
Fab Advances in Fabaceae for Abiotic Stress Resilience: From ‘Omics’ to Artificial Intelligence

 , , ,
, , ,
Abstract
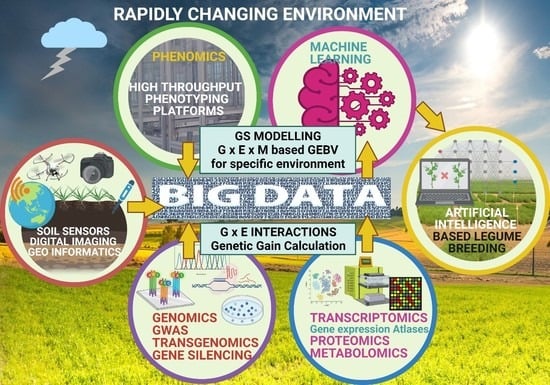
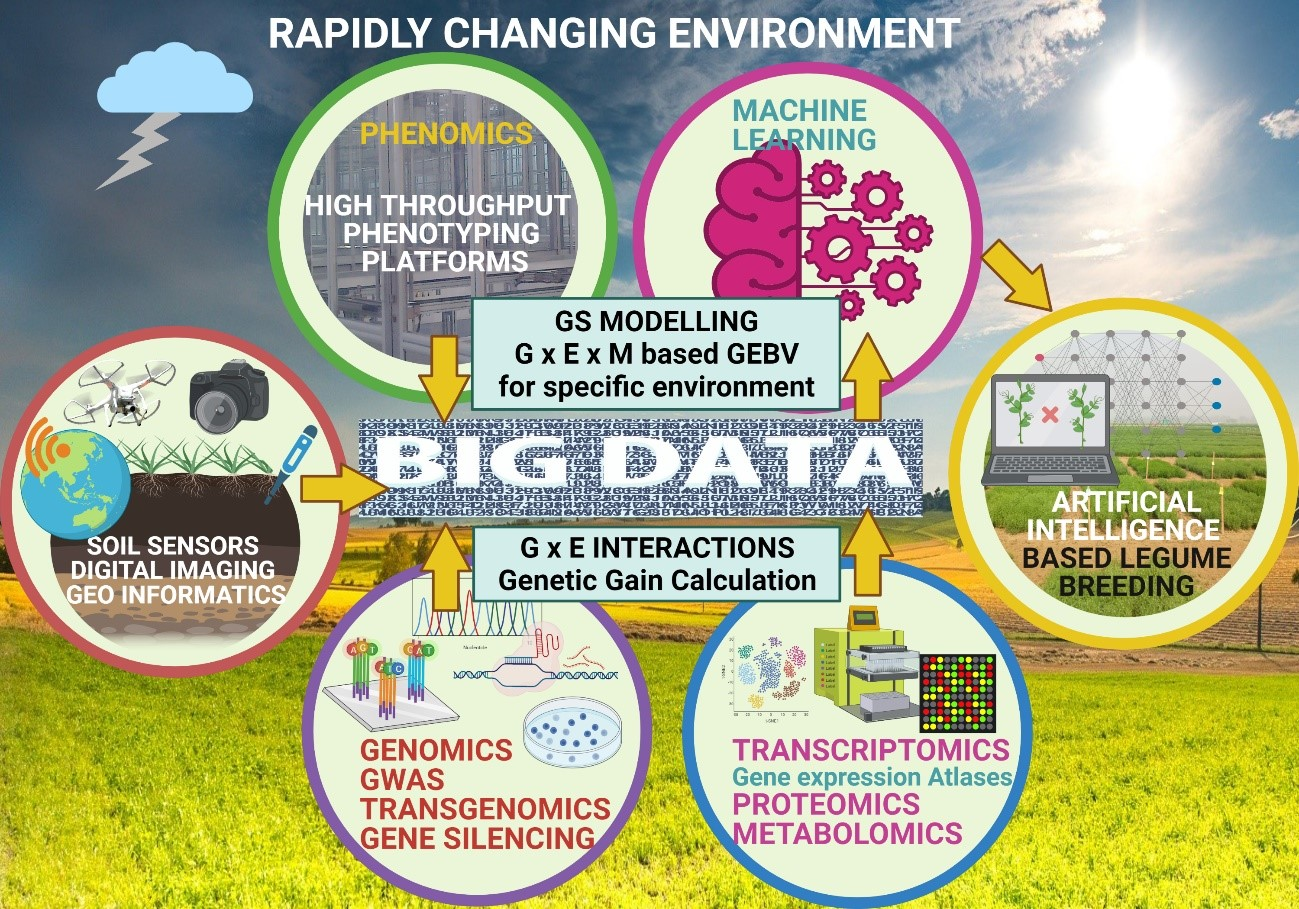
:
1. Introduction
1.1. Rationale
1.2. Objectives
2. Methods
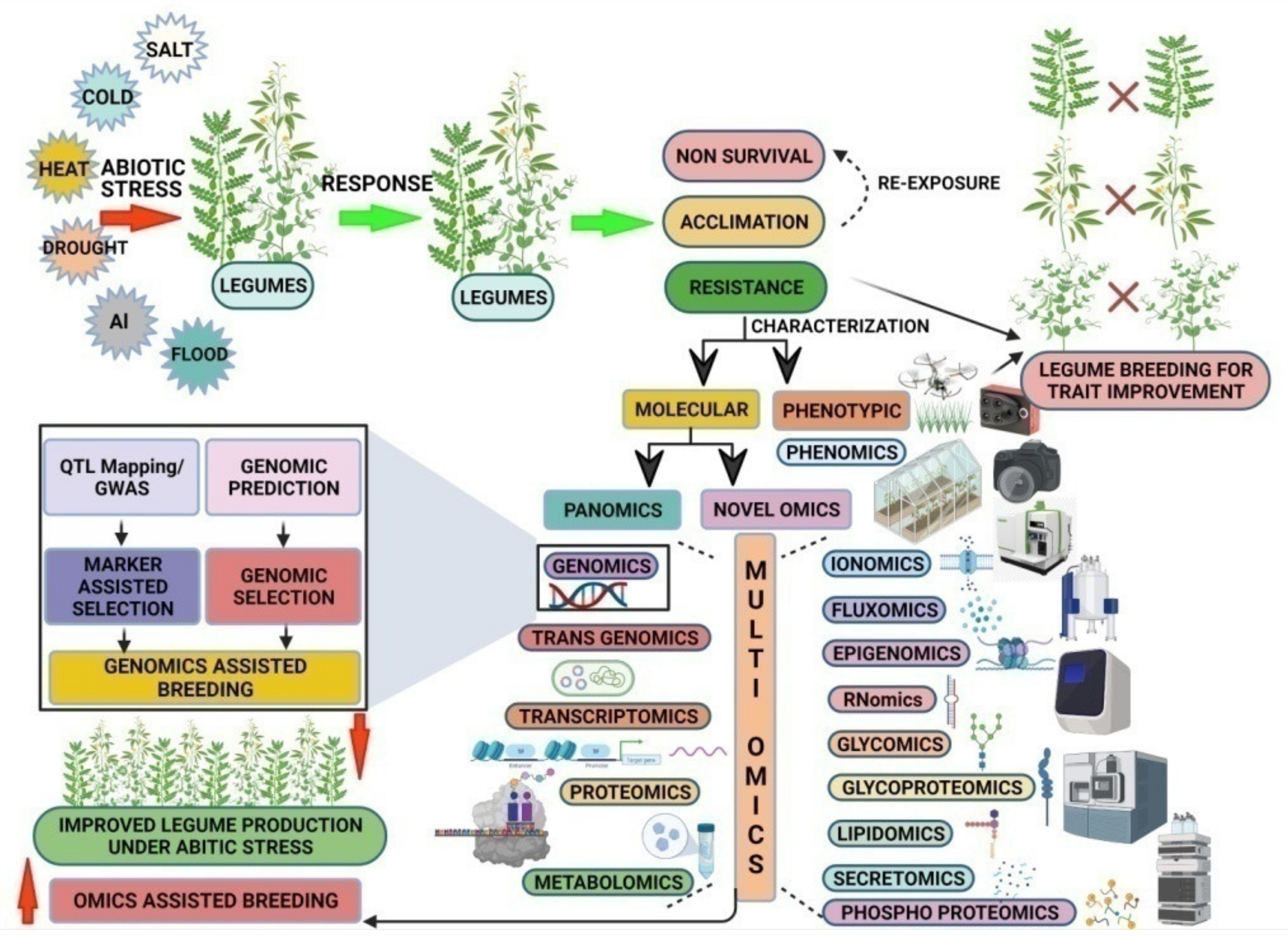
3. Novel ‘Omics’ Approaches for Future Pulse Breeding Programs
3.1. Ionomics
3.2. Epigenomics
3.3. Fluxomics
3.4. RNomics
3.5. Glycomics, Glycoproteomics, and Phosphoproteomics
3.6. Lipidomics, Regulomics, and Secretomics
4. Pan-Omics Approaches
4.1. Genomics
4.2. Transgenomics
4.3. Transcriptomics
4.4. Proteomics and Metabolomics in Abiotic Stress Mitigation
5. Multi-Omics Integration (MOI) for Future Pulse Breeding
6. Smart Farming: Artificial Intelligence (AI)-Based Pulse Breeding for Climate Resilience
6.1. Machine learning (ML)-Enabled Genomic Selection, QTL Mining, GWAS, and Functional Prediction for Pulse Breeding
6.2. Artificial Intelligence (AI)-Enabled Genome Editing
7. Challenges and Opportunities for Future Pulse Crop Breeding
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Maphosa, Y.; Jideani, V.A. The Role of Legumes in Human Nutrition, Functional Food. In Improve Health Through Adequate Food; Hueda, M.C., Ed.; IntechOpen: Rijeka, Croatia, 2017. [Google Scholar] [CrossRef] [Green Version]
- Bohra, A.; Sahrawat, K.L.; Kumar, S.; Joshi, R.; Parihar, A.K.; Singh, U.; Singh, D.; Singh, N.P. Genetics and genomics-based interventions for nutritional enhancement of grain legume crops: Status and outlook. J. Appl. Genet. 2015, 56, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Foyer, C.H.; Lam, H.M.; Nguyen, H.T.; Siddique, K.H.; Varshney, R.K.; Colmer, T.D.; Cowling, W.; Bramley, H.; Mori, T.A.; Hodgson, J.M.; et al. Neglecting legumes has compromised human health and sustainable food production. Nat. Plants 2016, 2, 16112. [Google Scholar] [CrossRef]
- Considine, M.J.; Siddique, K.H.M.; Foyer, C.H. Nature’s pulse power: Legumes, food security and climate change. J. Expt. Bot. 2017, 68, 1815–1818. [Google Scholar] [CrossRef] [PubMed]
- Graham, P.H.; Vance, C.P. Legumes: Importance and constraints to greater use. Plant. Physiol. 2003. 131, 872–877. [CrossRef] [Green Version]
- Kang, Y.J.; Kim, S.K.; Kim, M.Y.; Lestari, P.; Kim, K.H.; Ha, B.-K.; Jun, T.H.; Hwang, W.J.; Lee, T.; Lee, J.; et al. Genome sequence of mungbean and insights into evolution within Vigna species. Nat. Commun. 2014, 5, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varshney, R.K.; Gaur, P.M.; Chamarthi, S.K.; Krishnamurthy, L.; Tripathi, S.; Kashiwagi, J.; Samineni, S.; Singh, V.K.; Thudi, M.; Jaganathan, D. Fast-track introgression of “QTL-hotspot” for root traits and other drought tolerance traits in JG 11, an elite and leading variety of chickpea. Plant Genome 2013, 6, 3. [Google Scholar] [CrossRef] [Green Version]
- Schmutz, J.; McClean, P.E.; Mamidi, S.; Wu, G.A.; Cannon, S.B.; Grimwood, J.; Jenkins, J.; Shu, S.; Song, Q.; Chavarro, C.; et al. A reference genome for common bean and genome-wide analysis of dual domestications. Nat. Genet. 2014, 46, 707–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmutz, J.; Cannon, S.B.; Schlueter, J.; Ma, J.; Mitros, T.; Nelson, W.; Hyten, D.L.; Song, Q.; Thelen, J.J.; Cheng, J.; et al. Genome sequence of the palaeopolyploid soybean. Nature 2010, 463, 178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varshney, R.K.; Chen, W.; Li, Y.; Bharti, A.K.; Saxena, R.K.; Schlueter, J.A.; Donoghue, M.T.; Azam, S.; Fan, G.; Whaley, A.M.; et al. Draft genome sequence of pigeonpea (Cajanus cajan), an orphan legume crop of resource-poor farmers. Nat. Biotechnol. 2012, 30, 83. [Google Scholar] [CrossRef] [Green Version]
- Lonardi, S.; Muñoz-Amatriaín, M.; Liang, Q.; Shu, S.; Wanamaker, S.I.; Lo, S.; Tanskanen, J.; Schulman, A.H.; Zhu, T.; Luo, M.C.; et al. The genome of cowpea (Vigna unguiculata [L.] Walp.). Plant J. 2019, 98, 767–782. [Google Scholar] [CrossRef] [Green Version]
- Kreplak, J.; Madoui, M.A.; Cápal, P.; Novák, P.; Labadie, K.; Aubert, G.; Bayer, P.E.; Gali, K.K.; Syme, R.A.; Main, D.; et al. A reference genome for pea provides insight into legume genome evolution. Nat. Gen. 2019, 51, 1411–1422. [Google Scholar] [CrossRef]
- Jewell, M.C.; Campbell, B.C.; Godwin, I.D. Transgenic Plants for Abiotic Stress Resistance. In Transgenic Crop Plants; Springer: Berlin/Heidelberg, Germany, 2010; pp. 67–132. [Google Scholar]
- Raza, A.; Razzaq, A.; Mehmood, S.S.; Zou, X.; Zhang, X.; Lv, Y.; Xu, J. Impact of climate change on crops adaptation and strategies to tackle its outcome: A review. Plants 2019, 8, 34. [Google Scholar] [CrossRef] [Green Version]
- Le, B.H.; Wagmaister, J.A.; Kawashima, T.; Bui, A.Q.; Harada, J.J.; Goldberg, R.B. Using genomics to study legume seed development. Plant. Physiol. 2007, 144, 562–574. [Google Scholar] [CrossRef] [Green Version]
- Saito, K.; Matsuda, F. Metabolomics for functional genomics, systems biology and biotechnology. Annu. Rev. Plant. Biol. 2010, 61, 463–489. [Google Scholar] [CrossRef]
- Atkinson, N.J.; Urwin, P.E. The interaction of plant biotic and abiotic stresses: From genes to the field. J. Exp. Bot. 2012, 63, 3523–3543. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Wang, H.; Shao, H.; Tang, X. Recent advances in utilizing transcription factors to improve plant abiotic stress tolerance by transgenic technology. Front. Plant. Sci. 2016, 7, 67. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.K. Abiotic stress signalling and responses in plants. Cell 2016, 167, 313–324. [Google Scholar] [CrossRef] [Green Version]
- Varshney, R.K.; Roorkiwal, M.; Nguyen, T. Legume genomics: From genomic resources to molecular breeding. Plant. Genome 2013, 6, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Wheeler, J.A.; Cortes, A.J.; Sedlacek, J.; Karrenberg, S.; van Kleunen, M.; Wipf, S.; Hoch, G.; Bossdorf, O.; Rixen, C. The snow and the willows: Earlier spring snowmelt reduces performance in the low-lying alpine shrub Salix herbacea. J. Ecol. 2016, 104, 1041–1050. [Google Scholar] [CrossRef]
- Cortés, A.J.; Waeber, S.; Lexer, C.; Sedlacek, J.; Wheeler, J.A.; van Kleunen, M.; Boßdorf, O.; Hoch, G.; Rixen, C.; Wipf, S.; et al. Small-scale patterns in snowmelt timing affect gene flow and the distribution of genetic diversity in the alpine dwarf shrub Salix herbacea. Heredity 2014, 113, 233–239. [Google Scholar] [CrossRef] [Green Version]
- Wheeler, J.A.; Hoch, G.; Cortés, A.J.; Sedlacek, J.; Wipf, S.; Rixen, C. Increased spring freezing vulnerability for alpine shrubs under early snowmelt. Oecologia 2014, 175, 219–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wheeler, J.A.; Schnider, F.; Sedlacek, J.; Cortés, A.J.; Wipf, S.; Hoch, G.; Rixen, C. With a little help from my friends: Community facilitation increases performance in the dwarf shrub Salix herbacea. Basic Appl. Ecol. 2015, 16, 202–209. [Google Scholar] [CrossRef]
- Valencia, J.B.; Mesa, J.; León, J.G.; Madriñán, S.; Cortés, A.J. Climate vulnerability assessment of the espeletia complex on Páramo Sky Islands in the Northern Andes. Front. Ecol. Evol. 2020, 8, 309. [Google Scholar] [CrossRef]
- Sedlacek, J.; Cortés, A.J.; Wheeler, J.; Bossdorf, O.; Hoch, G.; Klápště, J.; Lexer, C.; Rixen, C.; Wipf, S.; Karrenberg, S.; et al. Evolutionary potential in the Alpine: Trait heritabilities and performance variation of the dwarf willow Salix herbacea from different elevations and microhabitats. Ecol. Evol. 2016, 6, 3940–3952. [Google Scholar] [CrossRef] [Green Version]
- Cortés, A.J.; Garzón, L.N.; Valencia, J.B.; Madriñán, S. On the causes of rapid diversification in the páramos: Isolation by ecology and genomic divergence in espeletia. Front. Plant. Sci. 2018, 9, 1700. [Google Scholar] [CrossRef] [PubMed]
- Little, C.J.; Wheeler, J.A.; Sedlacek, J.; Cortés, A.J.; Rixen, C. Small-scale drivers: The importance of nutrient availability and snowmelt timing on performance of the alpine shrub Salix herbacea. Oecologia 2016, 180, 1015–1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sedlacek, J.F.; Bossdorf, O.; Cortés, A.J.; Wheeler, J.A.; van Kleunen, M. What role do plant–soil interactions play in the habitat suitability and potential range expansion of the alpine dwarf shrub Salix herbacea? Basic Appl. Ecol. 2014, 15, 305–315. [Google Scholar] [CrossRef] [Green Version]
- Varshney, R.K. Application of Next Generation Sequencing and Genotyping Technologies to Develop Large-Scale Genomic Resources in SAT Legume Crops. In Genomics and Crop Improvement: Relevance and Reservations; Muralidharan, K., Siddiq., E.A., Acharya, N.G., Eds.; Ranga Agricultural University: Hyderabad, India, 2011; pp. 1–10. [Google Scholar]
- Varshney, R.K.; Singh, V.K.; Kumar, A.; Powell, W.; Sorrells, M.E. Can genomics deliver climate-change ready crops? Curr. Opin. Plant. Biol. 2018, 45, 205–211. [Google Scholar] [CrossRef]
- Abdelrahman, M.; Jogaiah, S.; Burritt, D.J.; Tran, L.S.P. Legume genetic resources and transcriptome dynamics under abiotic stress conditions. Plant. Cell Env. 2018, 41, 1972–1983. [Google Scholar] [CrossRef]
- Osakabe, Y.; Osakabe, K. Genome editing with engineered nucleases in plants. Plant. Cell Physiol. 2015, 56, 389–400. [Google Scholar] [CrossRef] [Green Version]
- Knowpulse website. Available online: http://knowpulse.usask.ca/ (accessed on 15 January 2021).
- Sato, S.; Nakamura, Y.; Kaneko, T.; Asamizu, E.; Kato, T.; Nakao, M.; Sasamoto, S.; Watanabe, A.; Ono, A.; Kawashima, K.; et al. Genome structure of the legume, Lotus japonicus. DNA Res. 2008, 15, 227–239. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, W.; Chen, H.; Yang, M.; Wang, J.; Pandey, M.K.; Zhang, C.; Chang, W.C.; Zhang, L.; Zhang, X.; Tang, R.; et al. The genome of cultivated peanut provides insight into legume karyotypes, polyploid evolution and crop domestication. Nature Genet. 2019, 51, 865–876. [Google Scholar] [CrossRef]
- Tripathi, P.; Rabara, R.C.; Rushton, P.J. A systems biology perspective on the role of WRKY transcription factors in drought responses in plants. Planta 2014, 239, 255–266. [Google Scholar] [CrossRef]
- Zhang, J.; Naik, H.S.; Assefa, T.; Sarkar, S.; Reddy, R.C.; Singh, A.; Ganapathysubramanian, B.; Singh, A.K. Computer vision and machine learning for robust phenotyping in genome-wide studies. Sci. Rep. 2017, 7, 44048. [Google Scholar] [CrossRef]
- Liberati, A.; Altman, D.G.; Tetzlaff, J.; Mulrow, C.; Gøtzsche, P.C.; Ioannidis, J.P.; Clarke, M.; Devereaux, P.J.; Kleijnen, J.; Moher, D. The PRISMA statement for reporting systematic reviews and meta-analyses of studies that evaluate health care interventions: Explanation and elaboration. J. Clinical Epidemiol. 2009, 62, e1–e34. [Google Scholar] [CrossRef] [Green Version]
- Lahner, B.; Gong, J.; Mahmoudian, M.; Smith, E.L.; Abid, K.B.; Rogers, E.E.; Guerinot, M.L.; Harper, J.F.; Ward, J.M.; McIntyre, L.; et al. Genomic scale profiling of nutrient and trace elements in Arabidopsis thaliana. Nat. Biotechnol. 2003, 21, 1215–1221. [Google Scholar] [CrossRef]
- Szpunar, J. Metallomics: A new frontier in analytical chemistry. Anal. Bioanal Chem. 2004, 378, 54–56. [Google Scholar] [CrossRef]
- Salt, D.E.; Baxter, I.; Lahner, B. Ionomics and the study of the plant ionome. Annu. Rev. Plant. Biol. 2008, 59, 709–733. [Google Scholar] [CrossRef] [Green Version]
- Baxter, I. Ionomics: The functional genomics of elements. Brief. Funct. Genom. 2010, 9, 14956. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.Y.; Salt, D.E. Plant ionomics: From elemental profiling to environmental adaptation. Mol. Plant 2016, 9, 787–797. [Google Scholar] [CrossRef] [Green Version]
- Ziegler, G.; Terauchi, A.; Becker, A.; Armstrong, P.; Hudson, K.; Baxter, I. Ionomic screening of field-grown soybean identifies mutants with altered seed elemental composition. Plant. Genome. 2013, 6. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Watanabe, T.; Shinano, T.; Okazaki, K.; Osaki, M. Rapid characterization of plant mutants with an altered ion-profile: A case study using Lotus japonicus. New Phytol. 2009, 181, 795–801. [Google Scholar] [CrossRef]
- Ziegler, G.; Nelson, R.; Granada, S.; Krishnan, H.B.; Gillman, J.D.; Baxter, I. Genome wide association study of ionomic traits on diverse soybean populations from germplasm collections. Plant. Direct. 2018, 15, e00033. [Google Scholar] [CrossRef]
- Hacisalihoglu, G.; Settles, A. Quantification of seed ionome variation in 90 diverse soybean (Glycine max) lines. J. Plant. Nutr. 2017, 40, 2808–2817. [Google Scholar] [CrossRef]
- Springer, N.M. Epigenetics and crop improvement. Trends Genet. 2013, 29, 241–247. [Google Scholar] [CrossRef]
- Labra, M.; Ghiani, A.; Citterio, S.; Sgorbati, S.; Sala, F.; Vannini, C.; Ruffini-Castiglione, M.; Bracale, M. Analysis of cytosine methylation pattern in response to water deficit in pea root tips. Plant. Biol. 2002, 4, 694–699. [Google Scholar] [CrossRef] [Green Version]
- Abid, G.; Mingeot, D.; Muhovski, Y.; Mergeai, G.; Aouida, M.; Abdelkarim, S.; Aroua, I.; El Ayed, M.; M’hamdi, M.; Sassi, K.; et al. Analysis of DNA methylation patterns associated with drought stress response in faba bean (Vicia faba L.) using methylation-sensitive amplification polymorphism (MSAP). Environ. Exp. Bot. 2017, 142, 34–44. [Google Scholar] [CrossRef]
- Rakei, A.; Maali-Amiri, R.; Zeinali, H.; Ranjbar, M. DNA methylation and physio-biochemical analysis of chickpea in response to cold stress. Protoplasma. 2015, 253, 61–76. [Google Scholar] [CrossRef]
- Song, Y.; Ji, D.; Li, S.; Wang, P.; Li, Q.; Xiang, F. The dynamic changes of DNA methylation and histone modifications of salt responsive transcription factor genes in soybean. PLoS ONE 2012, 7, e41274. [Google Scholar] [CrossRef]
- Liang, X.; Hou, X.; Li, J.; Han, Y.; Zhang, Y.; Feng, N.; Du, J.; Zhang, W.; Zheng, D.; Fang, S. High-resolution DNA methylome reveals that demethylation enhances adaptability to continuous cropping comprehensive stress in soybean. BMC Plant. Biol. 2019, 19, 79. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.; Pi, E.X.; Tsai, S.N.; Lam, H.M.; Sun, S.M.; Kwan, Y.W.; Ngai, S.M. GmPHD5 acts as an important regulator for crosstalk between histone H3K4 di-methylation and H3K14 acetylation in response to salinity stress in soybean. BMC Plant. Biol. 2011, 11, 178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sen, S.; Chakraborty, J.; Ghosh, P.; Basu, D.; Das, S. Chickpea WRKY70 regulates the expression of a homeodomain-leucine zipper (HD-Zip) I transcription factor CaHDZ12, which confers abiotic stress tolerance in transgenic tobacco and chickpea. Plant. Cell Physiol. 2017, 58, 1934–1952. [Google Scholar] [CrossRef] [PubMed]
- Awana, M.; Yadav, K.; Rani, K.; Gaikwad, K.; Praveen, S.; Kumar, S.; Singh, A. Insights into salt stress-induced biochemical, molecular and epigenetic regulation of spatial responses in pigeonpea (Cajanus cajan L.). J. Plant Growth Regul. 2019, 38, 1545–1561. [Google Scholar] [CrossRef]
- Chen, R.; Li, M.; Zhang, H.; Duan, L.; Sun, X.; Jiang, Q.; Zhang, H.; Hu, Z. Continuous salt stress-induced long non-coding RNAs and DNA methylation patterns in soybean roots. BMC Genomic. 2019, 20, 730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; Song, G.; Guo, W.; Wang, W.; Zhao, H.; Gao, T.; Lv, Q.; Yang, X.; Xu, F.; Dong, Y.; et al. Dynamic changes in genome-wide histone3 lysine27 trimethylation and gene expression of soybean roots in response to salt stress. Front. Plant. Sci. 2019, 10, 1031. [Google Scholar] [CrossRef] [PubMed]
- Gahlaut, V.; Zinta, G.; Jaiswal, V.; Kumar, S. Quantitative Epigenetics: A new avenue for crop improvement. Epigenomes 2020, 4, 25. [Google Scholar] [CrossRef]
- Schmitz, R.J.; He, Y.; Valdés-López, O.; Khan, S.M.; Joshi, T.; Urich, M.A.; Nery, J.R.; Diers, B.; Xu, D.; Stacey, G.; et al. Epigenome-wide inheritance of cytosine methylation variants in a recombinant inbred population. Genome Res. 2013, 23, 1663–1674. [Google Scholar] [CrossRef] [Green Version]
- Raju, S.K.K.; Shao, M.R.; Sanchez, R.; Xu, Y.Z.; Sandhu, A.; Graef, G.; Mackenzie, S. An epigenetic breeding system in soybean for increased yield and stability. Plant. Biotechnol. J. 2018, 16, 1836–1847. [Google Scholar] [CrossRef] [Green Version]
- Zhong, X. Comparative epigenomics: A powerful tool to understand the evolution of DNA methylation. New Phytol. 2016, 210, 76–80. [Google Scholar] [CrossRef]
- Junaid, A.; Singh, N.; Gaikwad, K. Patterns of gene-body-methylation conservation and its divergent association with gene expression in pigeonpea and soybean. bioRxiv 2020. [Google Scholar] [CrossRef]
- Kim, K.D.; El Baidouri, M.; Abernathy, B.; Iwata-Otsubo, A.; Chavarro, C.; Gonzales, M.; Libault, M.; Grimwood, J.; Jackson, S.A. A comparative epigenomic analysis of polyploidy-derived genes in soybean and common bean. Plant. Physiol. 2015, 168, 1433–1447. [Google Scholar] [CrossRef] [Green Version]
- Iyer, V.V.; Sriram, G.; Fulton, D.B.; Zhou, R.; Westgate, M.E.; Shanks, J.V. Metabolic flux maps comparing the effect of temperature on protein and oil biosynthesis in developing soybean cotyledons. Plant. Cell Environ. 2008, 31, 506–517. [Google Scholar] [CrossRef]
- Cocuron, J.C.; Koubaa, M.; Kimmelfield, R.; Ross, Z.; Alonso, A.P. A combined metabolomics and fluxomics analysis identifies steps limiting oil synthesis in maize embryos. Plant. Physiol. 2019, 181, 961–975. [Google Scholar] [CrossRef] [Green Version]
- Schwender, J.; Hay, J.O. Predictive modeling of biomass component tradeoffs in Brassica napus developing oilseeds based on in silico manipulation of storage metabolism. Plant. Physiol. 2012, 160, 1218–1236. [Google Scholar] [CrossRef] [Green Version]
- Salon, C.; Avice, J.C.; Colombié, S.; Dieuaide-Noubhani, M.; Gallardo, K.; Jeudy, C.; Ourry, A.; Prudent, M.; Voisin, A.S.; Rolin, D. Fluxomics links cellular functional analyses to whole-plant phenotyping. J. Expt. Bot. 2017, 68, 2083–2098. [Google Scholar] [CrossRef]
- Moreira, T.B.; Shaw, R.; Luo, X.; Ganguly, O.; Kim, H.S.; Coelho, L.G.F.; Cheung, C.Y.M.; Rhys Williams, T.C. A genome-scale metabolic model of soybean (Glycine max) highlights metabolic fluxes in seedlings. Plant. Physiol. 2019, 180, 1912–1929. [Google Scholar] [CrossRef] [Green Version]
- Kannan, K.; Wang, Y.; Lang, M.; Challa, G.S.; Long, S.P.; Marshall-Colon, A. Combining gene network, metabolic and leaf-level models shows means to future-proof soybean photosynthesis under rising CO2. In Silico Plants 2019, 1, diz008. [Google Scholar] [CrossRef] [Green Version]
- Kohli, D.; Joshi, G.; Deokar, A.A.; Bhardwaj, A.R.; Agarwal, M.; Katiyar-Agarwal, S.; Srinivasan, R.; Jain, P.K. Identification and characterization of Wilt and salt stress-responsive microRNAs in chickpea through high-throughput sequencing. PLoS ONE 2014, 9, e108851. [Google Scholar] [CrossRef]
- Barrera-Figueroa, B.E.; Gao, L.; Diop, N.N.; Wu, Z.; Ehlers, J.D.; Roberts, P.A.; Close, T.J.; Zhu, J.K.; Liu, R. Identification and comparative analysis of drought-associated microRNAs in two cowpea genotypes. BMC Plant. Biol. 2011, 11, 127. [Google Scholar] [CrossRef] [Green Version]
- Singh, U.; Khemka, N.; Rajkumar, M.S.; Garg, R.; Jain, M. PLncPRO for prediction of long non-coding RNAs (lncRNAs) in plants and its application for discovery of abiotic stress-responsive lncRNAs in rice and chickpea. Nucleic Acids Res. 2017, 45, e183. [Google Scholar] [CrossRef]
- Varki, A. Biological roles of glycans. Glycobiology 2017, 27, 3–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moller, I.E.; Pettolino, F.A.; Hart, C.; Lampugnani, E.R.; Willats, W.G.T.; Bacic, A. Glycan profiling of plant cell wall polymers using microarrays. J. Vis. Exp. 2012, 70, 4238. [Google Scholar] [CrossRef] [Green Version]
- Cummings, R.D.; Pierce, J.M. The challenge and promise of glycomics: Chem. Biol. 2014, 21, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Halim, A.; Nilsson, J.; Rüetschi, U.; Hesse, C.; Larson, G. Human urinary glycoproteomics attachment site specific analysis of N- and O-linked glycosylations by CID and ECD. Mol. Cell Proteomics. 2012, 11. [Google Scholar] [CrossRef] [Green Version]
- Mustafa, G.; Komatsu, S. Quantitative proteomics reveals the effect of protein glycosylation in soybean root under flooding stress. Front. Plant. Sci. 2014, 5, 627. [Google Scholar] [CrossRef] [Green Version]
- Subba, P.; Barua, P.; Kumar, R.; Datta, A.; Soni, K.K.; Chakraborty, S.; Chakraborty, N. Phosphoproteomic dynamics of chickpea (Cicer arietinum L.) reveals shared and distinct components of dehydration response. J. Proteome Res. 2013, 12, 5025–5047. [Google Scholar] [CrossRef]
- Subba, P.; Barua, P.; Kumar, R.; Datta, A.; Soni, K.K.; Chakraborty, S.; Chakraborty, N. Bioinformatic identification and analysis of hydroxyproline-rich glycoproteins in Populustrichocarpa. BMC Plant. Biol. 2016, 16, 229. [Google Scholar] [CrossRef] [Green Version]
- Balkir, P.; Kemahlioglu, K.; Yucel, U. Foodomics: A new approach in food quality and safety. Trends Food Sci. Technol. 2021, 108, 49–57. [Google Scholar] [CrossRef]
- Panzade, G.; Gangwar, I.; Awasthi, S.; Sharma, N.; Shankar, R. Plant Regulomics Portal (PRP): A comprehensive integrated regulatory information and analysis portal for plant genomes. Database 2019, 2019, baz130. [Google Scholar] [CrossRef]
- Ran, X.; Zhao, F.; Wang, Y.; Liu, J.; Zhuang, Y.; Ye, L.; Qi, M.; Cheng, J.; Zhang, Y. Plant Regulomics: A data-driven interface for retrieving upstream regulators from plant multi-omics data. Plant. J. Cell Mol. Biol. 2020, 101, 237–248. [Google Scholar] [CrossRef]
- Tanveer, T.; Shaheen, K.; Parveen, S.; Kazi, A.G.; Ahmad, P. Plant secretomics: Identification, isolation, and biological significance under environmental stress. Plant. Signal. Behav. 2014, 9, e29426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, S.; Wardhan, V.; Kumar, A.; Rathi, D.; Pandey, A.; Chakraborty, S.; Chakraborty, N. Secretome analysis of chickpea reveals dynamic extracellular remodeling and identifies a Bet v1-like protein, CaRRP1 that participates in stress response. Sci. Rep. 2015, 5, 18427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parveen, S.; Gupta, D.B.; Dass, S.; Kumar, A.; Pandey, A.; Chakraborty, S.; Chakraborty, N. Chickpea ferritin CaFer1 participates in oxidative stress response, and promotes growth and development. Sci. Rep. 2016, 6, 31218. [Google Scholar] [CrossRef] [Green Version]
- Fernandes, C.; Figueira, E.; Tauler, R. Exposure to chlorpyrifos induces morphometric, biochemical and lipidomic alterations in green beans (Phaseolus vulgaris). Ecotoxicol. Environ. Saf. 2018, 156, 25–33. [Google Scholar] [CrossRef]
- Narayanan, S.; Zoong-Lwe, Z.S.; Gandhi, N.; Welti, R.; Fallen, B.; Smith, J.R.; Rustgi, S. Comparative lipidomic analysis reveals heat stress responses of two soybean genotypes differing in temperature sensitivity. Plants 2020, 9, 457. [Google Scholar] [CrossRef] [Green Version]
- Okazaki, Y.; Takano, K.; Saito, K. Lipidomic analysis of soybean leaves revealed tissue-dependent difference in lipid remodeling under phosphorus-limited growth conditions. Plant. Biotechnol. 2017, 34, 57–63. [Google Scholar] [CrossRef] [Green Version]
- Yin, X.; Sakata, K.; Komatsu, S. Phosphoproteomics reveals the effect of ethylene in soybean root under flooding stress. J. Proteome Res. 2014, 13, 5618–5634. [Google Scholar] [CrossRef]
- Razzaq, M.K.; Aleem, M.; Mansoor, S.; Khan, M.A.; Rauf, S.; Iqbal, S.; Siddique, K.H.M. Omics and CRISPR-Cas9 approaches for molecular insight, functional gene analysis, and stress tolerance development in crops. Int. J. Mol. Sci. 2021, 22, 1292. [Google Scholar] [CrossRef]
- Young, N.D.; Cannon, S.B.; Sato, S.; Kim, D.; Cook, D.R.; Town, C.D.; Roe, B.A.; Tabata, S. Sequencing the gene spaces of Medicago truncatula and Lotus japonicus. Plant. Physiol. 2005, 137, 1174–1181. [Google Scholar] [CrossRef] [Green Version]
- Young, N.D.; Bharti, A.K. Genome-enabled insights into legume biology. Annu. Rev. Plant. Biol. 2012, 63, 283–305. [Google Scholar] [CrossRef]
- Bohra, A.; Pandey, M.K.; Jha, U.C.; Singh, B.; Singh, I.P.; Datta, D.; Chaturvedi, S.K.; Nadarajan, N.; Varshney, R.K. Genomics assisted breeding in four major pulse crops of developing countries: Present status and prospects. Theor. Appl. Genet. 2014, 127, 1263–1291. [Google Scholar] [CrossRef] [Green Version]
- Gilchrist, E.; Haughn, G. Reverse genetics techniques: Engineering loss and gain of gene function in plants. Brief. Funct. Genom. 2010, 9, 103–110. [Google Scholar] [CrossRef] [Green Version]
- Rehman, A.U.; Malhotra, R.S.; Bett, K.; Tar’An, B.; Bueckert, R.; Warkentin, T.D. Mapping QTL associated with traits affecting grain yield in chickpea (Cicer arietinum L.) under terminal drought stress. Crop. Sci. 2011, 51, 450–463. [Google Scholar] [CrossRef]
- Muchero, W.; Ehlers, J.D.; Close, T.J.; Roberts, P.A. Mapping QTL for drought stress-induced premature senescence and maturity in cowpea [Vigna unguiculata (L.) Walp.]. Theor. Appl. Genet. 2009, 118, 849–863. [Google Scholar] [CrossRef]
- Asfaw, A.; Blair, M.W.; Struik, P.C. Multienvironment quantitative trait loci analysis for photosynthate acquisition, accumulation, and remobilization traits in common bean under drought stress. G3 2012, 2, 579–595. [Google Scholar] [CrossRef] [Green Version]
- Nabateregga, M.; Mukankusi, C.; Raatz, B. Quantitative trait loci (QTL) mapping for intermittent drought tolerance in BRB 191 × SEQ 1027 Andean Intra-gene cross recombinant inbred line population of common bean (Phaseolus vulgaris L. African J. Biotechnol. 2019, 18, 452–461. [Google Scholar]
- y Teran, J.C.B.M.; Konzen, E.R.; Palkovic, A.; Tsai, S.M.; Rao, I.M.; Beebe, S.; Gepts, P. Effect of drought stress on the genetic architecture of photosynthate allocation and remobilization in pods of common bean (Phaseolus vulgaris L.), a key species for food security. BMC Plant. Biol. 2019, 19, 171. [Google Scholar] [CrossRef]
- Diaz, L.M.; Ricaurte, J.; Tovar, E.; Cajiao, C.; Teran, H.; Grajales, M.; Polania, J.; Rao, I.; Beebe, S.; Raatz, B. QTL analyses for tolerance to abiotic stresses in a common bean (Phaseolus vulgaris L.) population. PLoS ONE 2018, 13, e0202342. [Google Scholar] [CrossRef]
- Diaz, S.; Ariza-Suarez, D.; Izquierdo, P.; Lobaton, J.D.; de la Hoz, J.F.; Acevedo, F.; Duitama, J.; Guerrero, A.F.; Cajiao, C.; Mayor, V. Genetic mapping for agronomic traits in a MAGIC population of common bean (Phaseolus vulgaris L.) under drought conditions. BMC Genomics. 2020, 21, 799. [Google Scholar] [CrossRef]
- Idrissi, O.; Udupa, S.M.; De Keyser, E.; McGee, R.J.; Coyne, C.J.; Saha, G.C.; Muehlbauer, F.J.; Van Damme, P.; De Riek, J. Identification of quantitative trait loci controlling root and shoot traits associated with drought tolerance in a lentil (Lens culinaris Medik.) recombinant inbred line population. Front. Plant. Sci. 2016, 7, 1174. [Google Scholar] [CrossRef] [Green Version]
- Specht, J.E.; Chase, K.; Macrander, M.; Graef, G.L.; Chung, J.; Markwell, J.P.; Germann, M.; Orf, J.H.; Lark, K.G. Soybean response to water: A QTL analysis of drought tolerance. Crop. Sci. 2001, 41, 493–509. [Google Scholar] [CrossRef]
- Ye, H.; Song, L.; Schapaugh, W.T.; Ali, M.L.; Sinclair, T.R.; Riar, M.K.; Raymond, R.N.; Li, Y.; Vuong, T.; Valliyodan, B.; et al. The importance of slow canopy wilting in drought tolerance in soybean. J. Expt. Bot. 2020, 71, 642–652. [Google Scholar] [CrossRef]
- Sholihin, H.D.M. Molecular mapping of drought resistance in mungbean (Vigna radiata): 1. Linkage map in mungbean using AFLP markers. J.B. Pertanian. 2002, 7, 17–24. [Google Scholar]
- Liu, C.; Wu, J.; Wang, L. Quantitative trait locus mapping under irrigated and drought treatments based on a novel genetic linkage map in mungbean (Vigna radiata L.). Theor. Appl. Genet. 2017, 130, 2375–2393. [Google Scholar] [CrossRef] [PubMed]
- Iglesias-García, R.; Prats, E.; Fondevilla, S.; Satovic, Z.; Rubiales, D. Quantitative trait loci associated to drought adaptation in pea (Pisum sativum L.). Plant. Mol. Biol. Rep. 2015, 33, 1768–1778. [Google Scholar] [CrossRef] [Green Version]
- Paul, P.J.; Samineni, S.; Thudi, M.; Sajja, S.B.; Rathore, A.; Das, R.R.; Khan, A.W.; Chaturvedi, S.K.; Lavanya, G.R.; Varshney, R. Molecular mapping of QTLs for heat tolerance in chickpea. Int. J. Mol. Sci. 2018, 19, 2166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucas, M.R.; Ehlers, J.D.; Huynh, B.L.; Diop, N.N.; Roberts, P.A.; Close, T.J. Markers for breeding heat-tolerant cowpea. Mol. Breed. 2013, 31, 529–536. [Google Scholar] [CrossRef]
- Singh, D.; Singh, C.K.; Singh Tomar, R.S.; Pal, M. Genetics and molecular mapping of heat tolerance for seedling survival and pod set in lentil. Crop. Sci. 2017, 57, 3059–3067. [Google Scholar] [CrossRef]
- Mugabe, D.; Coyne, C.J.; Piaskowski, J.; Zheng, P.; Ma, Y.; Landry, E.; McGee, R.; Main, D.; Vandemark, G.; Zhang, H.; et al. Quantitative trait loci for cold tolerance in chickpea. Crop. Sci. 2019, 59, 573–582. [Google Scholar] [CrossRef] [Green Version]
- Sallam, A.; Arbaoui, M.; El-Esawi, M.; Abshire, N.; Martsch, R. Identification and verification of QTL associated with frost tolerance using linkage mapping and GWAS in winter faba bean. Front. Plant. Sci. 2016, 7, 1098. [Google Scholar] [CrossRef] [Green Version]
- Kahraman, A.; Kusmenoglu, I.; Aydin, N.; Aydogan, A.; Erskine, W.; Muehlbauer, F.J. QTL mapping of winter hardiness genes in Lentil. Crop. Sci. 2004, 44, 13. [Google Scholar] [CrossRef] [Green Version]
- Dumont, E.; Fontaine, V.; Vuylsteker, C.; Sellier, H.; Bodèle, S.; Voedts, N.; Devaux, R.; Frise, M.; Avia, K.; Hilbert, J.L. Association of sugar content QTL and PQL with physiological traits relevant to frost damage resistance in pea under field and controlled conditions. Theor. Appl. Genet. 2009, 118, 1561–1571. [Google Scholar] [CrossRef]
- Jähne, F.; Balko, C.; Hahn, V.; Würschum, T.; Leiser, W.L. Cold stress tolerance of soybeans during flowering: QTL mapping and efficient selection strategies under controlled conditions. Plant. Breed. 2019, 138, 708–720. [Google Scholar] [CrossRef]
- Vadez, V.; Krishnamurthy, L.; Thudi, M.; Anuradha, C.; Colmer, T.D.; Turner, N.C.; Siddique, K.H.; Gaur, P.M.; Varshney, R.K. Assessment of ICCV 2 × JG 62 chickpea progenies shows sensitivity of reproduction to salt stress and reveals QTL for seed yield and yield components. Mol. Breed. 2012, 30, 9–21. [Google Scholar] [CrossRef] [Green Version]
- Pushpavalli, R.; Krishnamurthy, L.; Thudi, M.; Gaur, P.M.; Rao, M.V.; Siddique, K.H.; Colmer, T.D.; Turner, N.C.; Varshney, R.K.; Vadez, V. Two key genomic regions harbour QTLs for salinity tolerance in ICCV 2 × JG 11 derived chickpea (Cicer arietinum L.) recombinant inbred lines. BMC Plant. Biol. 2015, 15, 124. [Google Scholar] [CrossRef] [Green Version]
- Chankaew, S.; Isemura, T.; Naito, K.; Ogiso-Tanaka, E.; Tomooka, N.; Somta, P.; Kaga, A.; Vaughan, D.A.; Srinives, P. QTL mapping for salt tolerance and domestication-related traits in Vigna marina subsp. oblonga, a halophytic species. Theor. Appl. Genet. 2013, 127, 691–702. [Google Scholar] [CrossRef]
- Leonforte, A.; Sudheesh, S.; Cogan, N.O.; Salisbury, P.A.; Nicolas, M.E.; Materne, M.; Forster, J.W.; Kaur, S. SNP marker discovery, linkage map construction and identification of QTLs for enhanced salinity tolerance in field pea (Pisum sativum L.). BMC Plant. Biol. 2013, 13, 161. [Google Scholar] [CrossRef] [Green Version]
- Lee, G.J.; Boerma, H.R.; Villagarcia, M.R.; Zhou, X.; Carter, T.E.; Li, Z.; Gibbs, M.O. A major QTL conditioning salt tolerance in S-100 soybean and descendent cultivars. Theor. Appl. Genet. 2004, 109, 1610–1619. [Google Scholar] [CrossRef]
- Wang, X.; Cheng, Y.; Yang, C.; Yang, C.; Mu, Y.; Xia, Q.; Ma, Q. QTL mapping for aluminum tolerance in RIL population of soybean (Glycine max L.) by RAD sequencing. PLoS ONE 2019, 14, e0223674. [Google Scholar] [CrossRef]
- Korir, P.C.; Zhang, J.; Wu, K.; Zhao, T.; Gai, J. Association mapping combined with linkage analysis for aluminum tolerance among soybean cultivars released in Yellow and Changjiang River Valleys in China. Theor. Appl. Genet. 2013, 126, 1659–1675. [Google Scholar] [CrossRef]
- Sharma, A.D.; Sharma, H.; Lightfoot, D.A. The genetic control of tolerance to aluminum toxicity in the ‘Essex’ by ‘Forrest’ recombinant inbred line population. Theor. Appl. Genet. 2011, 122, 687–694. [Google Scholar] [CrossRef]
- Correa, R.; Stanga, J.; Larget, B.; Roznowski, A.; Shu, G.; Dilkes, B.; Baum, D.A. An assessment of transgenomics as a tool for identifying genes involved in the evolutionary differentiation of closely related plant species. New Phytol. 2012, 193, 494–503. [Google Scholar] [CrossRef] [Green Version]
- Tzfira, T.; Weinthal, D.; Marton, I.; Zeevi, V.; Zuker, A.; Vainstein, A. Genome modifications in plant cells by custom-made restriction enzymes. Plant. Biotechnol. J. 2012, 10, 373–389. [Google Scholar] [CrossRef]
- Das Bhowmik, S.S.; Cheng, A.Y.; Long, H.; Tan, G.; Hoang, T.; Karbaschi, M.R.; Williams, B.; Higgins, T.; Mundree, S.G. Robust genetic transformation system to obtain non-chimeric transgenic chickpea. Front. Plant. Sci. 2019, 10, 524. [Google Scholar] [CrossRef] [Green Version]
- Das, A.; Basu, P.S.; Kumar, M.; Ansari, J.; Shukla, A.; Thakur, S.; Singh, P.; Datta, S.; Chaturvedi, S.K.; Sheshshayee, M.S.; et al. Transgenic chickpea (Cicer arietinum L.) harbouring AtDREB1a are physiologically better adapted to water deficit. BMC Plant. Biol. 2021, 21, 39. [Google Scholar] [CrossRef]
- Nguyen, Q.H.; Vu, L.T.K.; Nguyen, L.T.N.; Pham, N.T.T.; Nguyen, Y.T.H.; Van Le, S.; Chu, M.H. Overexpression of the GmDREB6 gene enhances proline accumulation and salt tolerance in genetically modified soybean plants. Sci. Rep. 2019, 9, 196630. [Google Scholar] [CrossRef]
- Meena, M.K.; Ghawana, S.; Dwivedi, V.; Roy, A.; Chattopadhyay, D. Expression of chickpea CIPK25 enhances root growth and tolerance to dehydration and salt stress in transgenic tobacco. Front. Plant. Sci. 2015, 6, 683. [Google Scholar] [CrossRef]
- Shukla, R.K.; Raha, S.; Tripathi, V.; Chattopadhyay, D. Expression of CAP2, an APETALA2-family transcription factor from chickpea, enhances growth and tolerance to dehydration and salt stress in transgenic tobacco. Plant. Physiol. 2006, 142, 113–123. [Google Scholar] [CrossRef] [Green Version]
- Jain, D.; Chattopadhyay, D. Promoter of CaZF, a chickpea gene that positively regulates growth and stress tolerance, Is activated by an AP2-family transcription factor CAP2. PLoS ONE 2013, 8, e56737. [Google Scholar] [CrossRef] [Green Version]
- Kumar, M.; Yusuf, M.A.; Yadav, P.; Narayan, S. Overexpression of chickpea defensin gene gonfers tolerance to water-deficit stress in Arabidopsis thaliana. Front. Plant. Sci. 2019, 10, 290. [Google Scholar] [CrossRef]
- Yu, X.; Peng, H.; Liu, Y.; Zhang, Y.; Shu, Y.; Chen, Q.; Shi, S.; Ma, L.; Ma, H.; Zhang, H. CarNAC2, a novel NAC transcription factor in chickpea (Cicer arietinum L.), is associated with drought-response and various developmental processes in transgenic Arabidopsis. J. Plant. Biol. 2014, 57, 55–66. [Google Scholar] [CrossRef]
- Figueroa-Balderas, R.E.; García-Ponce, B.; Rocha-Sosa, M. Hormonal and stress induction of the gene encoding common bean acetyl-coenzyme A carboxylase. Plant. Physiol. 2006, 142, 609–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niron, H.; Türet, M. A putative common bean Chalcone O-Methyltransferase improves salt tolerance in transgenic Arabidopsis thaliana. J. Plant. Growth Regul. 2020, 39, 957–969. [Google Scholar] [CrossRef]
- Chung, E.; Cho, C.W.; So, H.A.; Kang, J.S.; Chung, Y.S.; Lee, J.H. Overexpression of VrUBC1, a mungbean E2 ubiquitin-conjugating enzyme, enhances osmotic stress tolerance in Arabidopsis. PLoS ONE 2013, 8, e66056. [Google Scholar] [CrossRef] [Green Version]
- Banu, M.S.A.; Huda, K.M.K.; Sahoo, R.K.; Garg, B.; Tula, S.; Islam, S.S.; Tuteja, R.; Tuteja, N. Pea p68 imparts salinity stress tolerance in rice by scavenging of ROS-mediated H2O2 and interacts with argonaute. Plant. Mol. Biol. Rep. 2015, 33, 221–238. [Google Scholar] [CrossRef]
- Tuteja, N.; Banu, M.S.A.; Huda, K.M.K.; Gill, S.S.; Jain, P.; Pham, X.H.; Tuteja, R. Pea p68, a DEAD-Box helicase, provides salinity stress tolerance in transgenic tobacco by reducing oxidative stress and improving photosynthesis machinery. PLoS ONE 2014, 9, e98287. [Google Scholar] [CrossRef] [Green Version]
- Sahoo, R.K.; Gill, S.S.; Tuteja, N. Pea DNA helicase 45 promotes salinity stress tolerance in IR64 rice with improved yield. Plant. Signal. Behav. 2012, 7, 1042–1046. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, S.; Rahman, M.H.; Shah, S.; Kav, N.N. Constitutive expression of the pea ABA-responsive 17 (ABR17) cDNA confers multiple stress tolerance in Arabidopsis thaliana. Plant. Biotechnol. J. 2006, 4, 529–549. [Google Scholar] [CrossRef]
- Priyanka, B.; Sekhar, K.; Sunita, T.; Reddy, V.D.; Rao, K.V. Characterization of expressed sequence tags (ESTs) of pigeonpea (Cajanus cajan L.) and functional validation of selected genes for abiotic stress tolerance in Arabidopsis thaliana. Mol. Genet. Genomics 2010, 283, 273–287. [Google Scholar] [CrossRef]
- Tamirisa, S.; Vudem, D.R.; Khareedu, V.R. Overexpression of pigeonpea stress-induced cold and drought regulatory gene (CcCDR) confers drought, salt, and cold tolerance in Arabidopsis. J. Exp. Bot. 2014, 65, 4769–4781. [Google Scholar] [CrossRef] [Green Version]
- Sekhar, K.; Priyanka, B.; Reddy, V.D.; Rao, K.V. Isolation and characterization of a pigeonpeacyclophilin (CcCYP) gene, and its over-expression in Arabidopsis confers multiple abiotic stress tolerance. Plant. Cell Environ. 2010, 33, 1324–1338. [Google Scholar] [CrossRef]
- Sunitha, M.; Srinath, T.; Reddy, V.D.; Rao, K.V. Expression of cold and drought regulatory protein (CcCDR) of pigeonpea imparts enhanced tolerance to major abiotic stresses in transgenic rice plants. Planta 2017, 245, 1137–1148. [Google Scholar] [CrossRef]
- Rodrigues, S.M.; Andrade, M.O.; Gomes, A.P.S.; DaMatta, F.M.; Baracat-Pereira, M.C.; Fontes, E.P. Arabidopsis and tobacco plants ectopically expressing the soybean antiquitin-like ALDH7 gene display enhanced tolerance to drought, salinity, and oxidative stress. J. Expt. Bot. 2006, 57, 1909–1918. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Wang, Q.Y.; Cheng, X.G.; Xu, Z.S.; Li, L.C.; Ye, X.G.; Xia, L.Q.; Ma, Y.Z. GmDREB2, a soybean DRE-binding transcription factor, conferred drought and high-salt tolerance in transgenic plants. Biochem. Biophys. Res. Commun. 2007, 353, 299–305. [Google Scholar] [CrossRef]
- Zhang, G.; Chen, M.; Li, L.; Xu, Z.; Chen, X.; Guo, J.; Ma, Y. Overexpression of the soybean GmERF3 gene, an AP2/ERF type transcription factor for increased tolerances to salt, drought and diseases in transgenic tobacco. J. Exp. Bot. 2009, 60, 3781–3796. [Google Scholar] [CrossRef] [Green Version]
- Hajyzadeh, M.; Turktas, M.; Khawar, K.M.; Unver, T. miR408 overexpression causes increased drought tolerance in chickpea. Gene. 2015, 555, 186–193. [Google Scholar] [CrossRef]
- Khatib, F.; Makris, A.; Yamaguchi-Shinozaki, K.; Kumar, S.; Sarker, A.; Erskine, W.; Baum, M. Expression of the DREB1A gene in lentil (Lens culinaris Medik. subsp. culinaris) transformed with the Agrobacterium system. Crop. Pasture Sci. 2011, 62, 488–495. [Google Scholar] [CrossRef]
- Sahoo, D.P.; Kumar, S.; Mishra, S.; Kobayashi, Y.; Panda, S.K.; Sahoo, L. Enhanced salinity tolerance in transgenic mungbean overexpressing Arabidopsis antiporter (NHX1) gene. Mol. Breed. 2016, 36, 144. [Google Scholar] [CrossRef]
- Kumar, S.; Kalita, A.; Srivastava, R.; Sahoo, L. Co-expression of Arabidopsis NHX1 and bar improves the tolerance to salinity, oxidative stress, and herbicide in transgenic mungbean. Front. Plant. Sci. 2017, 8, 1896. [Google Scholar] [CrossRef] [Green Version]
- Seo, J.S.; Sohn, H.B.; Noh, K.; Jung, C.; An, J.H.; Donovan, C.M.; Somers, D.A.; Kim, D.I.; Jeong, S.C.; Kim, C.G.; et al. Expression of the Arabidopsis AtMYB44 gene confers drought/salt-stress tolerance in transgenic soybean. Mol. Breed. 2011, 29, 601–608. [Google Scholar] [CrossRef]
- Shanmugam, S.; Zhao, S.; Nandy, S.; Srivastava, V. and Khodakovskaya, M.;. Modification of soybean growth and abiotic stress tolerance by expression of truncated ERECTA Protein from Arabidopsis Thaliana. PLoS ONE 2020, 15, e0233383. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.; Behura, R.; Awasthi, J.P. Ectopic overexpression of a mungbean vacuolar Na+/H+ antiporter gene (VrNHX1) leads to increased salinity stress tolerance in transgenic Vigna unguiculata L. Walp. Mol. Breed. 2014, 34, 1345–1359. [Google Scholar] [CrossRef]
- Kwapata, K.; Nguyen, T.; Sticklen, M. Genetic transformation of common bean (Phaseolus vulgaris L.) with the guscolor Marker, the Bar herbicide resistance, and the barley (Hordeum vulgare) HVA1 drought tolerance genes. Int. J. Agron 2012, 2012. [Google Scholar] [CrossRef]
- Singh, R.; Sharma, S.; Kharb, P.; Saifi, S.; Tuteja, N. OsRuvB transgene induces salt tolerance in pigeon pea. J. Plant. Interactions. 2020, 15, 17–26. [Google Scholar] [CrossRef] [Green Version]
- Hanafy, M.S.; El-Banna, A.; Schumacher, H.M.; Jacobsen, H.-J.; Hassan, F.S. Enhanced tolerance to drought and salt stresses in transgenic faba bean (Vicia faba L.) plants by heterologous expression of the PR10a gene from potato. Plant. Cell Rep. 2013, 32, 663–674. [Google Scholar] [CrossRef]
- Lowe, R.; Shirley, N.; Bleackley, M.; Dolan, S.; Shafee, T. Transcriptomics technologies. PLoS Comput. Biol. 2017, 13, e1005457. [Google Scholar] [CrossRef] [Green Version]
- Libault, M.; Farmer, A.; Joshi, T.; Takahashi, K.; Langley, R.J.; Franklin, L.D.; He, J.; Xu, D.; May, G.; Stacey, G. An integrated transcriptome atlas of the crop model Glycine max, and its use in comparative analyses in plants. Plant. J. 2010, 63, 86–99. [Google Scholar] [CrossRef]
- Prince, S.J.; Joshi, T.; Mutava, R.N.; Syed, N.; Vitor, M.D.S.J.; Patil, G.; Song, L.; Wang, J.; Lin, L.; Chen, W.; et al. Comparative analysis of the drought-responsive transcriptome in soybean lines contrasting for canopy wilting. Plant. Sci. 2015, 240, 65–78. [Google Scholar] [CrossRef]
- Wang, L.; Dong, S.; Liu, L.; Ma, Y.; Li, S.; Zu, W. Transcriptome profiling reveals PEG-simulated drought, heat and combined stress response mechanisms in soybean. Comput. Biol. Chem. 2018, 77, 413–419. [Google Scholar] [CrossRef]
- Cortés, A.J.; Chavarro, M.C.; Blair, M.W. SNP marker diversity in common bean (Phaseolus vulgaris L.). Theor. Appl. Genet. 2011, 123, 827. [Google Scholar] [CrossRef]
- Blair, M.W.; Soler, A.; Cortes, A.J. Diversification and population structure in common beans (Phaseolus vulgaris L.). PLoS ONE 2012, 7, e49488. [Google Scholar] [CrossRef] [Green Version]
- Blair, M.W.; Cortés, A.J.; Penmetsa, R.V.; Farmer, A.; Carrasquilla-Garcia, N.; Cook, D.R. A high-throughput SNP marker system for parental polymorphism screening, and diversity analysis in common bean (Phaseolus vulgaris L.). Theor. Appl. Genet. 2013, 126, 535–548. [Google Scholar] [CrossRef]
- Galeano, C.H.; Cortés, A.J.; Fernández, A.C.; Soler, Á.; Franco-Herrera, N.; Makunde, G.; Vanderleyden, J.; Blair, M.W. Gene-based single nucleotide polymorphism markers for genetic and association mapping in common bean. BMC Genetics. 2012, 13, 1–11. [Google Scholar] [CrossRef]
- Das, A.; Rushton, P.J.; Rohila, J.S. Metabolomic profiling of soybeans (Glycine max L.) reveals the importance of sugar and nitrogen metabolism under drought and heat stress. Plants 2017, 6, 21. [Google Scholar] [CrossRef] [Green Version]
- Singh, D.; Singh, C.K.; Taunk, J.; Tomar, R.S.S.; Chaturvedi, A.K.; Gaikwad, K.; Pal, M. Transcriptome analysis of lentil (Lens culinaris Medikus) in response to seedling drought stress. BMC Genomics. 2017, 18, 206. [Google Scholar] [CrossRef] [Green Version]
- Pandey, A.; Chakraborty, S.; Datta, A.; Chakraborty, N. Proteomics approach to identify dehydration responsive nuclear proteins from chickpea (Cicer arietinum L.). Mol. Cell Proteomics. 2008, 7, 88–107. [Google Scholar] [CrossRef] [Green Version]
- Molina, C.; Rotter, B.; Horres, R.; Udupa, S.M.; Besser, B.; Bellarmino, L.; Baum, M.; Matsumura, H.; Terauchi, R.; Kahl, G.; et al. SuperSAGE: The drought stress-responsive transcriptome of chickpea roots. BMC Genomics 2008, 9, 553. [Google Scholar] [CrossRef] [Green Version]
- De Domenico, S.; Bonsegna, S.; Horres, R.; Pastor, V.; Taurino, M.; Poltronieri, P.; Imtiaz, M.; Kahl, G.; Flors, V.; Winter, P. Transcriptomic analysis of oxylipin biosynthesis genes and chemical profiling reveal an early induction of jasmonates in chickpea roots under drought stress. Plant. Physiol. Biochem. 2012, 61, 115–122. [Google Scholar] [CrossRef]
- Mahdavi Mashaki, K.; Garg, V.; Nasrollahnezhad Ghomi, A.A.; Kudapa, H.; Chitikineni, A.; Zaynali Nezhad, K.; Yamchi, A.; Soltanloo, H.; Varshney, R.K.; Thudi, M. RNA-Seq analysis revealed genes associated with drought stress response in kabuli chickpea (Cicer arietinum L.). PLoS ONE 2018, 13, e0199774. [Google Scholar] [CrossRef] [Green Version]
- Badhan, S.; Kole, P.; Ball, A.; Mantri, N. RNA sequencing of leaf tissues from two contrasting chickpea genotypes reveals mechanisms for drought tolerance. Plant. Physiol. Biochem. 2018, 129, 295–304. [Google Scholar] [CrossRef]
- Khandal, H.; Parween, S.; Roy, R.; Meena, M.K.; Chattopadhyay, D. MicroRNA profiling provides insights into post-transcriptional regulation of gene expression in chickpea root apex under salinity and water deficiency. Sci. Rep. 2017, 7, 4632. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Wang, L.; Li, L.; Wang, S. De novo assembly of the common bean transcriptome using short reads for the discovery of drought-responsive genes. PLoS ONE 2014, 9, e109262. [Google Scholar] [CrossRef] [Green Version]
- Pereira, W.J.; Melo, A.T.D.O.; Coelho, A.S.G.; Rodrigues, F.A.; Mamidi, S.; Alencar, S.A.D.; Lanna, A.C.; Valdisser, P.A.M.R.; Brondani, C.; Nascimento-Júnior, I.R.D.; et al. Genome-wide analysis of the transcriptional response to drought stress in root and leaf of common bean. Genet. Mol. Biol. 2020, 43, e20180259. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Zhang, W.J.; Yin, Z.G.; Li, W.J.; Zhao, H.H.; Zhang, S.; Zhuang, L.; Wang, Y.X.; Zhang, W.H.; Du, J.D. Genome—And transcriptome-wide identification of C3Hs in common bean (Phaseolus vulgaris L.) and structural and expression-based analyses of their functions during the sprout stage under salt-stress conditions. Front. Genet. 2020, 11, 564607. [Google Scholar] [CrossRef]
- Hiz, M.C.; Canher, B.; Niron, H.; Turet, M. Transcriptome analysis of salt tolerant common bean (Phaseolus vulgaris L.) under saline conditions. PLoS ONE 2014, 9, e92598. [Google Scholar] [CrossRef] [Green Version]
- Coetzer, N.; Gazendam, I.; Oelofse, D.; Berger, D.K. SSHscreen and SSHdb, generic software for microarray-based gene discovery: Application to the stress response in cowpea. Plant. Methods 2010, 6, 10. [Google Scholar] [CrossRef] [Green Version]
- Zuo, J.; Wang, Y.; Zhu, B.; Luo, Y.; Wang, Q.; Gao, L. sRNAome and transcriptome analysis provide insight into chilling response of cowpea pods. Gene 2018, 671. [Google Scholar] [CrossRef]
- Khan, M.A.; Alghamdi, S.S.; Ammar, M.H.; Sun, Q.; Teng, F.; Migdadi, H.M.; Al-Faifi, S.A. Transcriptome profiling of faba bean (Viciafaba L.) drought-tolerant variety hassawi-2 under drought stress using RNA sequencing. Electron. J. Biotechnol. 2019, 39, 15–29. [Google Scholar] [CrossRef]
- Alghamdi, S.S.; Khan, M.A.; Ammar, M.H.; Sun, Q.; Huang, L.; Migdadi, H.M.; El-Harty, E.H.; Al-Faifi, S.A. Characterization of drought stress-responsive root transcriptome of faba bean (Viciafaba L.) using RNA sequencing. 3 Biotech. 2018, 8, 502. [Google Scholar] [CrossRef]
- Yang, F.; Chen, H.; Liu, C.; Li, L.; Liu, L.; Han, X.; Wan, Z.; Sha, A. Transcriptome profile analysis of two Vicia faba cultivars with contrasting salinity tolerance during seed germination. Sci. Rep. 2020, 10, 7250. [Google Scholar] [CrossRef]
- Singh, D.; Singh, C.K.; Taunk, J.; Jadon, V.; Pal, M.; Gaikwad, K. Genome wide transcriptome analysis reveals vital role of heat responsive genes in regulatory mechanisms of lentil (Lens culinaris Medikus). Sci. Rep. 2019, 9, 12976. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Li, S.; Liu, Y.; Liu, X. Transcriptomic profiling reveals metabolic and regulatory pathways in the desiccation tolerance of mungbean (Vigna radiata [L.] R. Wilczek). Front. Plant. Sci. 2016, 7, 1921. [Google Scholar] [CrossRef] [Green Version]
- Severin, A.J.; Woody, J.L.; Bolon, Y.T.; Joseph, B.; Diers, B.W.; Farmer, A.D.; Muehlbauer, G.J.; Nelson, R.T.; Grant, D.; Specht, J.E.; et al. RNA-Seq Atlas of Glycine max: A guide to the soybean transcriptome. BMC Plant. Biol. 2010, 10, 160. [Google Scholar] [CrossRef] [Green Version]
- Dash, S.; Van Hemert, J.; Hong, L.; Wise, R.P.; Dickerson, J.A. PLEXdb: Gene expression resources for plants and plant pathogens. Nucleic Acids Res. 2012, 40, D1194–D1201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garg, R.; Patel, R.K.; Jhanwar, S.; Priya, P.; Bhattacharjee, A.; Yadav, G.; Bhatia, S.; Chattopadhyay, D.; Tyagi, A.K.; Jain, M. Gene discovery and tissue-specific transcriptome analysis in chickpea with massively parallel pyrosequencing and web resource development. Plant. Physiol. 2011, 156, 1661–1678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kudapa, H.; Garg, V.; Chitikineni, A.; Varshney, R.K. The RNA-Seq-based high resolution gene expression atlas of chickpea (Cicer arietinum L.) reveals dynamic spatio-temporal changes associated with growth and development. Plant. Cell Environ. 2018, 41, 2209–2225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Rourke, J.A.; Iniguez, L.P.; Fu, F.; Bucciarelli, B.; Miller, S.S.; Jackson, S.A.; McClean, P.E.; Li, J.; Dai, X.; Zhao, P.X.; et al. An RNA-Seq based gene expression atlas of the common bean. BMC Genomics 2014, 15, 866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, S.; Jiang, C.; Huang, Z.; Torres-Jerez, I.; Chang, J.; Zhang, H.; Udvardi, M.; Liu, R.; Verdier, J. The Vigna unguiculata Gene expression atlas (VuGEA) from de novo assembly and quantification of RNA-seq data provides insights into seed maturation mechanisms. Plant. J. 2016, 88, 318–327. [Google Scholar] [CrossRef]
- Alves-Carvalho, S.; Aubert, G.; Carrere, S.; Cruaud, C.; Brochot, A.L.; Jacquin, F.; Klein, A.; Martin, C.; Boucherot, K.; Kreplak, J.; et al. Full-length de novo assembly of RNA-seq data in pea (Pisumsativum L.) provides a gene expression atlas and gives insights into root nodulation in this species. Plant. J. 2015, 84, 1–19. [Google Scholar] [CrossRef]
- Pazhamala, L.T.; Purohit, S.; Saxena, R.K.; Garg, V.; Krishnamurthy, L.; Verdier, J.; Varshney, R.K. Gene expression atlas of pigeonpea and its application to gain insights into genes associated with pollen fertility implicated in seed formation. J. Exp. Bot. 2017, 68, 2037–2054. [Google Scholar] [CrossRef] [Green Version]
- Arikit, S.; Xia, R.; Kakrana, A.; Huang, K.; Zhai, J.; Yan, Z.; Valdés-López, O.; Prince, S.; Musket, T.A.; Nguyen, H.T.; et al. An atlas of soybean small RNAs identifies phased siRNAs from hundreds of coding genes. Plant. Cell 2014, 26, 4584–4601. [Google Scholar] [CrossRef] [Green Version]
- Arbona, V.; Manzi, M.; Ollas, C.D.; Gómez-Cadenas, A. Metabolomics as a tool to investigate abiotic stress tolerance in plants. Int. J. Mol. Sci. 2013, 14, 4885–4911. [Google Scholar] [CrossRef]
- Jorrín, J.V.; Maldonado, A.M.; Castillejo, M.A. Plant proteome analysis: A 2006 update. Proteomics. 2007, 7, 2947–2962. [Google Scholar] [CrossRef]
- Hakeem, K.R.; Chandna, R.; Ahmad, P.; Iqbal, M.; Ozturk, M. Relevance of proteomic investigations in plant abiotic stress physiology. OMICS 2012, 16, 621–635. [Google Scholar] [CrossRef]
- Wienkoop, S.; Morgenthal, K.; Wolschin, F.; Scholz, M.; Selbig, J.; Weckwerth, W. Integration of metabolomic and proteomic phenotypes analysis of data covariance dissects starch and RFO metabolism from low and high temperature compensation response in Arabidopsis thaliana. Mol. Cell Proteomics. 2008, 7, 1725–1736. [Google Scholar] [CrossRef] [Green Version]
- Vessal, S.; Arefian, M.; Siddique, K.H.M. Proteomic responses to progressive dehydration stress in leaves of chickpea seedlings. BMC Genomics 2020, 21, 523. [Google Scholar] [CrossRef]
- Gupta, S.; Mishra, S.K.; Misra, S.; Pandey, V.; Agrawal, L.; Nautiyal, C.S.; Chauhan, P.S. Revealing the complexity of protein abundance in chickpea root under drought-stress using a comparative proteomics approach. Plant. Physiol. Biochem. 2020, 151, 88–102. [Google Scholar] [CrossRef]
- Cevik, S.; Akpinar, G.; Yildizli, A.; Kasap, M.; Karaosmanoğlu, K.; Ünyayar, S. Comparative physiological and leaf proteome analysis between drought-tolerant chickpea Cicer reticulatum and drought-sensitive chickpea C. arietinum. J. Biosci. 2019, 44, 20. [Google Scholar] [CrossRef]
- Khan, N.; Bano, A.; Rahman, M.A.; Guo, J.; Kang, Z.; Babar, M.A. Comparative physiological and metabolic analysis reveals a complex mechanism involved in drought tolerance in chickpea (Cicer arietinum L.) induced by PGPR and PGRs. Sci. Rep. 2019, 9, 2097. [Google Scholar] [CrossRef]
- Goufo, P.; Moutinho-Pereira, J.M.; Jorge, T.F.; Correia, C.M.; Oliveira, M.R.; Rosa, E.A.; António, C.; Trindade, H. Cowpea (Vigna unguiculata L. Walp.) metabolomics: Osmoprotection as a physiological strategy for drought stress resistance and improved yield. Front. Plant. Sci. 2017, 8, 586. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Ruperao, P.; Batley, J.; Edwards, D.; Khan, T.; Colmer, T.D.; Pang, J.; Siddique, K.H.; Sutton, T. Investigating drought tolerance in chickpea using genome-wide association mapping and genomic selection based on whole-genome resequencing data. Front. Plant. Sci. 2018, 9, 190. [Google Scholar] [CrossRef] [Green Version]
- Arefian, M.; Vessal, S.; Malekzadeh-Shafaroudi, S.; Siddique, K.H.; Bagheri, A. Comparative proteomics and gene expression analyses revealed responsive proteins and mechanisms for salt tolerance in chickpea genotypes. BMC Plant. Biol. 2019, 19, 300. [Google Scholar] [CrossRef] [Green Version]
- Richter, J.A.; Behr, J.H.; Erban, A.; Kopka, J.; Zörb, C. Ion-dependent metabolic responses of Vicia faba L to salt stress. Plant Cell Environ. 2018, 42, 295–309. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Guo, R.; Jiao, Y.; Jin, X.; Zhang, H.; Shi, L. Comparison of salt tolerance in Soja based on metabolomics of seedling roots. Front. Plant. Sci. 2017, 8, 1101. [Google Scholar] [CrossRef] [PubMed]
- Parankusam, S.; Bhatnagar-Mathur, P.; Sharma, K.K. Heat responsive proteome changes reveal molecular mechanisms underlying heat tolerance in chickpea. Environ. Exp. Bot. 2017, 141, 132–144. [Google Scholar] [CrossRef] [Green Version]
- Duressa, D.; Soliman, K.; Taylor, R.; Senwo, Z. Proteomic analysis of soybean roots under aluminum stress. Int. J. Plant. Genomics 2011, 2011, 282531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langridge, P.; Fleury, D. Making the most of ‘omics’ for crop breeding. Trends Biotechnol. 2011, 29, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.K. Translational genomics and multi-omics integrated approaches as a useful strategy for crop breeding. Genes Genomics. 2019, 41, 133–146. [Google Scholar] [CrossRef] [Green Version]
- Varshney, R.K.; Kudapa, H.; Pazhamala, L.; Chitikineni, A.; Thudi, M.; Bohra, A.; Gaur, P.M.; Janila, P.; Fikre, A.; Kimurto, P.; et al. Translational genomics in agriculture: Some examples in grain legumes. Critical Rev. Plant. Sci. 2015, 34, 169–194. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Reif, J.C.; Jiang, Y.; Wen, Z.; Wang, D.; Liu, Z.; Guo, Y.; Wei, S.; Wang, S.; Yang, C.; et al. Potential of marker selection to increase prediction accuracy of genomic selection in soybean (Glycine max L.). Mol. Breed. 2016, 36, 113. [Google Scholar] [CrossRef] [Green Version]
- Sinha, A.; Haider, T.; Narula, K.; Ghosh, S.; Chakraborty, N.; Chakraborty, S. Integrated seed proteome and phosphoproteome analyses reveal interplay of nutrient dynamics, carbon-nitrogen partitioning, and oxidative signalling in chickpea. Proteomics 2020, 20, e1900267. [Google Scholar] [CrossRef]
- Yang, Z.B.; Eticha, D.; Führs, H.; Heintz, D.; Ayoub, D.; Van Dorsselaer, A.; Schlingmann, B.; Rao, I.M.; Braun, H.P.; Horst, W.J. Proteomic and phosphoproteomic analysis of polyethylene glycol-induced osmotic stress in root tips of common bean (Phaseolus vulgaris L.). J. Exp. Bot. 2013, 64, 5569–5586. [Google Scholar] [CrossRef] [Green Version]
- Tripathi, P.; Rabara, R.C.; Reese, R.N.; Miller, M.A.; Rohila, J.S.; Subramanian, S.; Shen, Q.J.; Morandi, D.; Bücking, H.; Shulaev, V. A toolbox of genes, proteins, metabolites and promoters for improving drought tolerance in soybean includes the metabolite coumestrol and stomatal development genes. BMC Genomics 2016, 17, 102. [Google Scholar] [CrossRef] [Green Version]
- Deshmukh, R.K.; Vivancos, J.; Guérin, V.; Sonah, H.; Labbé, C.; Belzile, F.; Bélanger, R.R. Identification and functional characterization of silicon transporters in soybean using comparative genomics of major intrinsic proteins in Arabidopsis and rice. Plant. Mol. Biol. 2013, 83, 303–315. [Google Scholar] [CrossRef]
- Pi, E.; Zhu, C.; Fan, W.; Huang, Y.; Qu, L.; Li, Y.; Zhao, Q.; Ding, F.; Qiu, L.; Wang, H.; et al. Quantitative phosphoproteomic and metabolomic analyses reveal GmMYB173 optimizes flavonoid metabolism in soybean under salt Stress. Mol. Cell Proteomics 2018, 17, 1209–1224. [Google Scholar] [CrossRef] [Green Version]
- Pi, E.; Qu, L.; Hu, J.; Huang, Y.; Qiu, L.; Lu, H.; Jiang, B.; Liu, C.; Peng, T.; Zhao, Y.; et al. Mechanisms of soybean roots’ tolerances to salinity revealed by proteomic and phosphoproteomic comparisons between two cultivars. Mol. Cell Proteomics 2016, 15, 266–288. [Google Scholar] [CrossRef] [Green Version]
- Valdés-López, O.; Batek, J.; Gomez-Hernandez, N.; Nguyen, C.T.; Isidra-Arellano, M.C.; Zhang, N.; Joshi, T.; Xu, D.; Hixson, K.K.; Weitz, K.K.; et al. Soybean roots grown under heat stress show global changes in their transcriptional and proteomic profiles. Front. Plant. Sci. 2016, 7, 517. [Google Scholar] [CrossRef] [Green Version]
- Harfouche, A.L.; Jacobson, D.A.; Kainer, D.; Romero, J.C.; Harfouche, A.H.; Mugnozza, G.S.; Moshelion, M.; Tuskan, G.A.; Keurentjes, J.J.; Altman, A. Accelerating climate resilient plant breeding by applying next-generation artificial intelligence. Trends Biotechnol. 2019, 37, 1217–1235. [Google Scholar] [CrossRef]
- Negin, B.; Moshelion, M. The advantages of functional phenotyping in pre-field screening for drought-tolerant crops. Funct. Plant. Biol. 2016, 44, 1–107. [Google Scholar] [CrossRef]
- Salter, W.T.; Shrestha, A.; Barbour, M.M. Open source 3D phenotyping of chickpea plant architecture across plant development. BioRxiv 2020. [Google Scholar] [CrossRef]
- Burridge, J.; Jochua, C.N.; Bucksch, A.; Lynch, J.P. Legume shovelomics: High—Throughput phenotyping of common bean (Phaseolus vulgaris L.) and cowpea (Vigna unguiculata subsp, unguiculata) root architecture in the field. Field Crops Res. 2016, 192, 21–32. [Google Scholar] [CrossRef]
- Nguyen, G.N.; Norton, S.L.; Rosewarne, G.M.; James, L.E.; Slater, A.T. Automated phenotyping for early vigour of field pea seedlings in controlled environment by colour imaging technology. PLoS ONE 2018, 13, e0207788. [Google Scholar] [CrossRef] [Green Version]
- Humplík, J.F.; Lazár, D.; Fürst, T.; Husičková, A.; Hýbl, M.; Spíchal, L. Automated integrative high-throughput phenotyping of plant shoots: A case study of the cold-tolerance of pea (Pisum sativum L.). Plant. Methods 2015, 11, 20. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Chen, H.; Zhou, J.; Fu, X.; Ye, H.; Nguyen, H.T. Development of an automated phenotyping platform for quantifying soybean dynamic responses to salinity stress in greenhouse environment. Comput. Electron. Agr. 2018, 151, 319–330. [Google Scholar] [CrossRef]
- Peirone, L.S.; PereyraIrujo, G.A.; Bolton, A. Assessing the efficiency of phenotyping early traits in a greenhouse automated platform for predicting drought tolerance of soybean in the field. Front. Plant. Sci. 2018, 9, 587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naik, H.S.; Zhang, J.; Lofquist, A.; Assefa, T.; Sarkar, S.; Ackerman, D.; Singh, A.; Singh, A.K.; Ganapathysubramanian, B. A real-time phenotyping framework using machine learning for plant stress severity rating in soybean. Plant. Methods. 2017, 13, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Libbrecht, M.W.; Noble, W.S. Machine learning applications in genetics and genomics. Nat. Rev. Genet. 2015, 16, 321–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrider, D.R.; Kern, A.D. Supervised machine learning for population genetics: A new paradigm. Trends Genet. 2018, 343, 301–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortés, A.J.; Restrepo-Montoya, M.; Bedoya-Canas, L.E. Modern strategies to assess and breed forest tree adaptation to changing climate. Front. Plant. Sci. 2020, 11, 1606. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, D.; He, F.; Wang, J.; Joshi, T.; Xu, D. Phenotype prediction and genome-wide association study using deep convolutional neural network of soybean. Front. Genet. 2019, 10, 1091. [Google Scholar] [CrossRef]
- Corrêa, A.M.; Teodoro, P.E.; Gonçalves, M.C.; Barroso, L.M.A.; Nascimento, M.; Santos, A.; Torres, F.E. Artificial intelligence in the selection of common bean genotypes with high phenotypic stability. Genet. Mol. Res. 2016, 15, gmr-15028230. [Google Scholar] [CrossRef]
- Cortés, A.J.; López-Hernández, F. Harnessing Crop Wild Diversity for Climate Change Adaptation. Genes 2021, 12, 783. [Google Scholar] [CrossRef]
- Falk, K.G.; Jubery, T.Z.; Mirnezami, S.V.; Parmley, K.A.; Sarkar, S.; Singh, A.; Ganapathysubramanian, B.; Singh, A.K. Computer vision and machine learning enabled soybean root phenotyping pipeline. Plant. Methods. 2020, 16, 5. [Google Scholar] [CrossRef]
- Cortés, A.J.; López-Hernández, F.; Osorio-Rodriguez, D. Predicting thermal adaptation by looking into populations’ genomic past. Front. Genet. 2020, 11, 1093. [Google Scholar] [CrossRef]
- Jenko, J.; Gorjanc, G.; Cleveland, M.A.; Varshney, R.K.; Whitelaw, C.B.A.; Woolliams, J.A.; Hickey, J.M. Potential of promotion of alleles by genome editing to improve quantitative traits in livestock breeding programs. Genet. Sel. Evol. 2015, 47, 55. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Cimen, E.; Singh, N.; Buckler, E. Deep learning for plant genomics and crop improvement. Curr. Opin. Plant. Biol. 2020, 54, 34–41. [Google Scholar] [CrossRef]
- Abadi, S.; Yan, W.X.; Amar, D.; Mayrose, I. A machine learning approach for predicting CRISPR-Cas9 cleavage efficiencies and patterns underlying its mechanism of action. PLoS Comput. Biol. 2017, 13, e1005807. [Google Scholar] [CrossRef]
- Lin, J.; Wong, K.C. Off-target predictions in CRISPR-Cas9 gene editing using deep learning. Bioinformatics 2018, 34, i656–i663. [Google Scholar] [CrossRef] [Green Version]
- Vakilian, K.A. Machine learning improves our knowledge about miRNA functions towards plant abiotic stresses. Sci. Rep. 2020, 10, 1–10. [Google Scholar]
- Varshney, R.K.; Pandey, M.K.; Bohra, A.; Singh, V.K.; Thudi, M.; Saxena, R.K. Toward the sequence-based breeding in legumes in the post-genome sequencing era. Theor. Appl. Genet. 2019, 132, 797–816. [Google Scholar] [CrossRef] [Green Version]
- Lenz, P.R.; Nadeau, S.; Mottet, M.J.; Perron, M.; Isabel, N.; Beaulieu, J.; Bousquet, J. Multi-trait genomic selection for weevil resistance, growth, and wood quality in Norway Spruce. Evol. Appl. 2020, 13, 76–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, H.; Nikoloski, Z. Machine learning approaches for crop improvement: Leveraging phenotypic and genotypic big data. J. Plant. Physiol. 2021, 257, 153354. [Google Scholar] [CrossRef] [PubMed]
- Cortés, A.J.; Liu, X.; Sedlacek, J.; Wheeler, J.A.; Lexer, C.; Karrenberg, S. Maintenance of Female-Bias in a Polygenic Sex Determination System is Consistent with Genomic Conflict. In On the Big Challenges of a Small Shrub: Ecological Genetics of Salix Herbacea, L.; Acta Universitatis Upsaliensis: Uppsala, Sweden, 2015. [Google Scholar]
- Crossa, J.; Martini, J.W.; Gianola, D.; Pérez-Rodríguez, P.; Jarquin, D.; Juliana, P.; Montesinos-López, O.; Cuevas, J. Deep kernel and deep learning for genome-based prediction of single traits in multienvironment breeding trials. Front. Genet. 2019, 10, 1168. [Google Scholar] [CrossRef] [PubMed]
- Abdollahiarpanahi, R.; Gianola, D.; Peñagaricano, F. Deep learning versus parametric and ensemble methods for genomic prediction of complex phenotypes. Genet. Sel. Evol. 2020, 52, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
| S. No. | Species | Strategy | Accession/Variety/Cultivar | Genome Size (Gbp) Coverage/Estimated | Reference |
|---|---|---|---|---|---|
| 1. | Chickpea (Cicer arietinum L.) | Illumina sequencing of 11 genomic libraries (180 bp to 20 kb) | CDC Frontier, a kabuli chickpea | 544.73 (738.09) | [7] |
| 2. | Pigeonpea (Cajanus cajan) | Illumina GA and HiSeq 2000 Sequencing system, Sanger-based bacterial artificial chromosome end sequencing | Pigeonpea genotype ICPL 87119 (Asha) | 605.78 (833.07) | [10] |
| 2. | Cowpea (Vigna unguiculata) | PacBio (Pacific Biosciences of California, Menlo Park, CA, USA) and Single-molecule real-time (SMRT) sequencing | Cowpea IT97K-499-35 | 519 (613) | [11] |
| 3. | Lentil (Lens culinaris) | Illumina sequencing | CDC cultivar Redberry | 2600 (4200) | [34] |
| 4. | Mungbean (Vigna radiata) | Illumina Hiseq2000 and GS FLX+, with five libraries of a 180-bp fragment, 5, 10, and 40-kb mate-pairs, and one single linear library | VC1973A | 543 (579) | [6] |
| 4. | Lotus (Lotus japonicus) | Clone-by-clone sequencing and shotgun sequencing | Miyakojima MG-20 | 315 (472) | [35] |
| 5. | Peanut (Arachis hypogaea) | Single-molecule real-time cells (204) run on PacBio RS II system, 14 cells run on the Sequel system, with P6/C4 chemistry | Peanut var. Shitouqi | 2540 (2890) | [36] |
| 7. | Soybean (Glycine max) | Whole-genome shotgun approach using Sanger sequencing protocols on ABI 3730XL capillary sequencing machines | Soybean var. Williams 82 | 950 (1115) | [9] |
| Pulse | Abiotic Stress | Omics Technology | Details | Reference |
|---|---|---|---|---|
| Chickpea | Drought | Phosphoproteomics | Phosphorylation of proteins triggered by progressive water deficit conditions. | [80] |
| Secretomics | Comprehensive analyses of dehydration, stress-responsive secretome, and highly complex metabolic network function in the extracellular matrix. | [86] | ||
| Oxidative | Secretomics | Role of CaFer1 in iron buffering and adaptation to oxidative stress under changing environmental conditions. | [87] | |
| Common bean | Chlorpyrifos | Lipidomics | Decrease in triacylglycerol levels in pods and seeds. | [88] |
| Soybean | Heat | Lipidomics | Decreased levels of lipids containing 18:3 acyl chains due to reduced expression of fatty acid desaturase. | [89] |
| Low phosphorus | Lipidomics | Lipid remodelling under limited phosphorus conditions. | [90] | |
| Flooding | Phosphoproteomics | Ethylene signaling pathway played an important role in protein phosphorylation in root tips during flood stress. | [91] | |
| Glycoproteomics | Flooding negatively impacted the N-glycosylation of proteins. | [81] |
| Abiotic Stress | Crop | Parental Lines/Mapping/Genetic Population | Population Type | Trait Studied | Associated Marker(s) | QTLs/Linkage Group(s) | Phenotypic Variation Explained (PVE) | Reference |
|---|---|---|---|---|---|---|---|---|
| Drought | Chickpea | ILC 588 × ILC 3279 | RILs | Harvest Index, early flowering, and early maturity | 97 SSRs | QTLs: Q3-1 and Q1-1 on LG-1 and LG-3, respectively | 38% | [97] |
| ICC 8261 × ICC 283 and ICC 4958 × ICC 1882 | RILs | Root traits | 322 SSRs | Main effect (M) QTLs and epistatic (E) QTLs on CaLG01, CaLG02, CaLG03, CaLG04, CaLG05, CaLG06, CaLG07, and CaLG08 | M-QTLs: 60% E-QTLs: 90% | [7] | ||
| Cowpea | IT93K503-1 × CB46 | RILs | Stem greenness (stg) and recovery dry weight (rdw) | 306 AFLP markers | QTL Dro-1-10 (10 QTLs) | For drought related QTLs: 4.7–24.2% For maturity: 14.4–28.9% | [98] | |
| Common Bean | DOR364 × BAT477 | RILs | Photosynthate acquisition, accumulation, remobilization, and other drought-stress-related traits | 165 markers (AFLP, RAPD, SSRs) | b03, b05, b06, b08, b09, and b10 | 37% | [99] | |
| BRB 191 × SEQ 1027 | RILs | Drought-stress-related traits | 53 SNPs | Pv10 | 21% | [100] | ||
| ICA Bunsi × SXB405 | RILs | Pod-wall weight, whole-seed weight, whole-pod weight, 100-seed weight | 721 SNPs | Pv07 | 17% | [101] | ||
| BAT 881 × G21212 | RILs | Yield components, plant vigor, dry matter redistribution, phenological traits, and mineral nutrients | 53 AFLP, 2 RAPD, 42 SSRs, and 127 SNPs | Pv01 and Pv08 | 12.14–17.24% for the differential stress response | [102] | ||
| SXB412 (A), INB827 (B), ALB213 (C), SEN56 (D), SCR2 (E), MIB778 (F), SCR9 (G), and INB841 (H); 8-way (ABCDEFGH) F1 | 8-way MAGIC population | Yield, 100-seed weight, iron and zinc accumulation, phenology, and pod harvest index | 20,615 SNPs and small indels (< 20 bp) | Pv01, Pv03, and Pv08 | 35.8 and 5.5% for the major QTL governing hotspot Pv01 | [103] | ||
| Lentil | ILL 6002 × ILL 5888 | RILs | Dry root weight, lateral root number, taproot length, specific root length, average tap root diameter, root surface area, dry shoot weight, shoot length at 12 and 22 days after sowing, growth rate, seedling vigor, chlorophyll content, root–shoot ratio, and wilting score | 220 SNPs and 180 AFLPs | QDRWVII: 21.93, QRSAVII: 21.94, QRSratioIX: 2.30, QLRNVII: 21.94, QSL12IV: 103.83, QSL12VI: 170.87, QSL12VII: 19.71, QDSWVII: 22.94, QSL22VII: 21.94, QLRNIII: 98.64, QSRLIV: 61.63, and QSPADVIII: 72.15. | 27.6 and 28.9% for the two consecutive seasons | [104] | |
| Soybean | Minsoy × Noir 1 | RILs | Yield | 665 markers (RFLP, SSR) | U14-L, U09-C2, and U11-M | U14-L (20–40%), U09-C2 (14%), and U11-M (23–29%) | [105] | |
| Pana × PI 567690 Magellan × PI 567731 | RILs | Slow wilting | 4117 SNPs | Gm05, Gm09, Gm12, Gm19 Gm06, and Gm10 | 7.8–10.4% for Gm05, Gm09, Gm12, and Gm19; 20–29.6% for Gm06, and Gm10. | [106] | ||
| Mungbean | Pagasa 7 × TC 1966 | RILs | Drought-related traits | 6 AFLPs | - | 13% | [107] | |
| VC2917 × ZL | RILs | Plant height, maximum leaf area, above-ground biomass, relative water content, days to flowering, seed yield, and drought tolerance index | 313 SSRs | qPH5A and qMLA2A | qPH5A (6.40–20.06%) and qMLA2A (6.97–7.94%) | [108] | ||
| Pea | P665 × cv Messire | RILs | Drought-related traits | 6 SSRs and 2 SNPs | A6, AA175, AC74, AD57, AB141, AB64, Psblox2, PsAAP2_SNP4, and DipeptIV_SNP1 | 20 to 57% | [109] | |
| Heat | Chickpea | ICC 4567 × ICC 15,614 | RILs | Number of filled pods/plot, grain yield/plot, total number of seeds/plot, and percentage of pods set | 271 SNPs | CaLG05 and CaLG06 | 50%< | [110] |
| Cowpea | CB27 × IT82E-18 | RILs | Heat-stress-related traits | 48 SNPs | Cht 5 | 11.5–18.1% | [111] | |
| Lentil | JL-3 × PDL-2 and E-153 × PDL-1 | F2 | Seedling survival and pods set | 7 SSRs | qHt_ss and qHt_ps | 12.1 and 9.23% for seedling survival and pods set, respectively. | [112] | |
| Cold/Frost | Chickpea | ICC 4958 and PI 489,777 | RILs | Cold-tolerance-related traits | 747 SNPs | CTCa3.1 and CTCa8.1 | 7.15 to 34.6% for CTCa3.1 and 11.5 to 48.4% for CTCa8.1 | [113] |
| Faba bean | Biparental population (BPP): Côte d’Or 1 (French landrace), Bean Pure Line 4628, and Gottingen Winter Bean population | RILs | Frost-tolerance-related traits | 5 SNPs | LGs (01, 02, 03, 04, 08, and 10) | 2.74 to 29.41% | [114] | |
| Lentil | WA8649090 × Precoz | RILs | Winter survival traits | 94 AFLP, 56 RAPD, 106 ISSR | LG4 | 22.9% | [115] | |
| Pea | Champagne × Terese | RILs | Frost tolerance and cold acclimation traits | 258 SNPs | LG5 and LG6 | 6.5 to 46.5% | [116] | |
| Soybean | Sigalia × Merlin | RILs | Pod number and cold-tolerance-specific traits | 7711 SNPs | Chr 11 | 20% | [117] | |
| Salinity | Chickpea | ICCV 2 × JG-62 | RILs | Seed yield, number, weight, flowering time, and shoot dry weight | 135 SSR | LG3 (QTL for seed number) LG6 (QTLs for seed number and seed weight) LG4 (QTLs for flowering and shoot dry weight) | 19% 14.8–49.7% 8.8–37.7% | [118] |
| ICCV 2 × JG 11 | RILs | Salinity- and yield-related traits | 28 SSRs and 28 SNPs | CaLG05 and CaLG07 | 12–17% | [119] | ||
| Cowpea | Vignaluteola × V. marina subsp. oblonga | F2 | Salt-tolerance- and domestication-related traits | 150 SSRs | LG1 | 20–50.7% | [120] | |
| Pea | Kaspa × Parafield | RILs | Salt tolerance traits | 705 SNPs | Ps III and VII | 12% (Ps III) and 19% (VII) | [121] | |
| Soybean | S-100 × Tokyo | F2:5 | Salt tolerance traits | 32 SSRs and 116 RFLPs | LG N | 29–45% | [122] | |
| Aluminum toxicity | Soybean | Zhonghuang 24 × Huaxia 3 | RIL | Al-tolerance-related traits | 2639 recombination bin markers (AFLP, RFLP, SSRs) | qRRE_04 and qAAC_04 | 7.09% (qRRE_04) and 8.98% (qAAC_04) | [123] |
| KF No.1 × NN1138-2 | RILs | Growth-related indicators for Al resistance, viz. relative total plant dry weight (RTDW), relative root dry weight (RRDW), and relative shoot dry weight (RSDW) | 11 SSRs | LG B1 | Four additive QTLs (29.39%), four epistatic QTLs (18.75%), and a collective unmapped minor QTL (43.07%) | [124] | ||
| Essex × Forrest | RILs | Physiological traits associated with Al tolerance | 14 DNA markers | LG F (Chr. 13) | 34% | [125] |
| Pulse Crop | (A)biotic Stress/Trait | Gene/TF | Gene/TF Family | Transgenic Plant | Reference |
|---|---|---|---|---|---|
| Chickpea | Drought and salinity | CaCIPK25 gene | CIPK | Tobacco | [131] |
| CAP2 TF | APETALA-2 | Tobacco | [132] | ||
| CaHDZ12 TF | HD-zip | Tobacco and Chickpea | [56] | ||
| Drought, salinity, and high temperature | CaZF gene | C2H2-zinc finger | Tobacco and Chickpea | [133] | |
| Drought | CaAFP gene | Defensin | Arabidopsis thaliana | [134] | |
| CarNAC2 TF | NAC | [135] | |||
| Common bean | ROS stress and wounding | PvACCase gene | Transferase enzyme family | Arabidopsis thaliana | [136] |
| Salinity | PvChOMT | O-methyltransferases | [137] | ||
| Mung bean | Osmotic stress | VrUBC1 gene | Mung Bean E2 Ubiquitin-Conjugating Enzyme | Arabidopsis thaliana | [138] |
| Pea | Salinity | p68 gene | DEAD-box protein family | Rice | [139] |
| Tobacco | [140] | ||||
| PDH45 gene | DNA helicase, initiation factor homologue | Rice and Tobacco | [141] | ||
| Cold, heat, salinity, drought, and freezing | ABR17 cDNA | Group 10 family of pathogenesis-related proteins (PR 10) | Arabidopsis thaliana | [142] | |
| Pigeonpea | PEG, NaCl, cold, and heat | Cajanus cajan cyclophilin (CcCYP), Cajanus cajan hybrid proline-rich protein (CcHyPRP), and Cajanus cajan cold and drought regulatory (CcCDR) genes | Cold- and drought-responsive gene; CYP gene family | Arabidopsis thaliana | [143] |
| Drought, salinity, and low temperature | C. cajan cold and drought regulatory (CcCDR) gene | Cold- and drought-responsive gene | [144] | ||
| Drought, salinity, and extreme temperatures | C. cajan cyclophilin (CcCYP) gene | CYP gene family | [145] | ||
| Drought, cold, and salt stress | C. cajan cold and drought regulatory (CcCDR) gene | Cold- and drought-responsive gene | Rice | [146] | |
| Soybean | Drought, salinity, and oxidative stress | GmTP55 gene | Antiquitin-like ALDH7 gene family | Arabidopsis thaliana and tobacco | [147] |
| Drought and high salinity | GmDREB2 gene | DREB TF family | [148] | ||
| Drought, high salinity, and resistance to Alternaria alternata, tobacco mosaic virus (TMV), and Ralstoniasola nacearum | GmERF3 gene | AP2/ERF TF family | Tobacco | [149] |
| Crop | Abiotic Stress | Tissue | Sequencing Platform | NCBI BioProject/Accession Number | Details | Reference |
|---|---|---|---|---|---|---|
| Chickpea | Drought | Root and Shoot | Illumina HiSeq 2500 | PRJNA396819 | TFs associated with drought tolerance were identified. | [173] |
| Root | Illumina HiSeq 2500 | PRJNA335939 | TFs (AP2-EREBP, bHLH, bZIP, C3H, MYB, NAC, WRKY, and MADS) associated with drought tolerance were identified. | [132] | ||
| Leaf | Illumina HiSeq 3000 | GSE104609 | RNA from leaf tissues at the leaf apical meristem stage was quantified and a total of 1562 genes were differentially expressed in the tolerant genotype. Drought-responsive genes were specifically upregulated in the tolerant genotype. | [174] | ||
| Salinity and drought | Root apex | Roche 454 FLX | PRJNA267525 | MiRNA-mediated post-transcriptional regulation of genes engaged in lateral root formation and re-patterning of root hair cells and with high affinity for K+ uptake under salinity and water deficiency conditions was dissected using root apex transcriptome profiling. | [175] | |
| Common bean | Drought | Leaf | Illumina GAIIx | SRR1523069 | Drought responsive genes differentially expressed during drought stress were identified. | [176] |
| Drought | Leaf and root | Illumina platforms (GAII and HiSeq 2000) | SRP077562 | Transcriptome data revealed new genes involved in response to drought stress. | [177] | |
| Salinity | Cotyledon, hypocotyl, and radicle | Illumina HiSeq 2500 PE 150 | PRJNA558376 | Role of zinc finger proteins (C3H) was elucidated during the sprouting stage under salinity stress. | [178] | |
| Root | Illumina HiSeq TM 2000 | SRP029243 | A total of 2678 TFs were identified from transcriptome data, 441 of which were responsible for salinity tolerance. | [179] | ||
| Cowpea | Drought | Leaf | Illumina deep sequencing technology | GSE26402 | Exclusive drought-responsive miRNAs were found. | [73] |
| Drought | Leaf | GSE20273 | A SSH database (http://sshdb.bi.up.ac.za/, accessed on 3 August 2021) was developed for drought-responsive genes. | [180] | ||
| Cold (Chilling) | Pods | Illumina HiSeq 2500 | - | sRNAomic and transcriptomic analysis revealed many sRNAs and miRNAs involved in response to chilling. | [181] | |
| Faba bean | Drought | Leaf | Illumina HiSeq 4000 | SRX3182042, SRX3182043, SRX3182046, SRX3182047 | A total of 538 and 642 putative TFs were identified during the vegetative and flowering stages, respectively. | [182] |
| Root | Illumina HiSeq 4000 | SRX3182040, SRX3182041, SRX3182044, SRX3182045 | Novel DEGs that showed a change in expression during drought were identified. | [183] | ||
| Salinity | Cotyledons | Illumina HiSeq 4000 | PRJNA591424 | A total of 1410 salinity-responsive genes were identified and significant up-regulation of these genes was observed in the salt-tolerant genotype. | [184] | |
| Lentil | Drought | Leaf | Illumina HiSeq 2500 | SRR3105360 | Genes involved in oxidation reduction processes, TCA cycle, organ senescence, and reduction of stomatal conductance were more severely upregulated in drought-tolerant genotypes than in drought-sensitive ones. | [92] |
| Heat | Leaf | Illumina HiSeq 2000 | SUB3390924 | Cell wall and secondary metabolite pathways were found to be majorly affected. | [185] | |
| Mung bean | Desiccation | Seed | Illumina HiSeq 2500 with PE125 | SRP077637 | Many TFs (MYB, AP2, and NAC), HSPs, LEA proteins, and genes encoding methyltransferase and histone were differentially expressed. | [186] |
| Crop | GEA | Details | Reference |
|---|---|---|---|
| Chickpea | CaGEA | The GEA was developed using tissues from 27 samples and RNA studies were done at five different stages, namely the germination, seedling, vegetative, reproductive, and senescence stages. Genes differentially expressed in drought QTL hotspots were also identified. | [190] |
| Common bean | PvGEA (http://plantgrn.noble.org/PvGEA/) | Regulation of nodulation, nitrogen use efficiency, etc. | [191] |
| Cowpea | VuGEA (http://vugea.noble.org/) | Conserved regulatory mechanism of miRNAs involved in drought stress and seed maturation. | [192] |
| Pea | Pea gene atlas portal PsCam (http://bios.dijon.inra.fr/FATAL/cgi/pscam.cgi) | The ‘Caméor’ (PsCam) unigene set allows for the identification of rare transcripts. It can be used to deduce the function of nodulation genes and genes responsible for abiotic stress tolerance. | [193] |
| Pigeonpea | CcGEA | An important resource for finding candidate genes responsible for specific developmental processes. | [194] |
| Soybean | http://www.soybase.org/soyseq | An important resource for studying seed filling and developmental genes. | [137] |
| http://digbio.missouri.edu/soybean_atlas | A database for comparative analyses with two model legume crops, i.e., Medicago truncatula and Lotus japonicus, together with the model plant Arabidopsis. | [107] | |
| Small RNA atlas | This atlas helps in identifying novel miRNAs and their targets in the genome. | [195] |
| Abiotic Stress | Crop | Omics Approach | Details | Reference |
|---|---|---|---|---|
| Drought | Chickpea | Proteomics | Potential resources for improving drought tolerance were identified. | [200] |
| Comparative proteomics | A total of 75 proteins were found to be differentially expressed in roots. | [201] | ||
| Comparative proteomics | MALDI-TOF/TOF-MS/MS analyses revealed 24 differently expressed proteins in leaves under drought stress. | [202] | ||
| Metabolomics | Effect of PGPRs under drought stress was identified using UPLC-HRMS analysis | [203] | ||
| Cowpea | Metabolomics | GC-TOF-MS profiling of primary metabolites and LC-DAD profiling of secondary metabolites under drought stress. Prolonged stress irrespective of the developmental stage affected the metabolome. | [204] | |
| Faba bean | Proteomics | Proteins including chitinase, Bet, and glutamate–glyoxylate aminotransferase were found to be upregulated in leaves under drought stress. | [205] | |
| Drought and Heat | Soybean | Metabolomics | Upregulation of nitrogen and metabolism under combined heat and drought stress. | [168] |
| Salinity | Chickpea | Comparative proteomics | Various proteins were found to be engaged in salinity tolerance. | [206] |
| Faba bean | Metabolomics | Molecules such as myo-inositol, allantoin, and glycerophosphoglycerol were found to be up-regulated in roots in response to salt stress. | [207] | |
| Soybean | Comparative metabolomics | A total of 47 different metabolites were found to be responsible for salt tolerance. | [208] | |
| Heat | Chickpea | Comparative proteomics | A total of 482 heat-responsive proteins were found to be engaged in heat stress tolerance. | [209] |
| Aluminium | Soybean | Comparative proteomics | MALDI TOF analysis revealed differential protein expression in roots under Al stress. | [210] |
| Crop | Stress/Trait/Genes | Pan-Omics Approach Used | Details | References |
|---|---|---|---|---|
| Chickpea | Abiotic stress | Proteomics and phosphoproteomics | Novel clues suggest that the ubiquitin–proteasome pathway regulates nutrient reallocation. An increased abundance of NAPs/NAPPs involved in redox sensing and signaling during seed development was observed. | [215] |
| Common bean | Osmotic stress | Proteomics and phosphoproteomics | Dehydrin played an important role in osmotic stress. | [216] |
| Soybean | Drought tolerance genes | Metabolomics, transcriptomics, and analyses of gene promoters | Metabolite coumestrol and stomatal development genes played important roles in drought tolerance. | [217] |
| Silicon transporter involved in (a)biotic stress tolerance | Comparative genomics, transcriptomics, and expression profiling | Two putative Si transporter genes, GmNIP2-1 and GmNIP2- 2, were identified. | [218] | |
| Salinity | Phosphoproteomics and metabolomics | Flavonoids were significantly upregulated after salt treatment. | [219] | |
| Salinity | Phosphoproteomics and proteomics | A total of 1163 differentially phosphorylated sites were found, of which ten MYB/MYB transcription factor-like proteins were identified, which were found to be involved in flavonol accumulation. | [220] | |
| Heat stress tolerance | Genome-wide transcriptomics and proteomics | Proteins involved in thermotolerance, chromatin remodelling, and post-transcriptional regulation under heat stress were identified. | [221] |
| Pulse Crop | Basis of Automated Platform | Details | Reference |
|---|---|---|---|
| Chickpea | Photogrammetry techniques | Open-source 3D phenotyping platform for plant architecture. | [224] |
| Common bean | Digital imaging techniques | Legume shovelomics—a high-throughput phenotyping platform for common bean and cowpea. | [225] |
| Cowpea | |||
| Pea | Color imaging technology | High-throughput phenotyping platform for early vigor detection of field pea seedlings responsible for water use efficiency and yield in changing environments. | [226] |
| RGB digital imaging | Advanced phenotyping platform for phenotyping pea shoots under cold stress. | [227] | |
| Soybean | Sensor-based technology | Automated phenotyping platform for assessment of salinity in soybean growing under greenhouse conditions. | [228] |
| Automated imaging combined with GlyPh | Automated phenotyping platform for predicting drought tolerance in soybeans growing in fields. | [229] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, D.; Chaudhary, P.; Taunk, J.; Singh, C.K.; Singh, D.; Tomar, R.S.S.; Aski, M.; Konjengbam, N.S.; Raje, R.S.; Singh, S.; et al. Fab Advances in Fabaceae for Abiotic Stress Resilience: From ‘Omics’ to Artificial Intelligence. Int. J. Mol. Sci. 2021, 22, 10535. https://doi.org/10.3390/ijms221910535
Singh D, Chaudhary P, Taunk J, Singh CK, Singh D, Tomar RSS, Aski M, Konjengbam NS, Raje RS, Singh S, et al. Fab Advances in Fabaceae for Abiotic Stress Resilience: From ‘Omics’ to Artificial Intelligence. International Journal of Molecular Sciences. 2021; 22(19):10535. https://doi.org/10.3390/ijms221910535
Chicago/Turabian StyleSingh, Dharmendra, Priya Chaudhary, Jyoti Taunk, Chandan Kumar Singh, Deepti Singh, Ram Sewak Singh Tomar, Muraleedhar Aski, Noren Singh Konjengbam, Ranjeet Sharan Raje, Sanjay Singh, and et al. 2021. "Fab Advances in Fabaceae for Abiotic Stress Resilience: From ‘Omics’ to Artificial Intelligence" International Journal of Molecular Sciences 22, no. 19: 10535. https://doi.org/10.3390/ijms221910535