubiquitin-dependent degradation of c-Jun after phosphorylation by MAP kinases. Science 1997, 275, 400–402. [Google Scholar] [CrossRef]
- Coso, O.; Chiariello, M.; Yu, J.; Teramoto, H.; Crespo, P.; Xu, N.; Miki, T.; Gutkind, J. The small GTP-binding proteins Rac1 and Cdc42 regulate the activity of the JNK/SAPK signaling pathway. Cell 1995, 81, 1137–1146. [Google Scholar] [CrossRef] [Green Version]
- Burke, A.C. Development of the turtle carapace: Implications for the evolution of a novel bauplan. J. Morphol. 1989, 199, 363–378. [Google Scholar] [CrossRef]
- SAUNDERS, J. The proximo-distal sequence of origin of the parts of the chick wing and the role of the ectoderm. J. Exp. Zool. 1948, 108, 363–403. [Google Scholar] [CrossRef]
- Sun, X.; Mariani, F.; Martin, G. Functions of FGF signalling from the apical ectodermal ridge in limb development. Nature 2002, 418, 501–508. [Google Scholar] [CrossRef]
- Logan, C.; Nusse, R. The Wnt signaling pathway in development and disease. Annu. Rev. Cell. Dev. Biol. 2004, 20, 781–810. [Google Scholar] [CrossRef] [Green Version]
- Ohuchi, H.; Nakagawa, T.; Itoh, N.; Noji, S. FGF10 can induce Fgf8 expression concomitantly with En1 and R-fng expression in chick limb ectoderm, independent of its dorsoventral specification. Dev. Growth Differ. 1999, 41, 665–673. [Google Scholar] [CrossRef]
- Stark, R.; Searls, R. A description of chick wing bud development and a model of limb morphogenesis. Dev. Biol. 1973, 33, 138–153. [Google Scholar] [CrossRef]
- Pearse, R.; Scherz, P.; Campbell, J.; Tabin, C. A cellular lineage analysis of the chick limb bud. Dev. Biol. 2007, 310, 388–400. [Google Scholar] [CrossRef] [Green Version]
- ten Berge, D.; Brugmann, S.; Helms, J.; Nusse, R. Wnt and FGF signals interact to coordinate growth with cell fate specification during limb development. Development 2008, 135, 3247–3257. [Google Scholar] [CrossRef] [Green Version]
- Solursh, M. Ectoderm as a determinant of early tissue pattern in the limb bud. Cell Differ. 1984, 15, 17–24. [Google Scholar] [CrossRef]
- Tokita, M.; Kuratani, S. Normal Embryonic Stages of the Chinese Softshelled Turtle Pelodiscus sinensis (Trionychidae). Zool. Sci. 2001, 18, 705–715. [Google Scholar] [CrossRef] [Green Version]
- Moustakas-Verho, J.; Zimm, R.; Cebra-Thomas, J.; Lempiäinen, N.; Kallonen, A.; Mitchell, K.; Hämäläinen, K.; Salazar-Ciudad, I.; Jernvall, J.; Gilbert, S. The origin and loss of periodic patterning in the turtle shell. Development 2014, 141, 3033–3039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Yu, P.; Zhou, Q.; Li, X.; Ding, S.; Su, S.; Zhang, X.; Yang, X.; Zhou, W.; Wan, Q.; et al. Screening and characterisation of sex differentiation-related long non-coding RNAs in Chinese soft-shell turtle (Pelodiscus sinensis). Sci. Rep.-UK 2018, 8, 8630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grabherr, M.; Haas, B.; Yassour, M.; Levin, J.; Thompson, D.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nature Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [Green Version]
- Bazin, J.; Baerenfaller, K.; Gosai, S.; Gregory, B.; Crespi, M.; Bailey-Serres, J. Global analysis of ribosome-associated noncoding RNAs unveils new modes of translational regulation. Proc. Natl. Acad. Sci. USA 2017, 114, E10018–E10027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, R.; Li, Z.; Lau Vetter, M.; Vishnivetskaya, T.; Zanina, O.; Lloyd, K.; Pfiffner, S.; Rivkina, E.; Wang, W.; Wiggins, J.; et al. Genomic reconstruction of fossil and living microorganisms in ancient Siberian permafrost. Microbiome 2021, 9, 110. [Google Scholar] [CrossRef] [PubMed]
- Langdon, W. Performance of genetic programming optimised Bowtie2 on genome comparison and analytic testing (GCAT) benchmarks. BioData Min. 2015, 8, 1. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef] [Green Version]
- Sendoel, A.; Dunn, J.; Rodriguez, E.; Naik, S.; Gomez, N.; Hurwitz, B.; Levorse, J.; Dill, B.; Schramek, D.; Molina, H.; et al. Translation from unconventional 5’ start sites drives tumour initiation. Nature 2017, 541, 494–499. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.; Pimentel, H.; Salzberg, S.; Rinn, J.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Dewey, C. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [Green Version]
- Shahriyari, L. Effect of normalization methods on the performance of supervised learning algorithms applied to HTSeq-FPKM-UQ data sets: 7SK RNA expression as a predictor of survival in patients with colon adenocarcinoma. Brief. Bioinform. 2019, 20, 985–994. [Google Scholar] [CrossRef]
- Cao, X.; Guo, Z.; Wang, H.; Dong, Y.; Lu, S.; He, Q.; Sun, X.; Zhang, G. Prorocentrum donghaienseAutoactivation of Translation Causes the Bloom of in Harmful Algal Blooms. J. Proteome Res. 2021, 20, 3179–3187. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.; Wang, Y.; Yang, W.; Li, Z.; Zhang, X.; Zhou, L.; Gui, J. Upregulation of the PPAR signaling pathway and accumulation of lipids are related to the morphological and structural transformation of the dragon-eye goldfish eye. Sci. China Life Sci. 2021, 64, 1031–1049. [Google Scholar] [CrossRef] [PubMed]
- Kronja, I.; Yuan, B.; Eichhorn, S.; Dzeyk, K.; Krijgsveld, J.; Bartel, D.; Orr-Weaver, T. Widespread changes in the posttranscriptional landscape at the Drosophila oocyte-to-embryo transition. Cell Rep. 2014, 7, 1495–1508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thisse, C.; Thisse, B. High-resolution in situ hybridization to whole-mount zebrafish embryos. Nat. Protoc. 2008, 3, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Juntawong, P.; Girke, T.; Bazin, J.; Bailey-Serres, J. Translational dynamics revealed by genome-wide profiling of ribosome footprints in Arabidopsis. Proc. Natl. Acad. Sci. USA 2014, 111, E203–E212. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Yu, P.; Liu, T.; Qiao, D.; Hu, Q.; Su, S. Identification and functional analysis of SOX transcription factors in the genome of the Chinese soft-shell turtle (Pelodiscus sinensis). Comp. Biochem. Phys. B 2020, 242, 110407. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Reed, A.; Chen, F.; Stewart, C. Statistical analysis of real-time PCR data. BMC Bioinform. 2006, 7, 85. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Forward Primer | Reverse Primer |
| Wnt5a | CGATGCCCTGAAGGAGAAAT | ATGGTGGGCGTGTTGAAAC |
| Dvl1 | GGATCCCACGCCTAGAAGTT | CCCGTCATGGCTGTGGTATGA |
| Daam1 | GGCTGCTAGAAAATCGCTGA | TCAAGCCGTCCAGGTCGATA |
| Mapk8 | GCGTGGTCATTTGTCCTACCT | TGACTTTGCCAAGGGTCACA |
| Mapk9 | ACCAGCCCTTCCCAGTCGT | GTCCCGTCAGGGCATCAAT |
| Ror2 | CAGCACAAGCCCCGTTAGTA | TGGGACCATTGGTCGGATC |
| Lef-1 | CAAATAAGGTGCCAGTGGTGC | AGGGATGTGTGAAGGGTGTGA |
| Daam2 | GGAATTCCGATTGCACCTGA | GCTTCGCAAGTTCCAGATCA |
| Dkk2 | AAGAAGCGTTGCCACAGAGA | CCAGCCCATGTCCTTGCTAG |
| Gapdh | GTGCTGCCCAGAACATCATT | GGGAGTTGGAACACGGAAAG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, J.; Yu, P.; Zhao, Y.; Zhou, Q.; Yang, J.; Hu, Q.; Liu, T.; Bao, C.; Su, S.; Gui, J.-F. Global Analysis of Transcriptome and Translatome Revealed That Coordinated WNT and FGF Regulate the Carapacial Ridge Development of Chinese Soft-Shell Turtle. Int. J. Mol. Sci. 2021, 22, 12441. https://doi.org/10.3390/ijms222212441
Zhang J, Yu P, Zhao Y, Zhou Q, Yang J, Hu Q, Liu T, Bao C, Su S, Gui J-F. Global Analysis of Transcriptome and Translatome Revealed That Coordinated WNT and FGF Regulate the Carapacial Ridge Development of Chinese Soft-Shell Turtle. International Journal of Molecular Sciences. 2021; 22(22):12441. https://doi.org/10.3390/ijms222212441
Chicago/Turabian StyleZhang, Jun, Peng Yu, Yang Zhao, Qinyan Zhou, Jiayu Yang, Qingtao Hu, Tiantian Liu, Chuanhe Bao, Shiping Su, and Jian-Fang Gui. 2021. "Global Analysis of Transcriptome and Translatome Revealed That Coordinated WNT and FGF Regulate the Carapacial Ridge Development of Chinese Soft-Shell Turtle" International Journal of Molecular Sciences 22, no. 22: 12441. https://doi.org/10.3390/ijms222212441