
MicroRNA 132-3p Is Upregulated in Laron Syndrome Patients and Controls Longevity Gene Expression





Abstract
:1. Introduction
2. Results
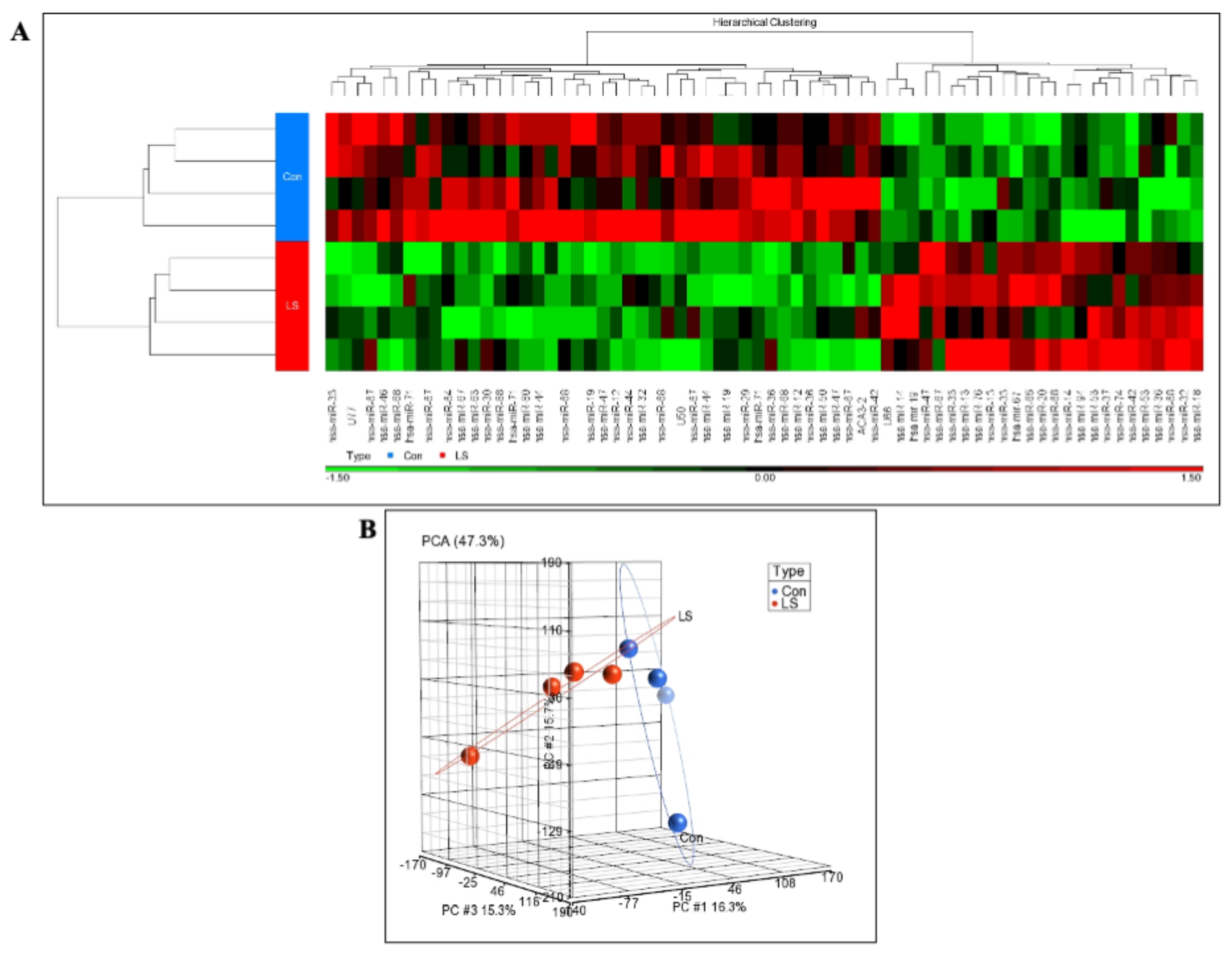
2.1. Genome-Wide Analysis of Differentially Expressed miRs in LS
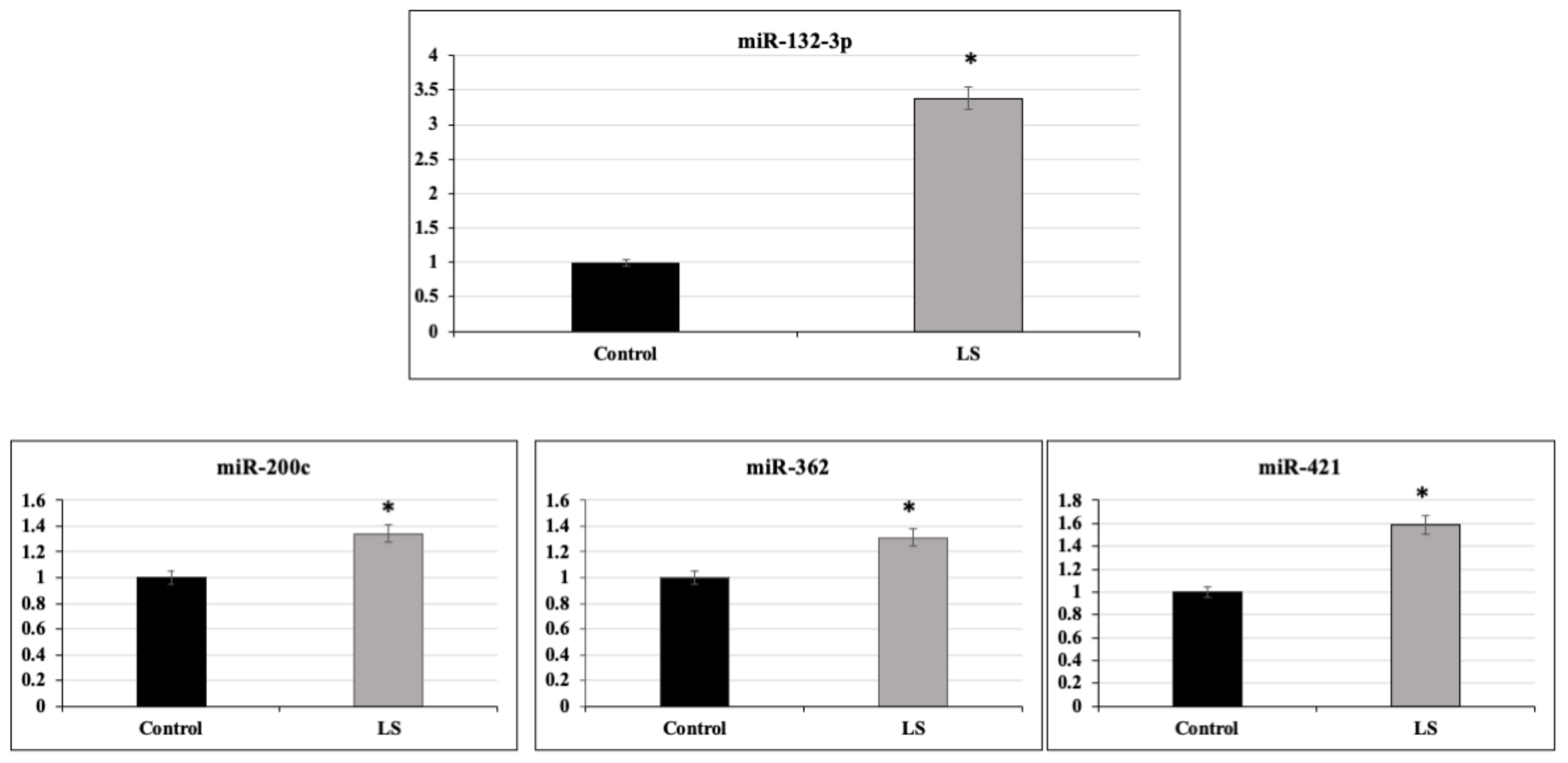
2.2. Identification of miR-132-3p as a Differentially Expressed miR in LS
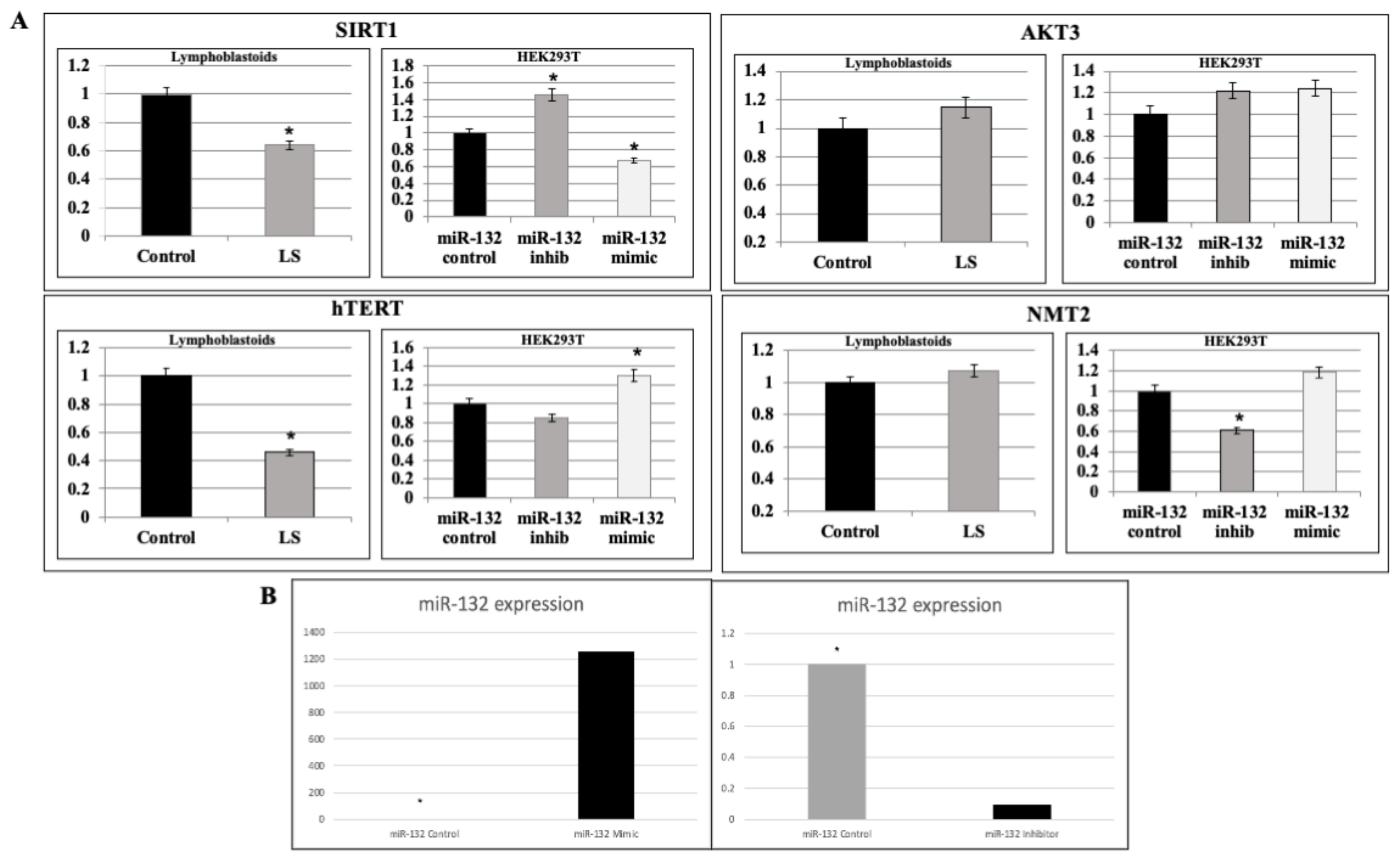
2.3. Analysis of miR-132-3p Target mRNAs Expression in LS Cells
2.4. Validation of miR-132-3p Target Genes by miR Manipulation
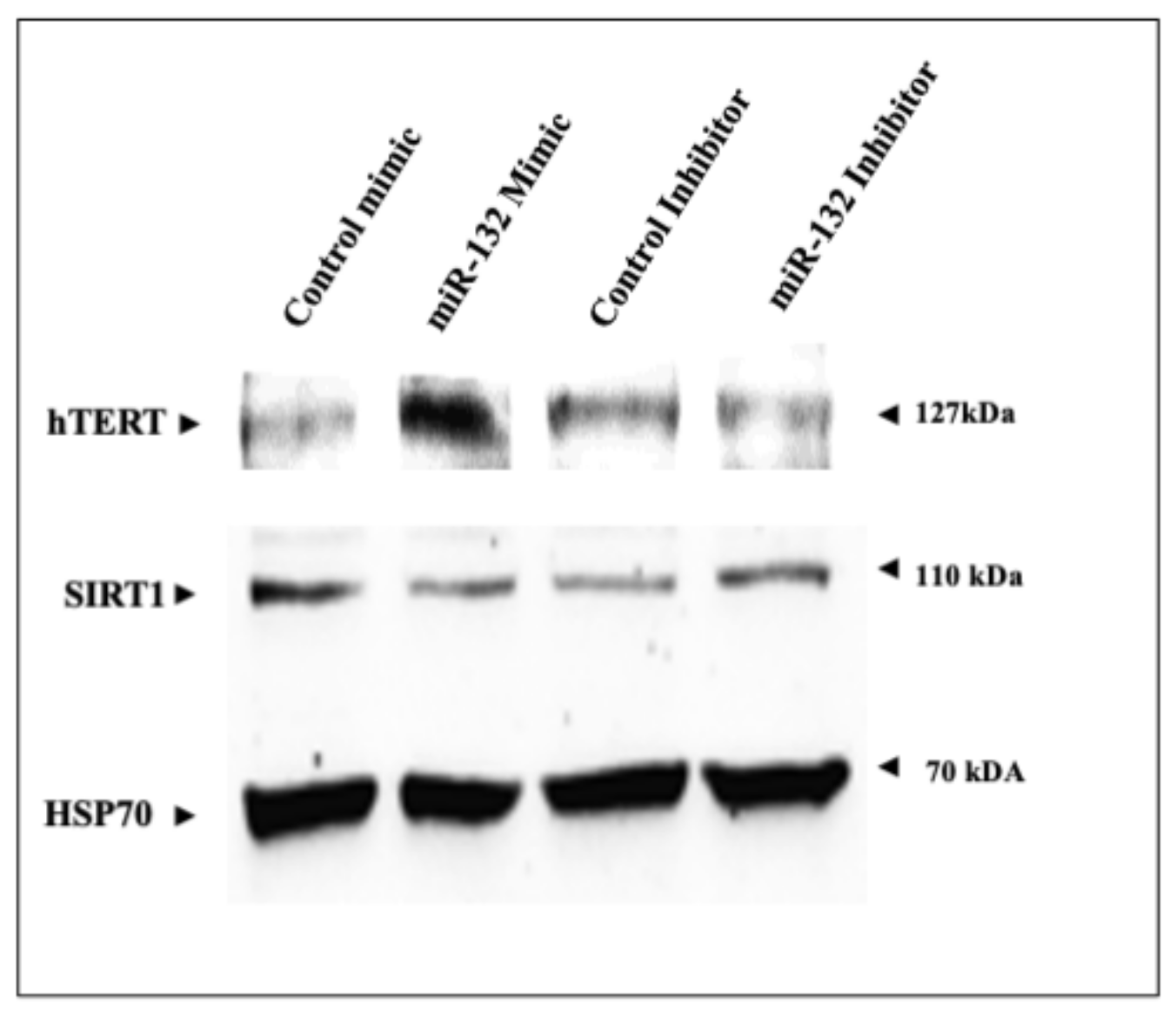
2.5. Regulation of SIRT1 and hTERT Protein Levels by miR-132-3p Mimics and Inhibitor
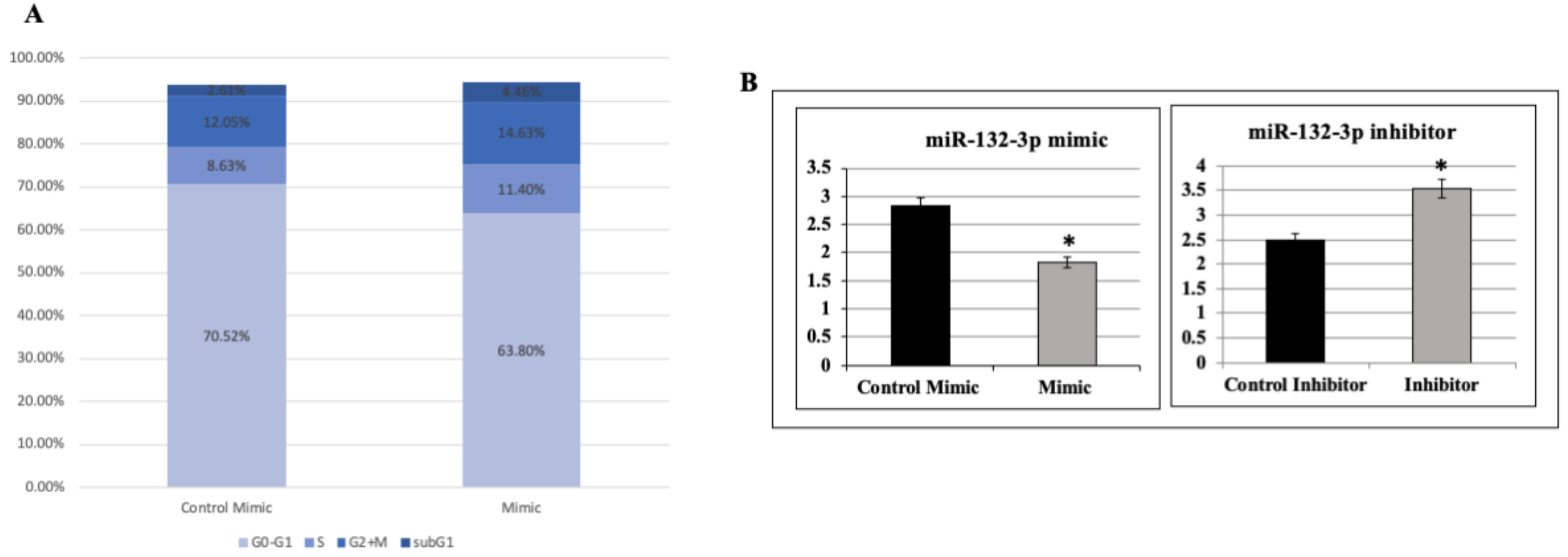
2.6. Effect of miR-132-3p on Cell Cycle Progression
2.7. Effect of miR-132-3p on Cell Proliferation
2.8. Correlations between IGF1, miR-132-3p and SIRT1
3. Discussion
4. Materials and Methods
4.1. Cell Cultures
4.2. Affymetrix GeneChip MicroRNA Analyses of LS-Derived Cells
4.3. Bioinformatics Analysis
4.4. Quantitative Real Time-Polymerase Chain Reaction for MicroRNA Expression Analysis
4.5. qRT-PCR for Gene Expression Analysis
4.6. MiR-132-3p Inhibitor and Mimics Experiments
4.7. Western Blot Analysis
4.8. Cell Cycle Analysis
4.9. Cell Proliferation Assays
4.10. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yakar, S.; Adamo, M.L. Insulin-like growth factor 1 physiology: Lessons from mouse models. Endocrinol. Metab. Clin. N. Am. 2012, 41, 231–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LeRoith, D.; Yakar, S. Mechanisms of disease: Metabolic effects of growth hormone and insulin-like growth factor-1. Nat. Clin. Pract. Endocrinol. Metab. 2007, 3, 302–310. [Google Scholar] [CrossRef] [PubMed]
- Bondy, C.A.; Werner, H.; Roberts, C.T., Jr.; LeRoith, D. Cellular pattern of insulin-like growth factor I (IGF-I) and type I IGF receptor gene expression in early organogenesis: Comparison with IGF-II gene expression. Mol. Endocrinol. 1990, 4, 1386–1398. [Google Scholar] [CrossRef]
- Werner, H.; Woloschak, M.; Adamo, M.; Shen-Orr, Z.; Roberts, C.T., Jr.; LeRoith, D. Developmental regulation of the rat insulin-like growth factor I receptor gene. Proc. Natl. Acad. Sci. USA 1989, 86, 7451–7455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yakar, S.; Werner, H.; Rosen, C.J. Insulin-like growth factors: Actions on the skeleton. J. Mol. Endocrinol 2018, 61, T115–T137. [Google Scholar] [CrossRef]
- Salmon, W.D.; Daughaday, W.H. A hormonally controlled serum factor which stimulates sulfate incorporation by cartilage in vitro. J. Lab. Clin. Med. 1957, 49, 825–836. [Google Scholar] [PubMed]
- Rosenfeld, R.G. Insulin-like growth factors and the basis of growth. N. Engl. J. Med. 2003, 349, 2184–2186. [Google Scholar] [CrossRef] [PubMed]
- Domené, S.; Domené, H.M. Genetic mutations in the GH/IGF axis. Pediatr. Endocrinol. Rev. 2018, 16, 39–62. [Google Scholar] [PubMed]
- Renehan, A.G.; Frystyk, J.; Flyvbjerg, A. Obesity and cancer risk: The role of the insulin-IGF axis. Trends Endocrinol. Metab. 2006, 17, 328–336. [Google Scholar] [CrossRef]
- Chan, J.M.; Stampfer, M.J.; Giovannucci, E.; Gann, P.H.; Ma, J.; Wilkinson, P.; Hennekens, C.H.; Pollak, M. Plasma insulin-like growth factor-I and prostate cancer risk: A prospective study. Science 1998, 279, 563–566. [Google Scholar] [CrossRef]
- Hankinson, S.E.; Willett, W.C.; Colditz, G.A.; Hunter, D.J.; Michaud, D.S.; Deroo, B.; Rosner, B.; Speizer, F.E.; Pollak, M. Circulating concentrations of insulin-like growth factor-I and risk of breast cancer. Lancet 1998, 351, 1393–1396. [Google Scholar] [CrossRef]
- Kaaks, R.; Lukanova, A. Energy balance and cancer: The role of insulin and insulin-like growth factors. Proc. Nutr. Soc. 2001, 60, 91–106. [Google Scholar] [CrossRef] [Green Version]
- Werner, H.; Bruchim, I. IGF-1 and BRCA1 signalling pathways in familial cancer. Lancet Oncol. 2012, 13, e537–e544. [Google Scholar] [CrossRef]
- Sarfstein, R.; Maor, S.; Reizner, N.; Abramovitch, S.; Werner, H. Transcriptional regulation of the insulin-like growth factor-1 receptor in breast cancer. Mol. Cell. Endocrinol. 2006, 252, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Pollak, M. The insulin and insulin-like growth factor receptor family in neoplasia: An update. Nat. Rev. Cancer 2012, 12, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Werner, H. Tumor suppressors govern insulin-like growth factor signaling pathways: Implications in metabolism and cancer. Oncogene 2012, 31, 2703–2714. [Google Scholar] [CrossRef] [PubMed]
- Baserga, R. The contradictions of the insulin-like growth factor 1 receptor. Oncogene 2000, 19, 5574–5581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holly, J.M.; Perks, C.M. Insulin-like growth factor physiology: What we have learned from human studies. Endocrinol. Metab. Clin. North Am. 2012, 41, 249–263. [Google Scholar] [CrossRef] [PubMed]
- LeRoith, D. Clinical relevance of systemic and local IGF-I: Lessons from animal models. Pediatr. Endocrinol. Rev. 2008, 5, 739–743. [Google Scholar]
- Laron, Z. Extensive personal experience. Laron syndrome (primary growth hormone resistance or insensitivity): The personal experience 1958-2003. J. Clin. Endocrinol. Metab. 2004, 89, 1031–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laron, Z.; Kopchik, J.J. Laron Syndrome—From Man to Mouse; Springer: Berlin/Heidelberg, Germany, 2011. [Google Scholar]
- Laron, Z.; Pertzelan, A.; Mannheimer, S. Genetic pituitary dwarfism with high serum concentration of growth hormone-a new inborn error of metabolism? Isr. J. Med. Sci. 1966, 2, 152–155. [Google Scholar] [PubMed]
- Eshet, R.; Werner, H.; Klinger, B.; Silbergeld, A.; Laron, Z.; LeRoith, D.; Roberts, C.T., Jr. Up-regulation of insulin-like growth factor-I (IGF-I) receptor gene expression in patients with reduced serum IGF-I levels. J. Mol. Endocrinol. 1993, 10, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Shevah, O.; Laron, Z. Genetic analysis of the pedigrees and molecular defects of the GH-receptor gene in the Israeli cohort of patients with Laron syndrome. Pediatr. Endocrinol. Rev. 2006, 3 (Suppl. S3), 489–497. [Google Scholar] [PubMed]
- Amselem, S.; Duquesnoy, P.; Attree, O.; Novelli, G.; Bousnina, S.; Postel-Vinay, M.C.; Goossens, M. Laron dwarfism and mutations of the growth hormone-receptor gene. N. Engl. J. Med. 1989, 321, 989–995. [Google Scholar] [CrossRef]
- Godowski, P.J.; Leung, D.W.; Meacham, L.R.; Galgani, J.P.; Hellmiss, R.; Keret, R.; Rotwein, P.S.; Parks, J.S.; Laron, Z.; Wood, W.I. Characterization of the human growth hormone receptor gene and demonstration of a partial gene deletion in two patients with Laron-type dwarfism. Proc. Natl. Acad. Sci. USA 1989, 86, 8083–8087. [Google Scholar] [CrossRef] [Green Version]
- Shevah, O.; Laron, Z. Patients with congenital deficiency of IGF-I seem protected from the development of malignancies: A preliminary report. Growth Horm. IGF Res. 2007, 17, 54–57. [Google Scholar] [CrossRef]
- Steuerman, R.; Shevah, O.; Laron, Z. Congenital IGF1 deficiency tends to confer protection against post-natal development of malignancies. Eur. J. Endocrinol. 2011, 164, 485–489. [Google Scholar] [CrossRef] [Green Version]
- Guevara-Aguirre, J.; Balasubramanian, P.; Guevara-Aguirre, M.; Wei, M.; Madia, F.; Cheng, C.W.; Hwang, D.; Martin-Montalvo, A.; Saavedra, J.; Ingles, S.; et al. Growth hormone receptor deficiency is associated with a major reduction in pro-aging signaling, cancer, and diabetes in humans. Sci. Transl. Med. 2011, 3, 70ra13. [Google Scholar] [CrossRef] [Green Version]
- Vitale, G.; Pellegrino, G.; Vollery, M.; Hofland, L.J. Role of IGF-1 system in the modulation of longevity: Controversies and new insights from a centenarian’s perspective. Front. Endocrinol. 2019, 10, 27. [Google Scholar] [CrossRef] [Green Version]
- Kimura, K.D.; Tissenbaum, H.A.; Liu, Y.; Ruvkun, G. daf-2, an insulin receptor-like gene that regulates longevity and diapause in Caenorhabditis elegans. Science 1997, 277, 942–946. [Google Scholar] [CrossRef]
- Tazearslan, C.; Huang, J.; Barzilai, N.; Suh, Y. Impaired IGF1R signaling in cells expressing longevity-associated human IGF1R alleles. Aging Cell 2011, 10, 551–554. [Google Scholar] [CrossRef] [Green Version]
- Coschigano, K.T.; Clemmons, D.; Bellush, L.L.; Kopchick, J.J. Assessment of growth parameters and life span of GHR/BP gene-disrupted mice. Endocrinology 2000, 141, 2608–2613. [Google Scholar] [CrossRef] [PubMed]
- Basu, R.; Qian, Y.; Kopchick, J.J. Lessons from growth hormone receptor gene-disrupted mice: Are there benefits of endocrine defects? Eur. J. Endocrinol. 2018, 178, R155–R181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carter, C.S.; Ramsey, M.M.; Sonntag, W.E. A critical analysis of the role of growth hormone and IGF-1 in aging and lifespan. Trends Genet. 2002, 18, 295–301. [Google Scholar] [CrossRef]
- Laron, Z.; Kauli, R.; Lapkina, L.; Werner, H. IGF-I deficiency, longevity and cancer protection of patients with Laron syndrome. Mutat. Res. Rev. Mutat. Res. 2017, 772, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Bartke, A. Can growth hormone (GH) accelerate aging? Evidence from GH-transgenic mice. Neuroendocrinology 2003, 78, 210–216. [Google Scholar] [CrossRef]
- Lapkina-Gendler, L.; Rotem, I.; Pasmanik-Chor, M.; Gurwitz, D.; Sarfstein, R.; Laron, Z.; Werner, H. Identification of signaling pathways associated with cancer protection in Laron syndrome. Endocr. Relat. Cancer 2016, 23, 399–410. [Google Scholar] [CrossRef] [Green Version]
- Nagaraj, K.; Lapkina-Gendler, L.; Sarfstein, R.; Gurwitz, D.; Pasmanik-Chor, M.; Laron, Z.; Yakar, S.; Werner, H. Identification of thioredoxin-interacting protein (TXNIP) as a downstream target for IGF1 action. Proc. Natl. Acad. Sci. USA 2018, 115, 1045–1050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Achlaug, L.; Sarfstein, R.; Nagaraj, K.; Lapkina-Gendler, L.; Bruchim, I.; Dixit, M.; Laron, Z.; Yakar, S.; Werner, H. Identification of ZYG11A as a candidate IGF1-dependent proto-oncogene in endometrial cancer. Oncotarget 2019, 10, 4437–4448. [Google Scholar] [CrossRef]
- Sarfstein, R.; Lapkina-Gendler, L.; Nagaraj, K.; Laron, Z.; Werner, H. Identification of nephronectin as a new target for IGF1 action. Eur. J. Cancer 2020, 141, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Shibel, R.; Sarfstein, R.; Nagaraj, K.; Lapkina-Gendler, L.; Laron, Z.; Dixit, M.; Yakar, S.; Werner, H. The olfactory receptor gene product, OR5H2, modulates endometrial cancer cells proliferation via interaction with the IGF1 signaling pathway. Cells 2021, 10, 1483. [Google Scholar] [CrossRef] [PubMed]
- Werner, H.; Sarfstein, R.; Nagaraj, K.; Laron, Z. Laron syndrome research paves the way for new insights in oncological investigation. Cells 2020, 9, 2446. [Google Scholar] [CrossRef]
- Guarnieri, D.J.; DiLeone, R.J. MicroRNAs: A new class of gene regulators. Ann. Med. 2008, 40, 197–208. [Google Scholar] [CrossRef]
- Zalts, H.; Shomron, N. The impact of microRNAs on endocrinology. Pediatr. Endocrinol. Rev. 2011, 8, 354–362. [Google Scholar]
- Hobert, O. Gene regulation by transcription factors and microRNAs. Science 2008, 319, 1785–1786. [Google Scholar] [CrossRef] [PubMed]
- Hornstein, E.; Shomron, N. Canalization of development by microRNAs. Nat. Genet. 2006, 38, S20–S24. [Google Scholar] [CrossRef]
- Nana-Sinkam, S.P.; Croce, C.M. MicroRNAs as therapeutic targets in cancer. Transl. Res. 2011, 157, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Liu, X.; Chen, Z.; Jin, Y.; Heidbreder, C.E.; Kolokythas, A.; Wang, A.; Dai, Y.; Zhou, X. MicroRNA-7 targets IGF1R (insulin-like growth factor 1 receptor) in tongue squamous cell carcinoma cells. Biochem J. 2010, 432, 199–205. [Google Scholar] [CrossRef]
- Shu, S.; Liu, X.; Xu, M.; Gao, X.; Chen, S.; Zhang, L.; Li, R. MicroRNA-320a acts as a tumor suppressor in endometrial carcinoma by targeting IGF-1R. Int. J. Mol. Med. 2019, 43, 1505–1512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKinsey, E.L.; Parrish, J.K.; Irwin, A.E.; Niemeyer, B.F.; Kern, H.B.; Birks, D.K.; Jedlicka, P. A novel oncogenic mechanism in Ewing sarcoma involving IGF pathway targeting by EWS/Fli1-regulated microRNAs. Oncogene 2011, 30, 4910–4920. [Google Scholar] [CrossRef] [Green Version]
- Dobre, M.; Herlea, V.; Vladut, C.; Ciocirlan, M.; Balaban, V.D.; Constantinescu, G.; Diculescu, M.; Milanesi, E. Dysregulation of miRNAs targeting the IGF-1R pathway in pancreatic ductal adenocarcinoma. Cells 2021, 10, 1856. [Google Scholar] [CrossRef]
- Catellani, C.; Ravegnini, G.; Sartori, C.; Angelini, S.; Street, M.E. GH and IGF system: The regulatory role of miRNAs and lncRNAs in cancer. Front. Endocrinol. 2021, 12, 701246. [Google Scholar] [CrossRef]
- Cirillo, F.; Catellani, C.; Lazzeroni, P.; Sartori, C.; Street, M.E. The role of microRNAs in influencing body growth and development. Horm. Res. Paediatr. 2020, 93, 7–15. [Google Scholar] [CrossRef]
- Anand, S.; Majeti, B.K.; Acevedo, L.M.; Murphy, E.A.; Mukthavaram, R.; Scheppke, L.; Huang, M.; Shields, D.J.; Lindquist, J.N.; Lapinski, P.E.; et al. MicroRNA-132-mediated loss of p120RasGAP activates the endothelium to facilitate pathological angiogenesis. Nat. Med. 2010, 16, 909–914. [Google Scholar] [CrossRef]
- Li, D.; Wang, A.; Liu, X.; Meisgen, F.; Grünler, J.; Botusan, I.R.; Narayanan, S.; Erikci, E.; Li, X.; Blomqvist, L.; et al. MicroRNA-132 enhances transition from inflammation to proliferation during wound healing. J. Clin. Investig. 2015, 125, 3008–3026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hadar, A.; Milanesi, E.; Walczak, M.; Puzianowska-Kuźnicka, M.; Kuźnicki, J.; Squassina, A.; Niola, P.; Chillotti, C.; Attems, J.; Gozes, I.; et al. SIRT1, miR-132 and miR-212 link human longevity to Alzheimer’s Disease. Sci. Rep. 2018, 8, 8465. [Google Scholar] [CrossRef] [PubMed]
- Deelen, J.; Uh, H.W.; Monajemi, R.; van Heemst, D.; Thijssen, P.E.; Böhringer, S.; van den Akker, E.B.; de Craen, A.J.; Rivadeneira, F.; Uitterlinden, A.G.; et al. Gene set analysis of GWAS data for human longevity highlights the relevance of the insulin/IGF-1 signaling and telomere maintenance pathways. Age 2013, 35, 235–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackey, J.R.; Lai, J.; Chauhan, U.; Beauchamp, E.; Dong, W.F.; Glubrecht, D.; Sim, Y.W.; Ghosh, S.; Bigras, G.; Lai, R.; et al. N-myristoyltransferase proteins in breast cancer: Prognostic relevance and validation as a new drug target. Breast Cancer Res. Treat. 2021, 186, 79–87. [Google Scholar] [CrossRef]
- Werner, H.; Laron, Z. Role of the GH-IGF1 system in progression of cancer. Mol. Cell. Endocrinol. 2020, 518, 111003. [Google Scholar] [CrossRef]
- Klammt, J.; Pfaffle, R.; Werner, H.; Kiess, W. IGF signaling defects as causes of growth failure and IUGR. Trends Endocrinol. Metab. 2008, 19, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Laron, Z. Lessons from 50 years of study of Laron syndrome. Endocr. Pract. 2015, 21, 1395–1402. [Google Scholar] [CrossRef] [PubMed]
- Werner, H.; Lapkina-Gendler, L.; Laron, Z. Fifty years on: New lessons from Laron syndrome. Isr. Med. Assoc. J. 2017, 19, 6–7. [Google Scholar]
- Werner, H.; Sarfstein, R. Transcriptional and epigenetic control of IGF1R gene expression: Implications in metabolism and cancer. Growth Horm. IGF Res. 2014, 24, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Baserga, R. The decline and fall of the IGF-I receptor. J. Cell. Physiol. 2013, 28, 675–679. [Google Scholar] [CrossRef] [PubMed]
- Kenyon, C. A conserved regulatory system for aging. Cell 2001, 105, 165–168. [Google Scholar] [CrossRef] [Green Version]
- Juul, A. Serum levels of insulin-like growth factor I and its binding proteins in health and disease. Growth Horm IGF Res. 2003, 13, 113–170. [Google Scholar] [CrossRef]
- Anisimov, V.N.; Bartke, A. The key role of growth hormone-insulin-IGF-1 signaling in aging and cancer. Crit. Rev. Oncol. Hematol. 2013, 87, 201–223. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Lu, C.; Qian, Y.; Li, H.; Tan, Y.; Cai, L.; Weng, H. Urinary miR-21 as a potential biomarker of hypertensive kidney injury and fibrosis. Sci. Rep. 2017, 7, 17737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akgör, U.; Ayaz, L.; Çayan, F. Expression levels of maternal plasma microRNAs in preeclamptic pregnancies. J. Obstet. Gynecol. 2021, 41, 910–914. [Google Scholar] [CrossRef] [PubMed]
- Kariba, Y.; Yoshizawa, T.; Sato, Y.; Tsuyama, T.; Araki, E.; Yamagata, K. Brown adipocyte-derived exosomal miR-132-3p suppress hepatic Srebf1 expression and thereby attenuate expression of lipogenic genes. Biochem. Biophys. Res. Commun. 2020, 530, 500–507. [Google Scholar] [CrossRef]
- Qian, Y.; Song, J.; Ouyang, Y.; Han, Q.; Chen, W.; Zhao, X.; Xie, Y.; Chen, Y.; Yuan, W.; Fan, C. Advances in roles of miR-132 in the nervous system. Front. Pharmacol. 2017, 8, 770. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Rapp, J.; Rainone, S.; Goupil, C.; Dorval, V.; Smith, P.Y.; Saint-Pierre, M.; Vallée, M.; Planel, E.; Droit, A.; Calon, F.; et al. microRNA-132/212 deficiency enhances Aβ production and senile plaque deposition in Alzheimer’s disease triple transgenic mice. Sci. Rep. 2016, 6, 30953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, B.L. Sirt1 and the mitochondria. Mol. Cells 2016, 39, 87–95. [Google Scholar] [PubMed] [Green Version]
- Leite, J.A.; Ghirotto, B.; Targhetta, V.P.; de Lima, J.; Câmara, N.O.S. Sirtuins as pharmacological targets in neurodegenerative and neuropsychiatric disorders. Br. J. Pharmacol. 2021. Online ahead of print. [Google Scholar] [CrossRef]
- Strum, J.C.; Johnson, J.H.; Ward, J.; Xie, H.; Feild, J.; Hester, A.; Alford, A.; Waters, K.M. MicroRNA 132 regulates nutritional stress-induced chemokine production through repression of SirT1. Mol. Endocrinol. 2009, 23, 1876–1884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, F.; Wu, A. The protective mechanism of SIRT1 in the regulation of mitochondrial biogenesis and mitochondrial autophagy in Alzheimer’s Disease. J. Alzheimers Dis. 2021, 82, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.; Mittal, A.K.; Kalonia, H.; Madan, S.; Ghosh, S.; Sinha, J.K.; Rajput, S.K. SIRT1 promotes neuronal fortification in neurodegenerative diseases through attenuation of pathological hallmarks and enhancement of cellular lifespan. Curr. Neuropharmacol. 2021, 19, 1019–1037. [Google Scholar] [CrossRef] [PubMed]
- Tran, D.; Bergholz, J.; Zhang, H.; He, H.; Wang, Y.; Zhang, Y.; Li, Q.; Kirkland, J.L.; Xiao, Z.X. Insulin-like growth factor-1 regulates the SIRT1-p53 pathway in cellular senescence. Aging Cell 2014, 13, 669–678. [Google Scholar] [CrossRef] [PubMed]
- Burgers, A.M.G.; Biermasz, N.R.; Schoones, J.W.; Pereira, A.M.; Renehan, A.G.; Zwahlen, M.; Egger, M.; Dekkers, O.M. Meta-analysis and dose-response metaregression: Circulating insulin-like growth factor I (IGF-I) and mortality. J. Clin. Endocrinol. Metab. 2011, 96, 2912–2920. [Google Scholar] [CrossRef]
- Friedrich, N.; Haring, R.; Nauck, M.; Lüdemann, J.; Rosskopf, D.; Spilcke-Liss, E.; Felix, S.B.; Dörr, M.; Brabant, G.; Völzke, H.; et al. Mortality and serum insulin-like growth factor (IGF)-I and IGF binding protein 3 concentrations. J. Clin. Endocrinol. Metab. 2009, 94, 1732–1739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milman, S.; Huffman, D.M.; Barzilai, N. The somatotropic axis in human aging: Framework for the current state of knowledge and future research. Cell Metab. 2016, 23, 980–989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.B.; Ye, K.; Barzilai, N.; Milman, S. The antagonistic pleiotropy of insulin-like growth factor 1. Aging Cell 2021, 313443. [Google Scholar]
- Aguiar-Oliveira, M.H.; Bartke, A. Growth hormone deficiency: Health and longevity. Endocr. Rev. 2019, 40, 575–601. [Google Scholar] [CrossRef] [PubMed]
- Bartke, A. Pleiotropic effects of growth hormone signaling in aging. Trends Endocrinol. Metab. 2011, 22, 437–442. [Google Scholar] [CrossRef] [Green Version]
- Bartke, A.; Sun, L.Y.; Longo, V. Somatotropic signaling: Trade-offs between growth, reproductive development, and longevity. Physiol. Rev. 2013, 93, 571–598. [Google Scholar] [CrossRef]
- Broughton, S.J.; Piper, M.D.W.; Ikeya, T.; Bass, T.M.; Jacobson, J.; Driege, Y.; Martinez, P.; Hafen, E.; Withers, D.J.; Leevers, S.J.; et al. Longer lifespan, altered metabolism, and stress resistance in Drosophila from ablation of cells making insulin-like ligands. Proc. Natl. Acad. Sci. USA 2005, 102, 3105–3110. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.S.; Irizarry, R.A.; Gentleman, R.; Martines-Murillo, F.; Spencer, F. A model-based background adjustment for oligonucleotide expression arrays. J. Am. Stat. Assoc. 2004, 99, 909–917. [Google Scholar] [CrossRef] [Green Version]
- Bolstad, B.M.; Irizarry, R.A.; Astrand, M.; Speed, T.P. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 2003, 19, 185–193. [Google Scholar] [CrossRef] [Green Version]
- Irizarry, R.A.; Hobbs, B.; Collin, F.; Beazer-Barclay, Y.D.; Antonellis, K.J.; Scherf, U.; Speed, T.P. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 2003, 4, 249–264. [Google Scholar] [CrossRef] [Green Version]
- Irizarry, R.A.; Bolstad, B.M.; Collin, F.; Cope, L.M.; Hobbs, B.; Speed, T.P. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 2003, 31, e15. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Transcript ID (Array Design) | p-Value (LS vs. Con) | Fold-Change (LS vs. Con) | Gene_Assignment | Gene Symbol | RefSeq | p-Value (LS vs. Con) | Fold-Change (LS vs. Con) | Pearson’s Correlation | p-Value |
|---|---|---|---|---|---|---|---|---|---|
| hsa-miR-132-3p | 0.027634 | 3.20613 | AY005799 // AKT3 // v-akt murine thymoma viral oncogene homolog 3 (protein kinase B, ga) | AKT3 | AY005799 | 0.034986 | −1.51126 | −0.76 | 0.027 |
| hsa-miR-132-3p | 0.027634 | 3.20613 | AF043325 // NMT2 // N-myristoyltransferase 2 | NMT2 | AF043325 | 0.007468 | −1.53421 | −0.76 | 0.027 |
| hsa-miR-362-5p | 0.031091 | 2.43203 | AY005799 // AKT3 // v-akt murine thymoma viral oncogene homolog 3 (protein kinase B, ga) | AKT3 | AY005799 | 0.034986 | −1.51126 | −0.75 | 0.031 |
| hsa-miR-421 | 0.027239 | 2.39773 | AB015706 // IL6ST // interleukin 6 signal transducer (gp130, oncostatin M receptor) | IL6ST | AB015706 | 0.022031 | −1.63661 | −0.76 | 0.027 |
| hsa-miR-421 | 0.027239 | 2.39773 | BC035128 // MXI1 // MAX interactor 1 // 10q24-q25 | MXI1 | BC035128 | 0.005779 | −1.62721 | −0.76 | 0.027 |
| hsa-miR-200c-3p | 0.047791 | 2.00074 | AB015706 // IL6ST // interleukin 6 signal transducer (gp130, oncostatin M receptor) | IL6ST | AB015706 | 0.022031 | −1.63661 | −0.71 | 0.047 |
| hsa-miR-200c-3p | 0.047791 | 2.00074 | BC035128 // MXI1 // MAX interactor 1 // 10q24-q25 | MXI1 | BC035128 | 0.005779 | −1.62721 | −0.71 | 0.047 |
| hsa-miR-200c-3p | 0.047791 | 2.00074 | AK302477 // NPNT // nephronectin // 4q24 // 255743 | NPNT | AK302477 | 0.013197 | −3.14456 | −0.71 | 0.047 |
| hsa-miR-200c-3p | 0.047791 | 2.00074 | BC000261 // THRA // thyroid hormone receptor, alpha | THRA | BC000261 | 0.007158 | −1.52116 | −0.71 | 0.047 |
| hsa-miR-532-5p | 0.029735 | 1.65873 | AB015706 // IL6ST // interleukin 6 signal transducer (gp130, oncostatin M receptor) | IL6ST | AB015706 | 0.022031 | −1.63661 | −0.75 | 0.029 |
| hsa-miR-532-5p | 0.029735 | 1.65873 | AF252872 // SLC7A11 // solute carrier family 7 (anionic amino acid transporter light ch) | SLC7A11 | AF252872 | 0.023959 | −1.632 | −0.75 | 0.029 |
| hsa-miR-199a-3p | 0.033323 | −1.58718 | AB073146 // DCBLD2 // discoidin, CUB and LCCL domain containing 2 | DCBLD2 | AB073146 | 0.00852 | 1.58136 | −0.74 | 0.033 |
| hsa-miR-30b-5p | 0.032193 | −1.79613 | AF091433 // CCNE2 // cyclin E2 // 8q22.1 // 9134 | CCNE2 | AF091433 | 0.000984 | 1.55079 | −0.74 | 0.032 |
| hsa-miR-30b-5p | 0.032193 | −1.79613 | BC017338 // FUCA1 // fucosidase, alpha-L- 1, tissue | FUCA1 | BC017338 | 0.00248 | 2.34712 | −0.74 | 0.032 |
| hsa-miR-30b-5p | 0.032193 | −1.79613 | AB039723 // FZD3 // frizzled family receptor 3 // 8p21 | FZD3 | AB039723 | 0.014296 | 1.59799 | −0.75 | 0.032 |
| hsa-miR-30b-5p | 0.032193 | −1.79613 | AF186780 // RGL1 // ral guanine nucleotide dissociation stimulator-like 1 | RGL1 | AF186780 | 0.037572 | 1.8744 | −0.75 | 0.032 |
| hsa-miR-30b-5p | 0.032193 | −1.79613 | AF111801 // TRERF1 // transcriptional regulating factor 1 | TRERF1 | AF111801 | 0.041722 | 1.9583 | Not in table | |
| hsa-miR-30b-5p | 0.032193 | −1.79613 | AM404182 // TRERF1 // transcriptional regulating factor | TRERF1 | AM404182 | 0.036958 | 1.71707 | Not in table |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yaron-Saminsky, D.; Nagaraj, K.; Sarfstein, R.; Laron, Z.; Pasmanik-Chor, M.; Werner, H. MicroRNA 132-3p Is Upregulated in Laron Syndrome Patients and Controls Longevity Gene Expression. Int. J. Mol. Sci. 2021, 22, 11861. https://doi.org/10.3390/ijms222111861
Yaron-Saminsky D, Nagaraj K, Sarfstein R, Laron Z, Pasmanik-Chor M, Werner H. MicroRNA 132-3p Is Upregulated in Laron Syndrome Patients and Controls Longevity Gene Expression. International Journal of Molecular Sciences. 2021; 22(21):11861. https://doi.org/10.3390/ijms222111861
Chicago/Turabian StyleYaron-Saminsky, Danielle, Karthik Nagaraj, Rive Sarfstein, Zvi Laron, Metsada Pasmanik-Chor, and Haim Werner. 2021. "MicroRNA 132-3p Is Upregulated in Laron Syndrome Patients and Controls Longevity Gene Expression" International Journal of Molecular Sciences 22, no. 21: 11861. https://doi.org/10.3390/ijms222111861