
A New Paradigm for KIM-PTP Drug Discovery: Identification of Allosteric Sites with Potential for Selective Inhibition Using Virtual Screening and LEI Analysis


Abstract
:1. Introduction
2. Results and Discussion
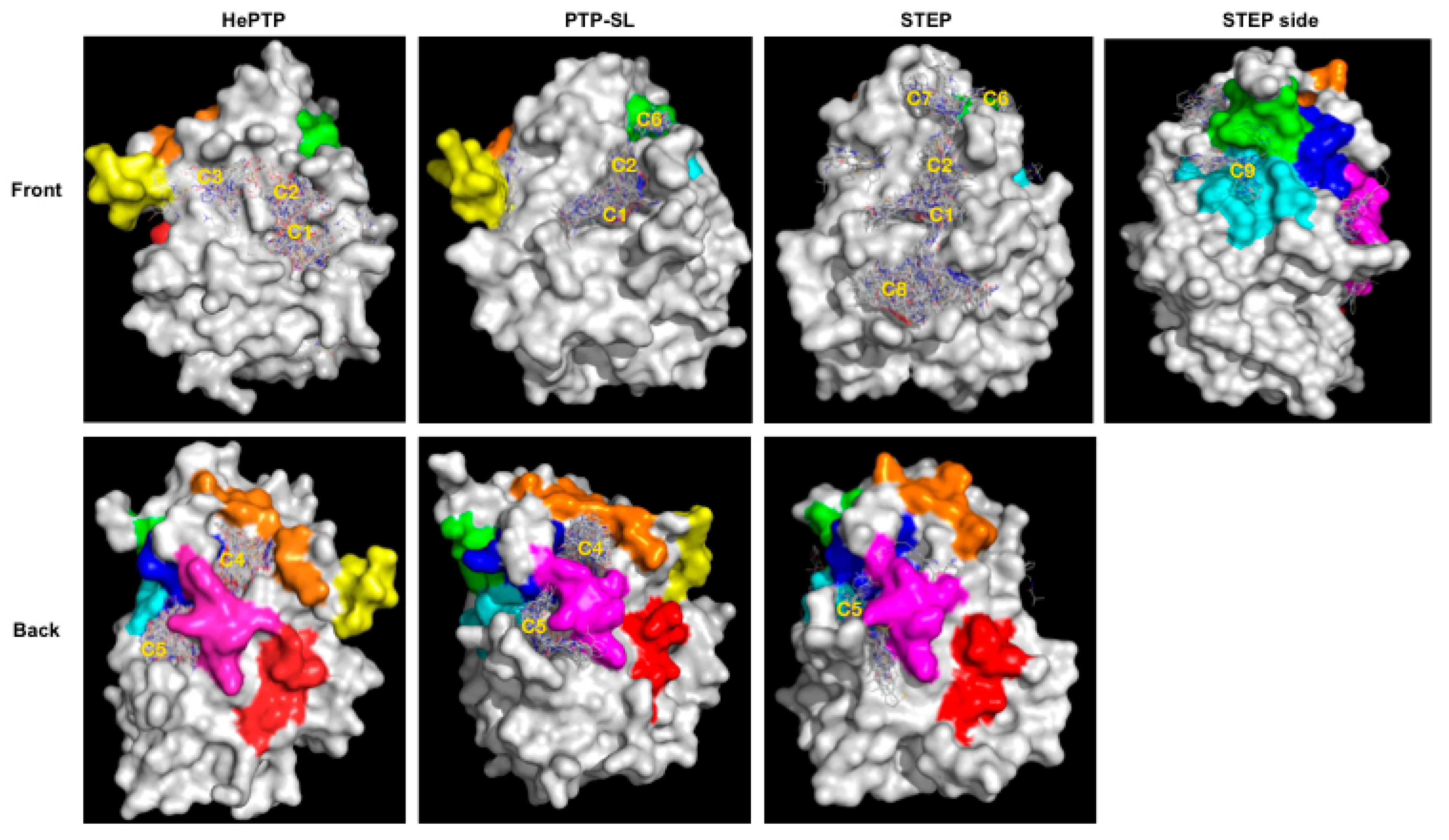
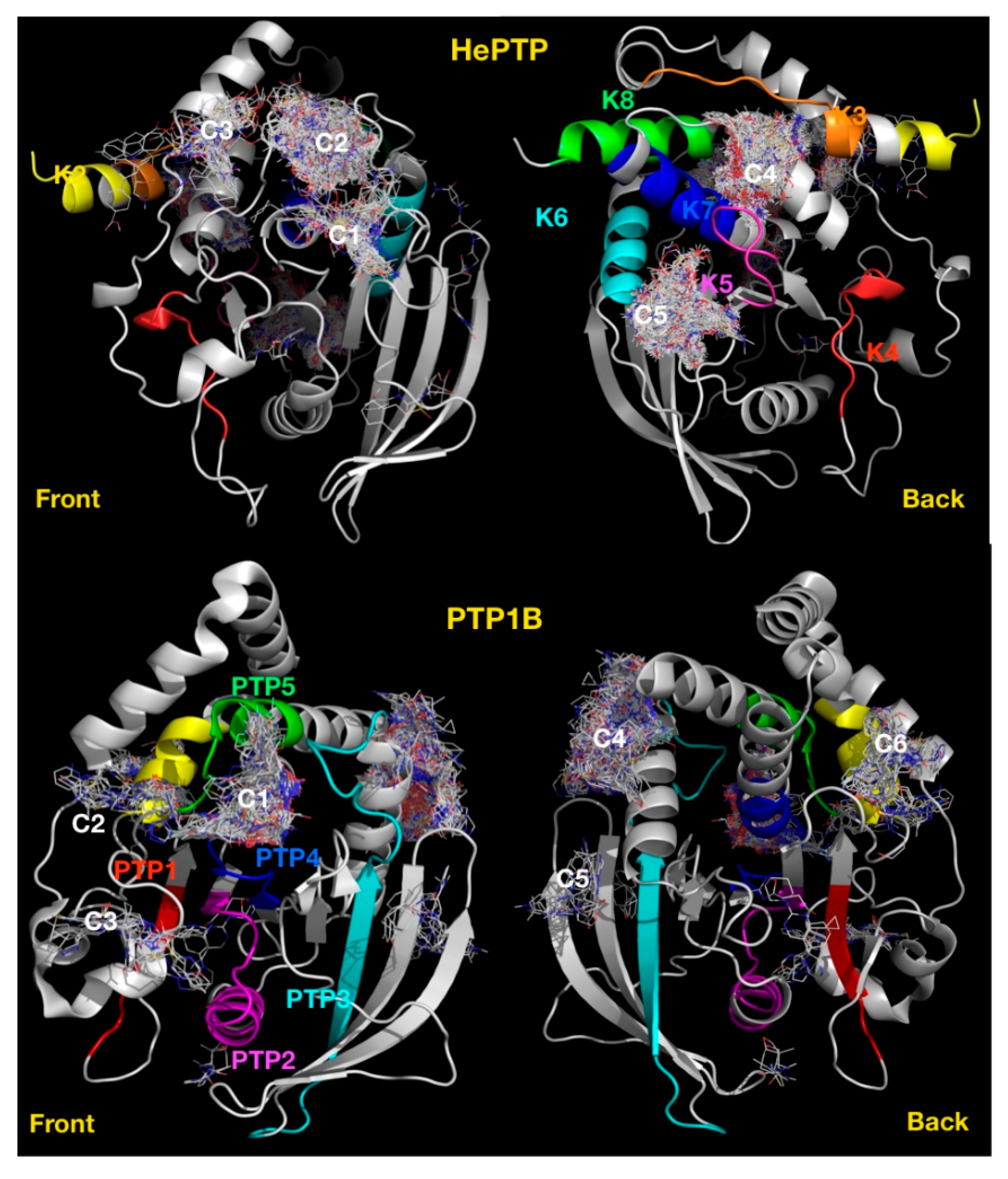
2.1. VSpipe Blind Docking Comparison of the KIM-PTPs
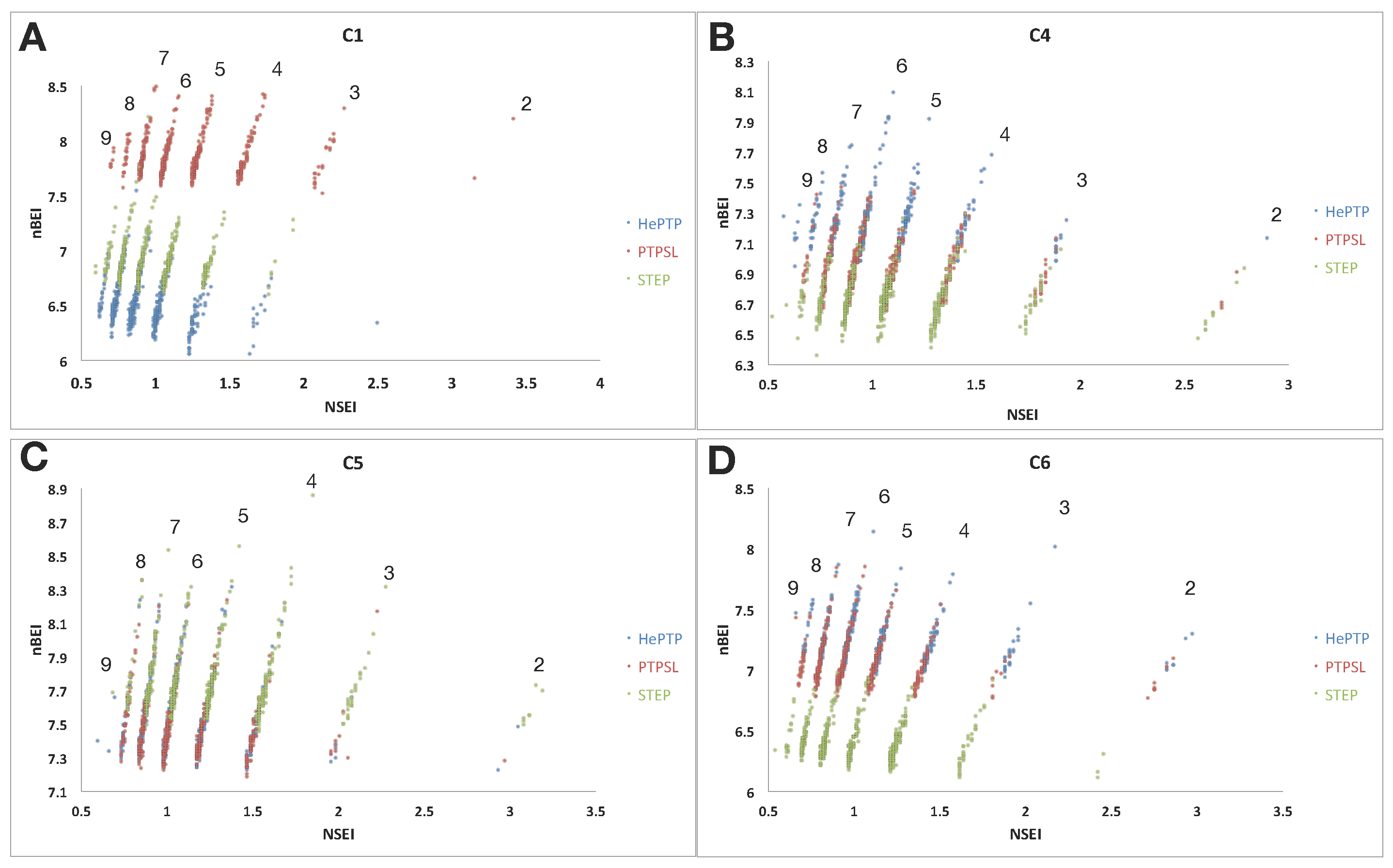
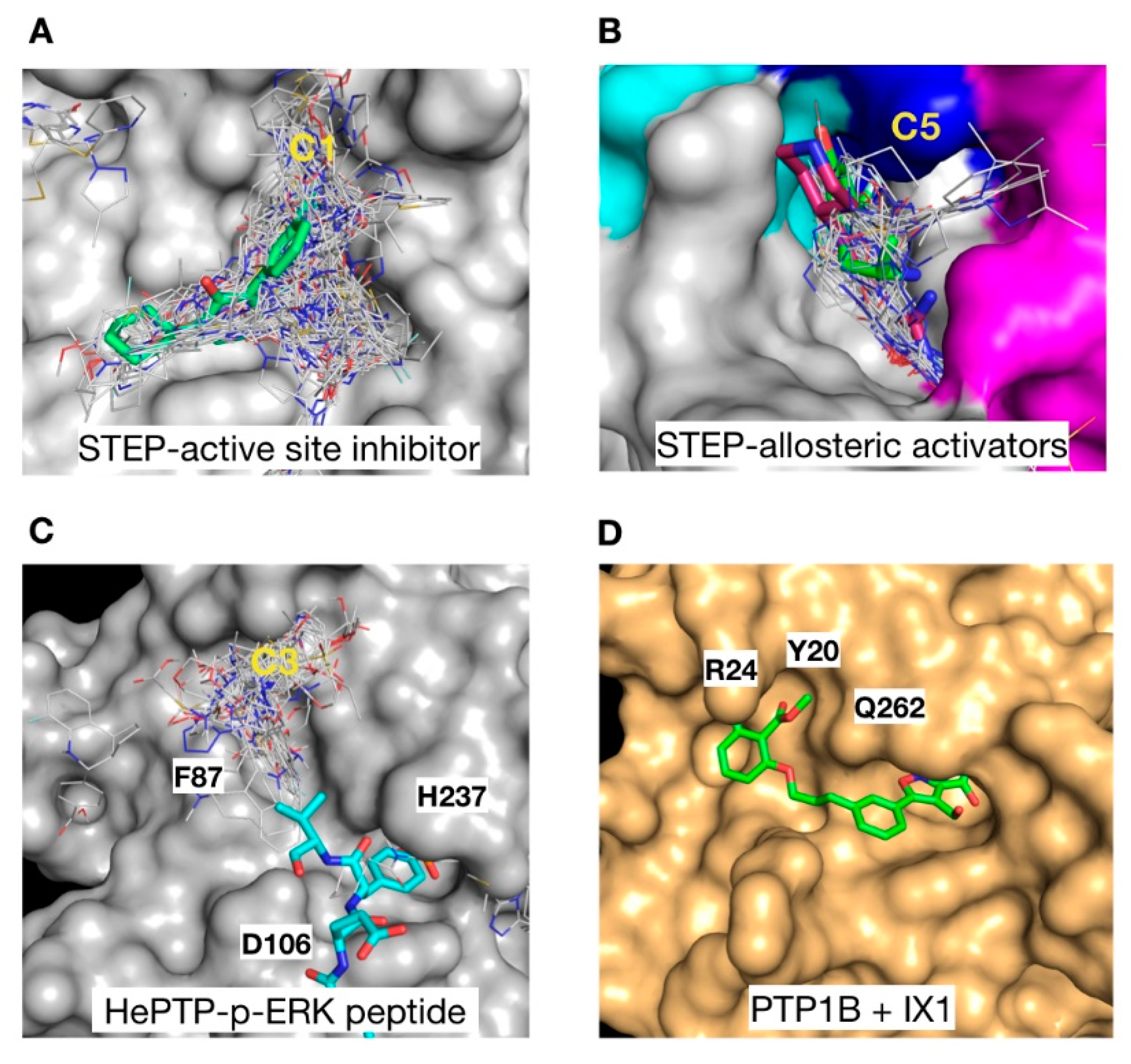
2.2. Ligand Efficiency Analysis of Binders at Clusters 1, 4, 5 and 6
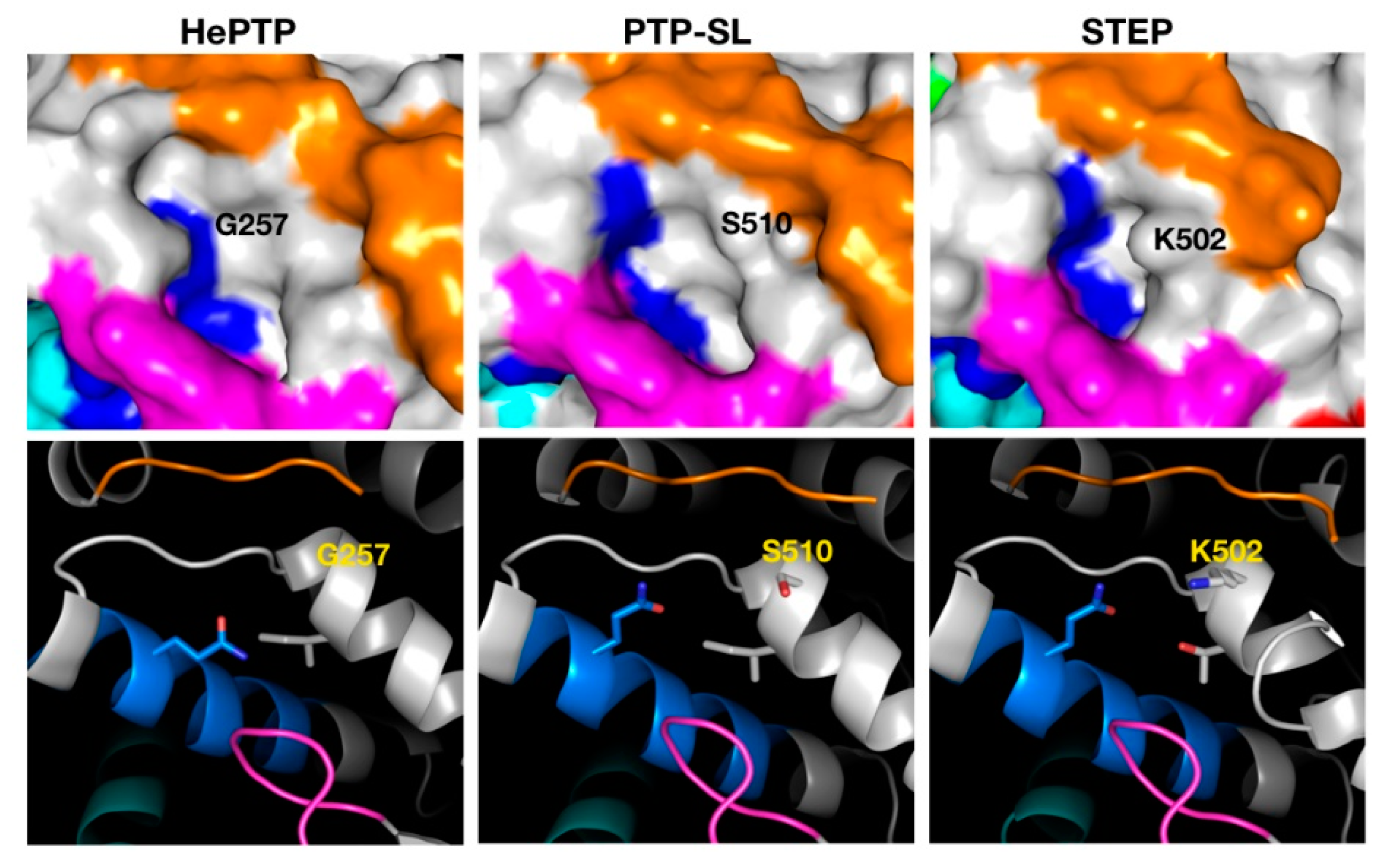
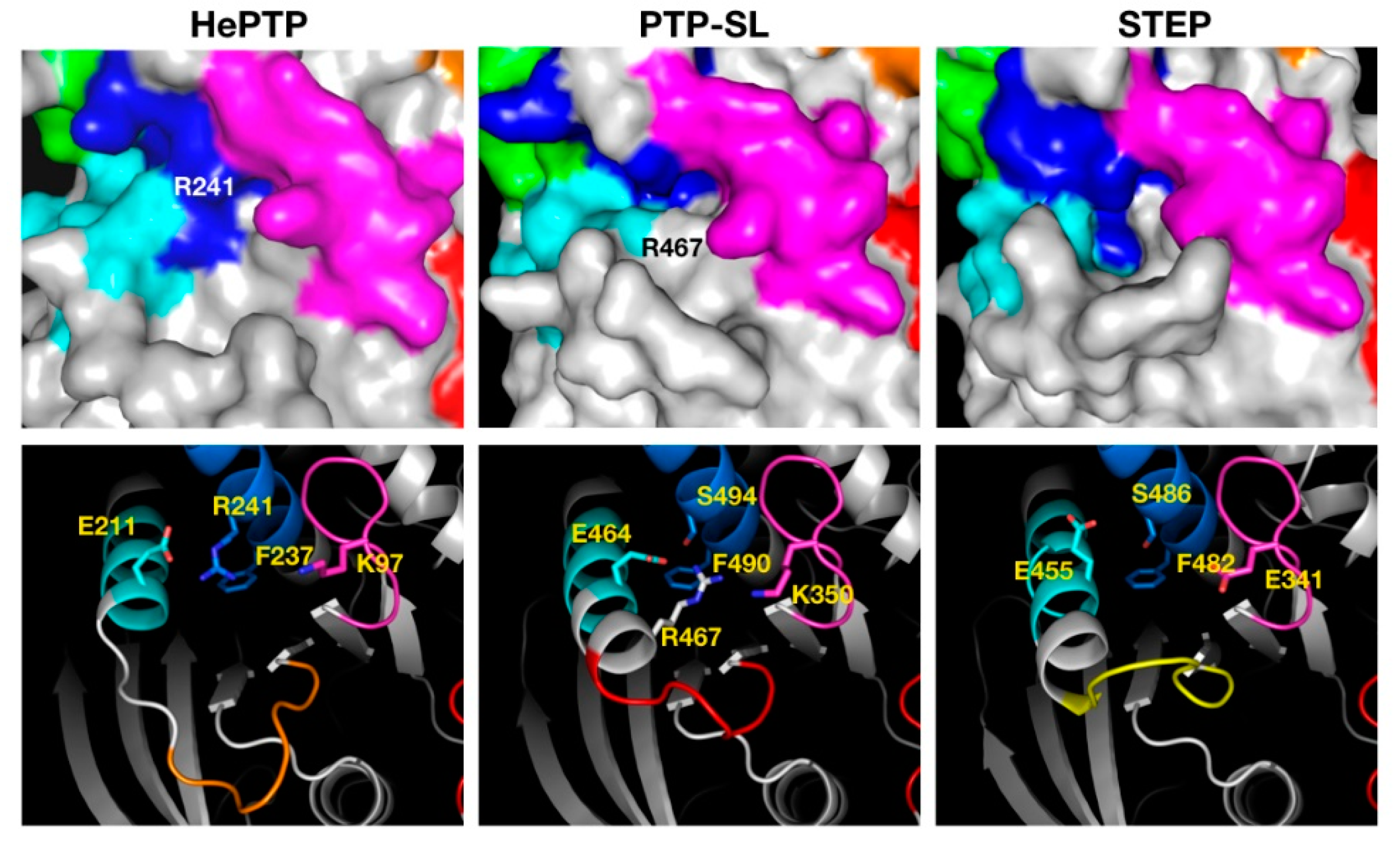
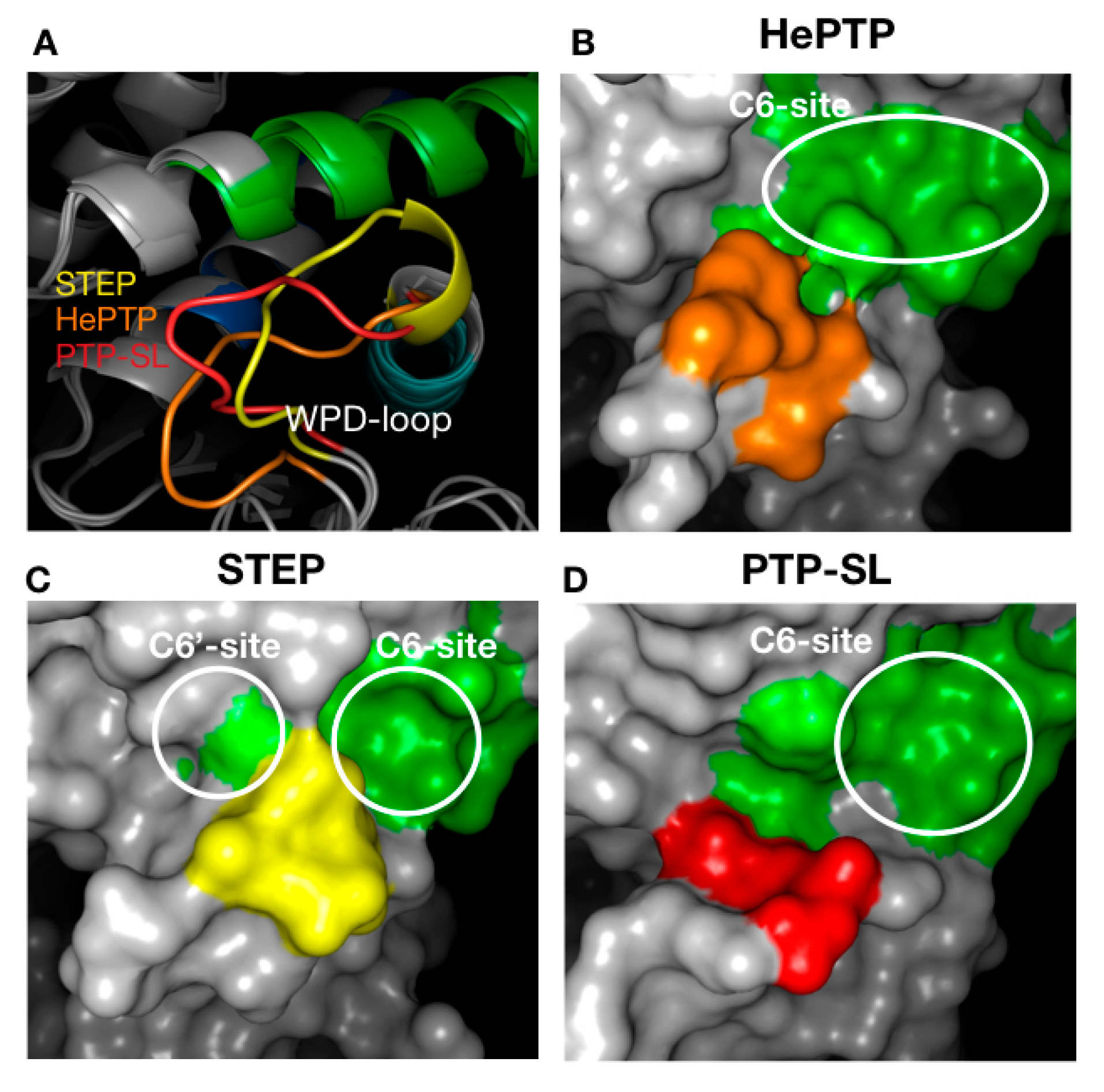
2.3. Structural Analysis of the Cluster Binding Sites
2.4. Blind Docking Clusters Are Subfamily Specific
2.5. Functional Correlation with Cluster Distribution
3. Materials and Methods
3.1. Preparation of the Models
3.2. Virtual Screening with VSpipe
3.3. Ligand Efficiency (LEI) Plots
3.4. Pocket Druggability Predictions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mustelin, T.; Vang, T.; Bottini, N. Protein tyrosine phosphatases and the immune response. Nat. Rev. Immunol. 2005, 5, 43–57. [Google Scholar] [CrossRef]
- Ostman, A.; Hellberg, C.; Bohmer, F.D. Protein-tyrosine phosphatases and cancer. Nat. Rev. Cancer 2006, 6, 307–320. [Google Scholar] [CrossRef]
- Bohmer, F.; Szedlacsek, S.; Tabernero, L.; Ostman, A.; den Hertog, J. Protein tyrosine phosphatase structure-function relationships in regulation and pathogenesis. FEBS J. 2013, 280, 413–431. [Google Scholar] [CrossRef]
- Tonks, N.K. Special issue: Protein phosphatases: From molecules to networks: Introduction. FEBS J. 2013, 280, 323. [Google Scholar] [CrossRef]
- Mazharian, A.; Mori, J.; Wang, Y.J.; Heising, S.; Neel, B.G.; Watson, S.P.; Senis, Y.A. Megakaryocyte-specific deletion of the protein-tyrosine phosphatases Shp1 and Shp2 causes abnormal megakaryocyte development, platelet production, and function. Blood 2013, 121, 4205–4220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butterworth, S.; Overduin, M.; Barr, A.J. Targeting protein tyrosine phosphatase SHP2 for therapeutic intervention. Future Med. Chem. 2014, 6, 1423–1437. [Google Scholar] [CrossRef]
- Pulido, R.; Zuniga, A.; Ullrich, A. PTP-SL and STEP protein tyrosine phosphatases regulate the activation of the extracellular signal-regulated kinases ERK1 and ERK2 by association through a kinase interaction motif. EMBO J. 1998, 17, 7337–7350. [Google Scholar] [CrossRef] [Green Version]
- Zuniga, A.; Torres, J.; Ubeda, J.; Pulido, R. Interaction of mitogen-activated protein kinases with the kinase interaction motif of the tyrosine phosphatase PTP-SL provides substrate specificity and retains ERK2 in the cytoplasm. J. Biol. Chem. 1999, 274, 21900–21907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saxena, M.; Williams, S.; Brockdorff, J.; Gilman, J.; Mustelin, T. Inhibition of T cell signaling by mitogen-activated protein kinase-targeted hematopoietic tyrosine phosphatase (HePTP). J. Biol. Chem. 1999, 274, 11693–11700. [Google Scholar] [CrossRef] [Green Version]
- Paul, S.; Nairn, A.C.; Wang, P.; Lombroso, P.J. NMDA-mediated activation of the tyrosine phosphatase STEP regulates the duration of ERK signaling. Nat. Neurosci. 2003, 6, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Zanke, B.; Squire, J.; Griesser, H.; Henry, M.; Suzuki, H.; Patterson, B.; Minden, M.; Mak, T.W. A hematopoietic protein tyrosine phosphatase (HePTP) gene that is amplified and overexpressed in myeloid malignancies maps to chromosome 1q32.1. Leukemia 1994, 8, 236–244. [Google Scholar] [PubMed]
- Chen, J.; Zhao, X.; Yuan, Y.; Jing, J.J. The expression patterns and the diagnostic/prognostic roles of PTPN family members in digestive tract cancers. Cancer Cell Int. 2020, 20, 238. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Forn, M.; Martinez-Torres, S.; Garcia-Diaz Barriga, G.; Alberch, J.; Mila, M.; Azkona, G.; Perez-Navarro, E. Pharmacogenetic modulation of STEP improves motor and cognitive function in a mouse model of Huntington’s disease. Neurobiol. Dis. 2018, 120, 88–97. [Google Scholar] [CrossRef]
- Chatterjee, M.; Kurup, P.K.; Lundbye, C.J.; Hugger Toft, A.K.; Kwon, J.; Benedict, J.; Kamceva, M.; Banke, T.G.; Lombroso, P.J. STEP inhibition reverses behavioral, electrophysiologic, and synaptic abnormalities in Fmr1 KO mice. Neuropharmacology 2018, 128, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Eswaran, J.; von Kries, J.P.; Marsden, B.; Longman, E.; Debreczeni, J.E.; Ugochukwu, E.; Turnbull, A.; Lee, W.H.; Knapp, S.; Barr, A.J. Crystal structures and inhibitor identification for PTPN5, PTPRR and PTPN7: A family of human MAPK-specific protein tyrosine phosphatases. Biochem. J. 2006, 395, 483–491. [Google Scholar] [CrossRef]
- Bobkova, E.V.; Liu, W.H.; Colayco, S.; Rascon, J.; Vasile, S.; Gasior, C.; Critton, D.A.; Chan, X.; Dahl, R.; Su, Y.; et al. Inhibition of the Hematopoietic Protein Tyrosine Phosphatase by Phenoxyacetic Acids. ACS Med. Chem. Lett. 2011, 2, 113–118. [Google Scholar] [CrossRef]
- Sergienko, E.; Xu, J.; Liu, W.H.; Dahl, R.; Critton, D.A.; Su, Y.; Brown, B.T.; Chan, X.; Yang, L.; Bobkova, E.V.; et al. Inhibition of hematopoietic protein tyrosine phosphatase augments and prolongs ERK1/2 and p38 activation. ACS Chem. Biol. 2012, 7, 367–377. [Google Scholar] [CrossRef] [Green Version]
- Witten, M.R.; Wissler, L.; Snow, M.; Geschwindner, S.; Read, J.A.; Brandon, N.J.; Nairn, A.C.; Lombroso, P.J.; Kack, H.; Ellman, J.A. X-ray Characterization and Structure-Based Optimization of Striatal-Enriched Protein Tyrosine Phosphatase Inhibitors. J. Med. Chem. 2017, 60, 9299–9319. [Google Scholar] [CrossRef]
- Tautermann, C.S.; Binder, F.; Buttner, F.H.; Eickmeier, C.; Fiegen, D.; Gross, U.; Grundl, M.A.; Heilker, R.; Hobson, S.; Hoerer, S.; et al. Allosteric Activation of Striatal-Enriched Protein Tyrosine Phosphatase (STEP, PTPN5) by a Fragment-like Molecule. J. Med. Chem. 2019, 62, 306–316. [Google Scholar] [CrossRef] [PubMed]
- Bialy, L.; Waldmann, H. Inhibitors of protein tyrosine phosphatases: Next-generation drugs? Angew. Chem. Int. Ed. Engl. 2005, 44, 3814–3839. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.Y. Drugging the Undruggable: Therapeutic Potential of Targeting Protein Tyrosine Phosphatases. ACC Chem. Res. 2017, 50, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Stanford, S.M.; Bottini, N. Targeting Tyrosine Phosphatases: Time to End the Stigma. Trends Pharmacol. Sci. 2017, 38, 524–540. [Google Scholar] [CrossRef] [PubMed]
- Salamoun, J.M.; Wipf, P. Allosteric Modulation of Phosphatase Activity May Redefine Therapeutic Value. J. Med. Chem. 2016, 59, 7771–7772. [Google Scholar] [CrossRef]
- Sheriff, S.; Beno, B.R.; Zhai, W.; Kostich, W.A.; McDonnell, P.A.; Kish, K.; Goldfarb, V.; Gao, M.; Kiefer, S.E.; Yanchunas, J.; et al. Small molecule receptor protein tyrosine phosphatase gamma (RPTPgamma) ligands that inhibit phosphatase activity via perturbation of the tryptophan-proline-aspartate (WPD) loop. J. Med. Chem. 2011, 54, 6548–6562. [Google Scholar] [CrossRef] [PubMed]
- Wiesmann, C.; Barr, K.J.; Kung, J.; Zhu, J.; Erlanson, D.A.; Shen, W.; Fahr, B.J.; Zhong, M.; Taylor, L.; Randal, M.; et al. Allosteric inhibition of protein tyrosine phosphatase 1B. Nat. Struct. Mol. Biol. 2004, 11, 730–737. [Google Scholar] [CrossRef]
- Krishnan, N.; Konidaris, K.F.; Gasser, G.; Tonks, N.K. A potent, selective, and orally bioavailable inhibitor of the protein-tyrosine phosphatase PTP1B improves insulin and leptin signaling in animal models. J. Biol. Chem. 2018, 293, 1517–1525. [Google Scholar] [CrossRef] [Green Version]
- Garcia Fortanet, J.; Chen, C.H.; Chen, Y.N.; Chen, Z.; Deng, Z.; Firestone, B.; Fekkes, P.; Fodor, M.; Fortin, P.D.; Fridrich, C.; et al. Allosteric Inhibition of SHP2: Identification of a Potent, Selective, and Orally Efficacious Phosphatase Inhibitor. J. Med. Chem. 2016, 59, 7773–7782. [Google Scholar] [CrossRef]
- Bai, Y.; Yu, Z.H.; Liu, S.; Zhang, L.; Zhang, R.Y.; Zeng, L.F.; Zhang, S.; Zhang, Z.Y. Novel Anticancer Agents Based on Targeting the Trimer Interface of the PRL Phosphatase. Cancer Res. 2016, 76, 4805–4815. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Carretero, S.; Pavlopoulou, N.; Adams, J.; Gilsenan, J.; Tabernero, L. VSpipe, an Integrated Resource for Virtual Screening and Hit Selection: Applications to Protein Tyrosine Phospahatase Inhibition. Molecules 2018, 23, 353. [Google Scholar] [CrossRef] [Green Version]
- Thornton, B.P.; Johns, A.; Al-Shidhani, R.; Alvarez-Carretero, S.; Storer, I.S.R.; Bromley, M.J.; Tabernero, L. Identification of Functional and Druggable Sites in Aspergillus fumigatus Essential Phosphatases by Virtual Screening. Int. J. Mol. Sci. 2019, 20, 4636. [Google Scholar] [CrossRef] [Green Version]
- Hopkins, A.L.; Groom, C.R.; Alex, A. Ligand efficiency: A useful metric for lead selection. Drug Discov. Today 2004, 9, 430–431. [Google Scholar] [CrossRef]
- Abad-Zapatero, C.; Metz, J.T. Ligand efficiency indices as guideposts for drug discovery. Drug Discov. Today 2005, 10, 464–469. [Google Scholar] [CrossRef]
- Nordle, A.K.; Rios, P.; Gaulton, A.; Pulido, R.; Attwood, T.K.; Tabernero, L. Functional assignment of MAPK phosphatase domains. Proteins 2007, 69, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.N.; Mortensen, O.H.; Peters, G.H.; Drake, P.G.; Iversen, L.F.; Olsen, O.H.; Jansen, P.G.; Andersen, H.S.; Tonks, N.K.; Moller, N.P. Structural and evolutionary relationships among protein tyrosine phosphatase domains. Mol. Cell Biol. 2001, 21, 7117–7136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Critton, D.A.; Tautz, L.; Page, R. Visualizing active-site dynamics in single crystals of HePTP: Opening of the WPD loop involves coordinated movement of the E loop. J. Mol. Biol. 2011, 405, 619–629. [Google Scholar] [CrossRef] [Green Version]
- Fiser, A.; Šali, A. Modeller: Generation and refinement of homology-based protein structure models. Methods Enzymol. 2003, 374, 461–491. [Google Scholar] [CrossRef] [PubMed]
- Critton, D.A.; Tortajada, A.; Stetson, G.; Peti, W.; Page, R. Structural basis of substrate recognition by hematopoietic tyrosine phosphatase. Biochemistry 2008, 47, 13336–13345. [Google Scholar] [CrossRef] [Green Version]
- Volkamer, A.; Kuhn, D.; Rippmann, F.; Rarey, M. DoGSiteScorer: A web server for automatic binding site prediction, analysis and druggability assessment. Bioinformatics 2012, 28, 2074–2075. [Google Scholar] [CrossRef] [Green Version]
- Abad-Zapatero, C.; Perisic, O.; Wass, J.; Bento, A.P.; Overington, J.; Al-Lazikani, B.; Johnson, M.E. Ligand efficiency indices for an effective mapping of chemico-biological space: The concept of an atlas-like representation. Drug Discov. Today 2010, 15, 804–811. [Google Scholar] [CrossRef]
- Puius, Y.A.; Zhao, Y.; Sullivan, M.; Lawrence, D.S.; Almo, S.C.; Zhang, Z.-Y. Identification of a second aryl phosphate-binding site in protein-tyrosine phosphatase 1B: A paradigm for inhibitor design. Proc. Natl. Acad. Sci. USA 1997, 94, 13420–13425. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Liu, G.; Xin, Z.; Serby, M.D.; Pei, Z.; Szczepankiewicz, B.G.; Hajduk, P.J.; Abad-Zapatero, C.; Hutchins, C.W.; Lubben, T.H.; et al. Isoxazole carboxylic acids as protein tyrosine phosphatase 1B (PTP1B) inhibitors. Bioorganic Med. Chem. Lett. 2004, 14, 5543–5546. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PTP | Pocket (Site of Cluster) | Druggability Probability | Vol. Hull/Å3 | % Polar Residues |
|---|---|---|---|---|
| HePTP | C1/C2 | 0.83 | 617 | 39 |
| HePTP | C4 | 0.61 | 324 | 39 |
| HePTP | C5 | 0.81 | 522 | 29 |
| HePTP | C6 | 0.27 | 135 | 10 |
| PTP-SL | C1 | 0.61 | 428 | 45 |
| PTP-SL | C4 | 0.55 | 259 | 50 |
| PTP-SL | C5 | 0.74 | 604 | 29 |
| PTP-SL | C6 | 0.37 | 177 | 20 |
| STEP | C1 | 0.51 | 280 | 53 |
| STEP | C4 | 0.24 | 187 | 67 |
| STEP | C5 | 0.72 | 475 | 37 |
| STEP | C6 | 0.34 | 155 | 50 |
| STEP | C6′ | 0.23 | 134 | 50 |
| PTP | Cluster | ΔG Low (kcal/mol) | ΔG High (kcal/mol) | ΔG Average (kcal/mol) |
|---|---|---|---|---|
| HePTP | 1 | −6.7 | −8.3 | −6.9 |
| HePTP | 4 | −7.7 | −9.0 | −7.9 |
| HePTP | 5 | −8.0 | −9.4 | −8.2 |
| HePTP | 6 | −7.7 | −9.1 | −7.9 |
| PTP-SL | 1 | −8.5 | −9.8 | −8.7 |
| PTP-SL | 4 | −7.3 | −8.2 | −7.5 |
| PTP-SL | 5 | −8.0 | −9.3 | −8.2 |
| PTP-SL | 6 | −7.4 | −8.7 | −7.6 |
| STEP | 1 | −7.2 | −9.1 | −7.4 |
| STEP | 4 | −7.0 | −8.0 | −7.2 |
| STEP | 5 | −8.3 | −10.1 | −8.5 |
| STEP | 6 | −6.6 | −7.5 | −6.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adams, J.; Thornton, B.P.; Tabernero, L. A New Paradigm for KIM-PTP Drug Discovery: Identification of Allosteric Sites with Potential for Selective Inhibition Using Virtual Screening and LEI Analysis. Int. J. Mol. Sci. 2021, 22, 12206. https://doi.org/10.3390/ijms222212206
Adams J, Thornton BP, Tabernero L. A New Paradigm for KIM-PTP Drug Discovery: Identification of Allosteric Sites with Potential for Selective Inhibition Using Virtual Screening and LEI Analysis. International Journal of Molecular Sciences. 2021; 22(22):12206. https://doi.org/10.3390/ijms222212206
Chicago/Turabian StyleAdams, James, Benjamin P. Thornton, and Lydia Tabernero. 2021. "A New Paradigm for KIM-PTP Drug Discovery: Identification of Allosteric Sites with Potential for Selective Inhibition Using Virtual Screening and LEI Analysis" International Journal of Molecular Sciences 22, no. 22: 12206. https://doi.org/10.3390/ijms222212206