
Universal Properties and Specificities of the β2-Adrenergic Receptor-Gs Protein Complex Activation Mechanism Revealed by All-Atom Molecular Dynamics Simulations


Abstract
:1. Introduction
2. Results and Discussion
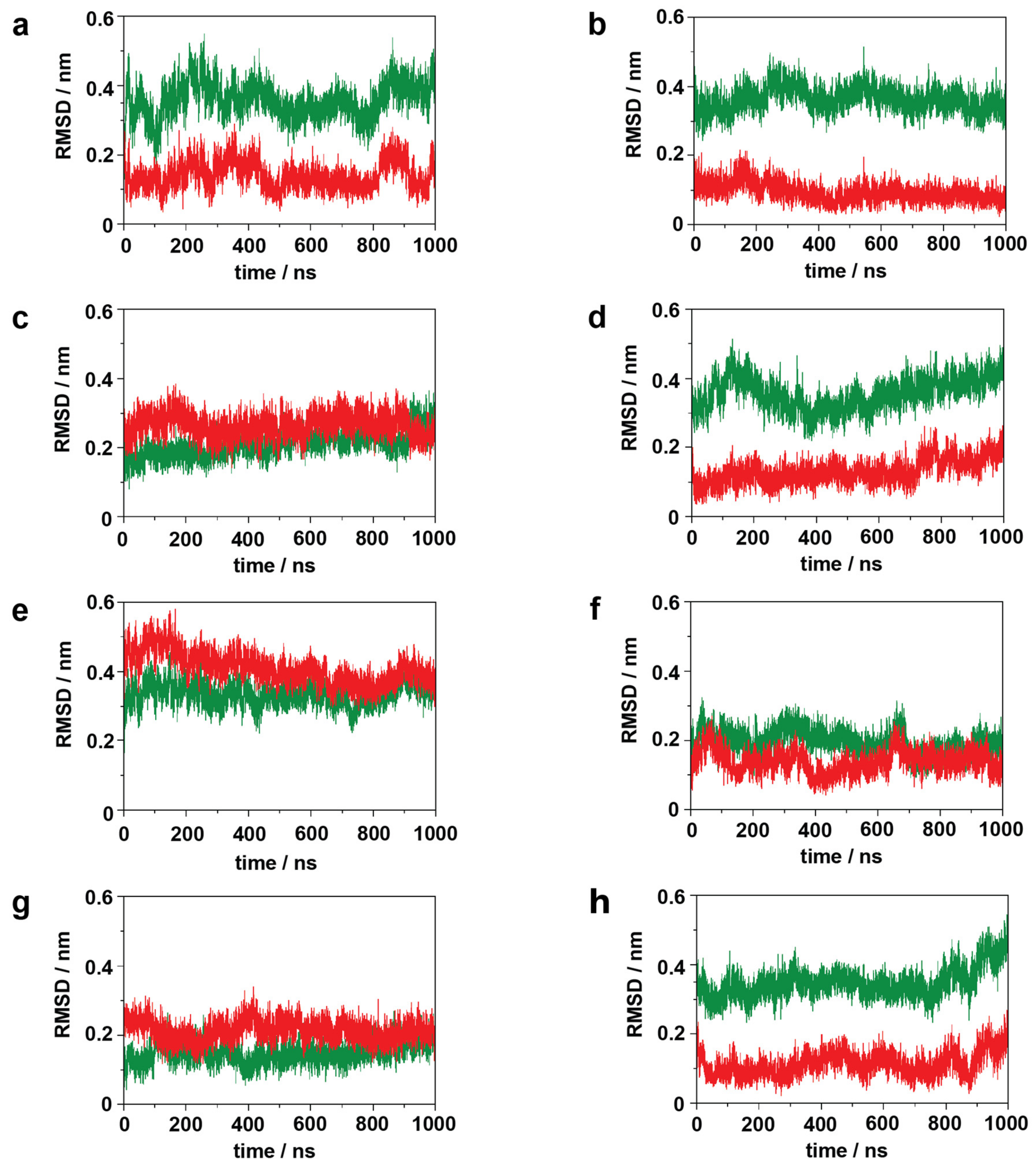
2.1. Simulation System Integrity
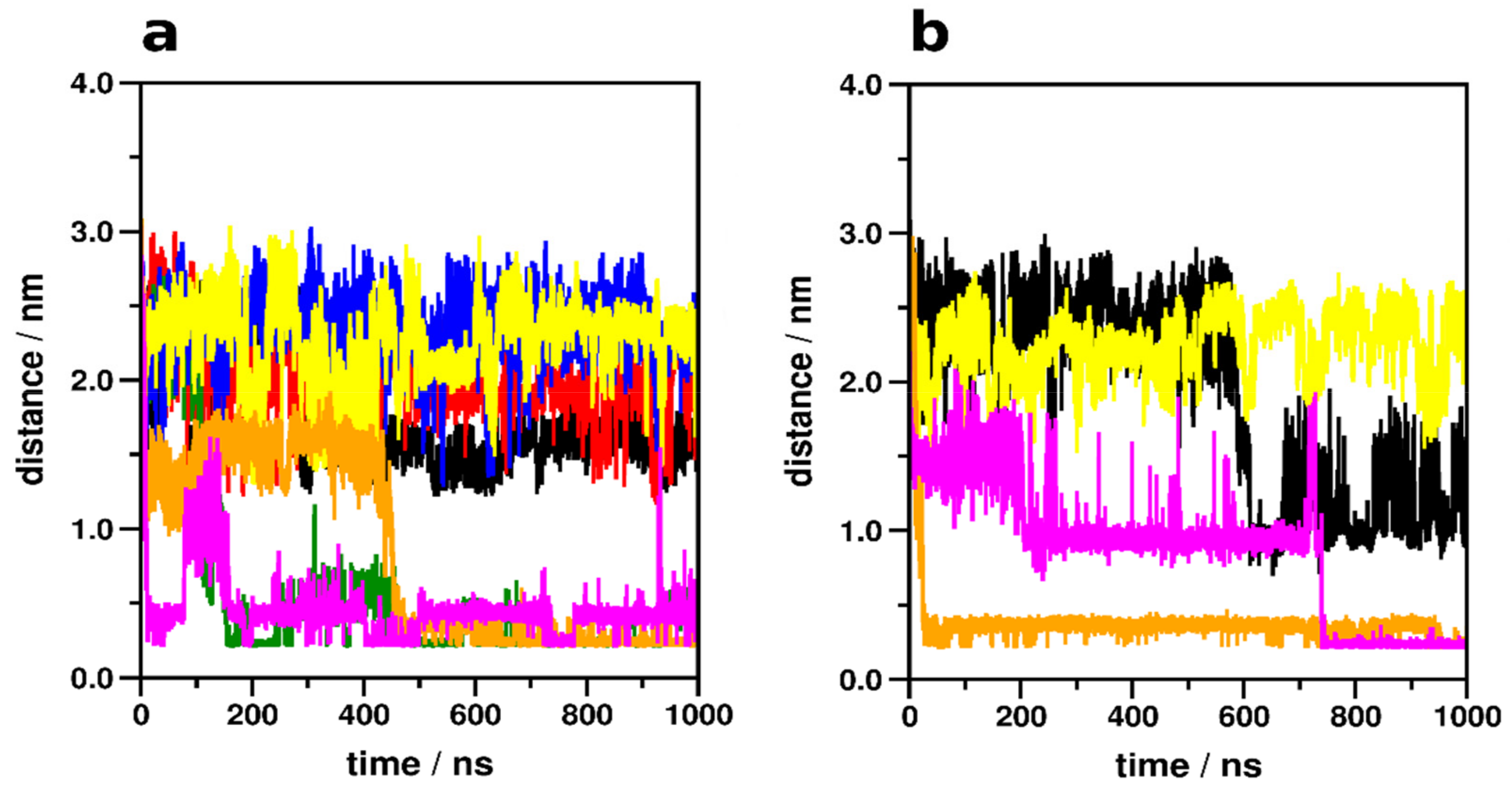
2.2. Allosteric Na+ Binding
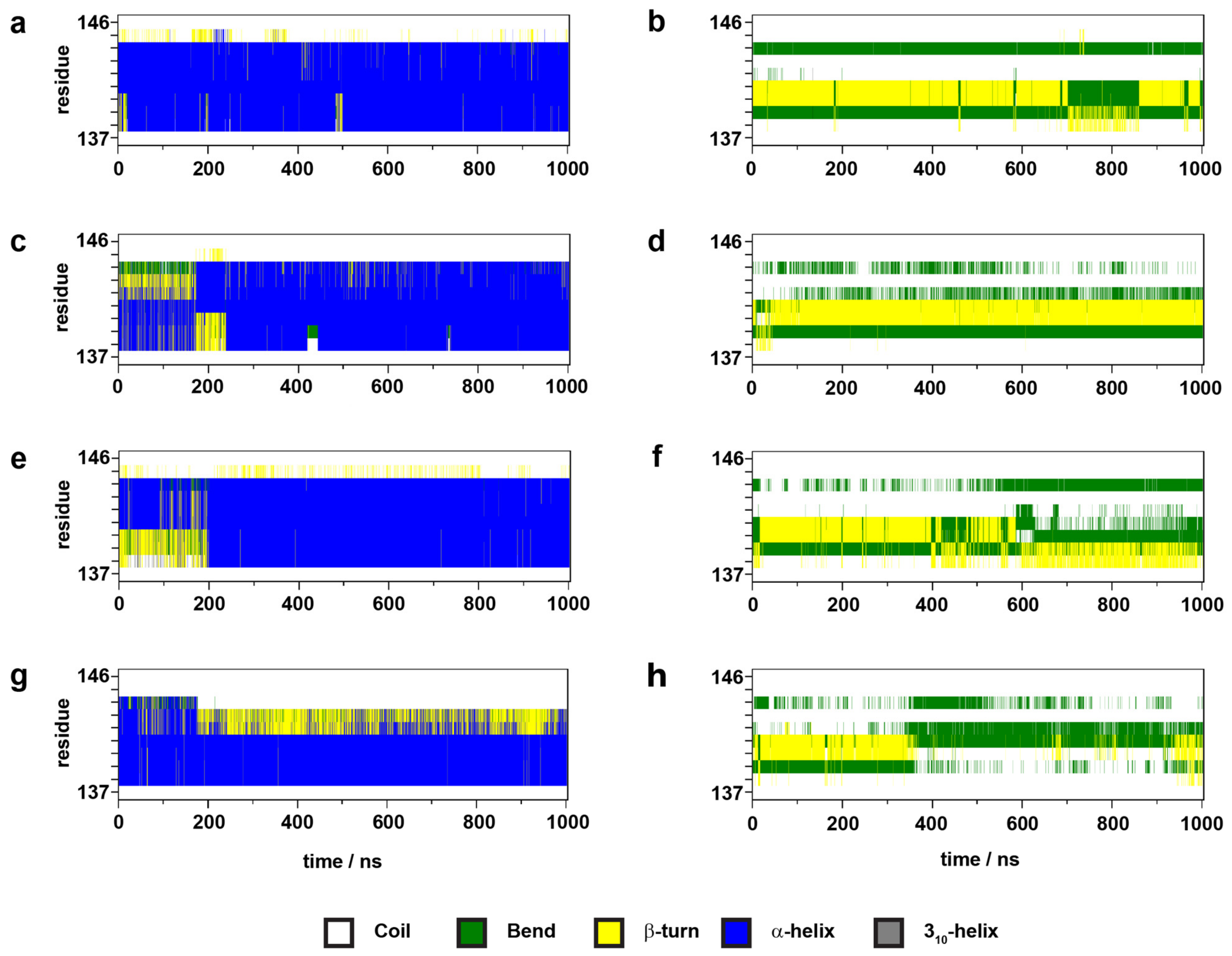
2.3. Transmembrane Helix and Loop Dynamics
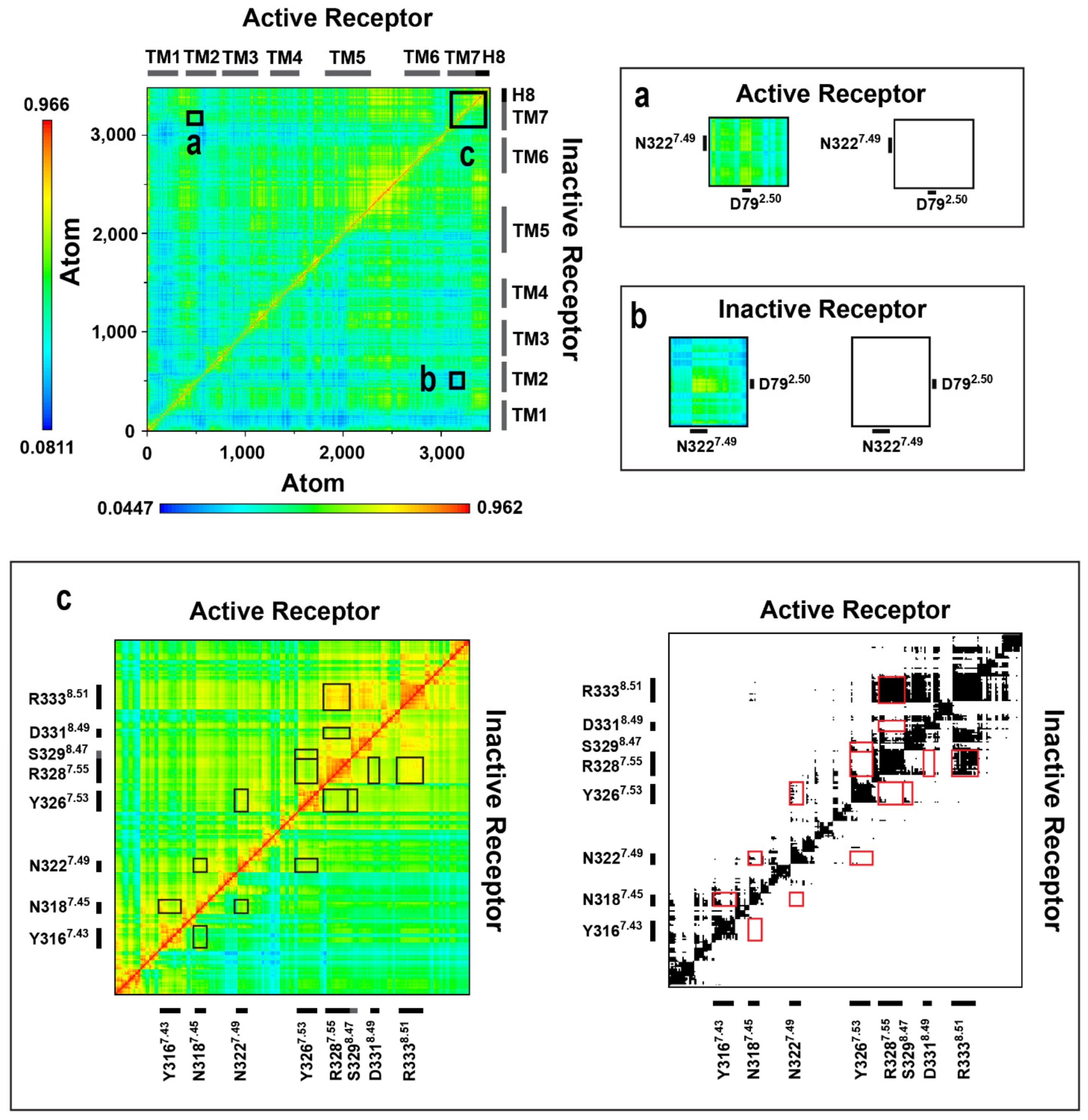
2.4. Correlated Side-Chain Motions in the Transmembrane Domain
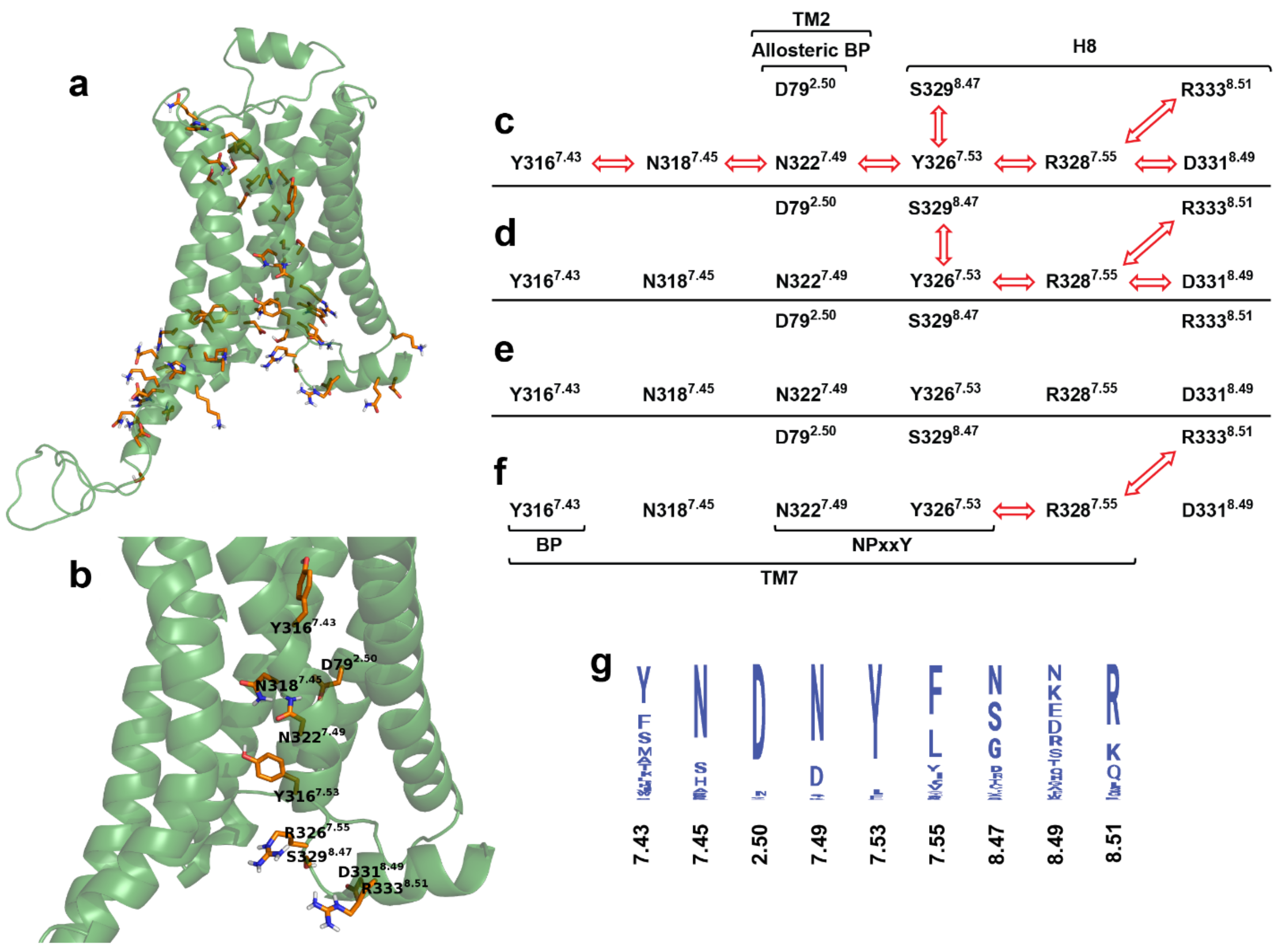
2.5. Intramolecular Interactions
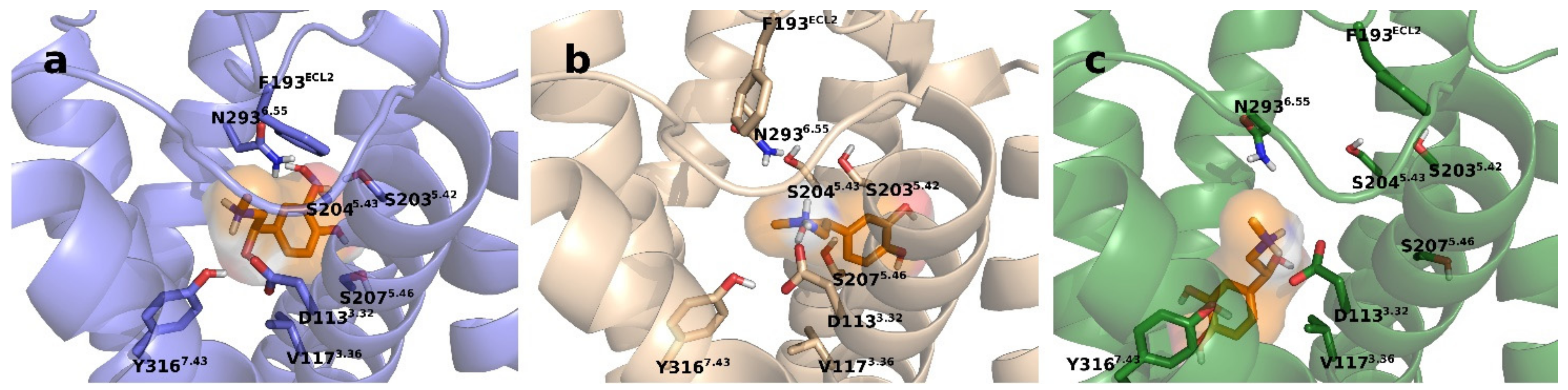
2.6. Intermolecular Interactions
3. Methods
3.1. System Building
3.2. MD Simulations
3.3. MD Trajectory Analysis
3.4. Sequence Alignment and Conservation Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Venter, J.C.; Adams, M.D.; Myers, E.W.; Li, P.W.; Mural, R.J.; Sutton, G.G.; Smith, H.O.; Yandell, M.; Evans, C.A.; Holt, R.A.; et al. The sequence of the human genome. Science 2001, 291, 1304–1351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hauser, A.; Attwood, M.M.; Rask-Andersen, M.; Schiöth, H.B.; Gloriam, D. Trends in GPCR drug discovery: New agents, targets and indications. Nat. Rev. Drug Discov. 2017, 16, 829–842. [Google Scholar] [CrossRef] [PubMed]
- Congreve, M.; de Graaf, C.; Swain, N.A.; Tate, C.G. Impact of GPCR Structures on Drug Discovery. Cell 2020, 181, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Brandt, D.R.; Asano, T.; Pedersen, S.E.; Ross, E.M. Reconstitution of catecholamine-stimulated guanosinetriphosphatase activity. Biochemistry 1983, 22, 4357–4362. [Google Scholar] [CrossRef]
- Cherezov, V.; Rosenbaum, D.M.; Hanson, M.A.; Rasmussen, S.G.; Thian, F.S.; Kobilka, T.S.; Choi, H.J.; Kuhn, P.; Weis, W.I.; Kobilka, B.K.; et al. High-resolution crystal structure of an engineered human β2-adrenergic G protein-coupled receptor. Science 2007, 318, 1258–1265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanson, M.A.; Cherezov, V.; Griffith, M.T.; Roth, C.B.; Jaakola, V.P.; Chien, E.Y.; Velasquez, J.; Kuhn, P.; Stevens, R.C. A specific cholesterol binding site is established by the 2.8 A structure of the human beta2-adrenergic receptor. Structure 2008, 16, 897–905. [Google Scholar] [CrossRef] [Green Version]
- Rasmussen, S.G.F.; DeVree, B.T.; Zou, Y.; Kruse, A.C.; Chung, K.Y.; Kobilka, T.S.; Thian, F.S.; Chae, P.S.; Pardon, E.; Calinski, D.; et al. Crystal structure of the beta2-adrenergic receptor-Gs protein complex. Nature 2011, 477, 549–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ring, A.; Manglik, A.; Kruse, A.C.; Enos, M.D.; Weis, W.; Garcia, K.C.; Kobilka, B.K. Adrenaline-activated structure of β2-adrenoceptor stabilized by an engineered nanobody. Nature 2013, 502, 575–579. [Google Scholar] [CrossRef] [Green Version]
- Weichert, D.; Kruse, A.C.; Manglik, A.; Hiller, C.; Zhang, C.; Hübner, H.; Kobilka, B.K.; Gmeiner, P. Covalent agonists for studying G protein-coupled receptor activation. Proc. Natl. Acad. Sci. USA 2014, 111, 10744–10748. [Google Scholar] [CrossRef] [Green Version]
- Zou, Y.; Weis, W.I.; Kobilka, B.K. N-Terminal T4 Lysozyme Fusion Facilitates Crystallization of a G Protein Coupled Receptor. PLoS ONE 2012, 7, e46039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wacker, D.; Fenalti, G.; Brown, M.A.; Katritch, V.; Abagyan, R.; Cherezov, V.; Stevens, R.C. Conserved binding mode of hu-man beta2-adrenergic receptor inverse agonists and antagonist revealed by X-ray crystallography. J. Am. Chem. Soc. 2010, 132, 11443–11445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasmussen, S.G.F.; Choi, H.J.; Rosenbaum, D.M.; Kobilka, T.S.; Thian, F.S.; Edwards, P.C.; Burghammer, M.; Ratnala, V.R.; Sanishvili, R.; Fischetti, R.F.; et al. Crystal structure of the human beta2-adrenergic G-protein-coupled receptor. Nature 2007, 450, 383–387. [Google Scholar] [CrossRef]
- Rasmussen, S.; Choi, H.-J.; Fung, J.J.; Pardon, E.; Casarosa, P.; Chae, P.S.; DeVree, B.; Rosenbaum, D.M.; Thian, F.S.; Kobilka, T.S.; et al. Structure of a nanobody-stabilized active state of the β2 adrenoceptor. Nature 2011, 469, 175–180. [Google Scholar] [CrossRef] [Green Version]
- Rosenbaum, D.M.; Zhang, C.; Lyons, J.; Holl, R.; Aragao, D.; Arlow, D.H.; Rasmussen, S.; Choi, H.-J.; DeVree, B.; Sunahara, R.K.; et al. Structure and function of an irreversible agonist-β2 adrenoceptor complex. Nature 2011, 469, 236–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munk, C.; Mutt, E.; Isberg, V.; Nikolajsen, L.F.; Bibbe, J.M.; Flock, T.; Hanson, M.A.; Stevens, R.C.; Deupi, X.; Gloriam, D.E. An online resource for GPCR structure determination and analysis. Nat. Methods 2019, 16, 151–162. [Google Scholar] [CrossRef] [PubMed]
- Latorraca, N.R.; Venkatakrishnan, A.J.; Dror, R.O. GPCR Dynamics: Structures in Motion. Chem. Rev. 2016, 117, 139–155. [Google Scholar] [CrossRef]
- Dror, R.O.; Arlow, D.H.; Maragakis, P.; Mildorf, T.J.; Pan, A.C.; Xu, H.; Borhani, D.W.; Shaw, D.E. Activation mechanism of the beta2-adrenergic receptor. Proc. Natl. Acad. Sci. USA 2011, 108, 18684–18689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nygaard, R.; Zou, Y.; Dror, R.O.; Mildorf, T.J.; Arlow, D.H.; Manglik, A.; Pan, A.C.; Liu, C.W.; Fung, J.J.; Bokoch, M.P.; et al. The dynamic process of β(2)-adrenergic receptor activation. Cell 2013, 152, 532–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dror, R.O.; Arlow, D.H.; Borhani, D.W.; Jensen, M.Ø.; Piana, S.; Shaw, D.E. Identification of two distinct inactive confor-mations of the beta2-adrenergic receptor reconciles structural and biochemical observations. Proc. Natl. Acad. Sci. USA 2009, 106, 4689–4694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marino, K.A.; Shang, Y.; Filizola, M. Insights into the function of opioid receptors from molecular dynamics simulations of available crystal structures. Br. J. Pharmacol. 2017, 175, 2834–2845. [Google Scholar] [CrossRef] [PubMed]
- Palczewski, K.; Kumasaka, T.; Hori, T.; Behnke, C.; Motoshima, H.; Fox, B.; Le Trong, I.; Teller, D.C.; Okada, T.; Stenkamp, R.E.; et al. Crystal Structure of Rhodopsin: A G Protein-Coupled Receptor. Science 2000, 289, 739–745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoddart, L.A.; Kellam, B.; Briddon, S.J.; Hill, S.J. Effect of a toggle switch mutation in TM6 of the human adenosine A3 receptor on Gi protein-dependent signalling and Gi-independent receptor internalization. Br. J. Pharmacol. 2014, 171, 3827–3844. [Google Scholar] [CrossRef]
- Pert, C.B.; Pasternak, G.; Snyder, S.H. Opiate Agonists and Antagonists Discriminated by Receptor Binding in Brain. Science 1973, 182, 1359–1361. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Chun, E.; Thompson, A.A.; Chubukov, P.; Xu, F.; Katritch, V.; Han, G.W.; Roth, C.B.; Heitman, L.; Ijzerman, A.; et al. Structural Basis for Allosteric Regulation of GPCRs by Sodium Ions. Science 2012, 337, 232–236. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.; Yang, D.; Wu, M.; Guo, Y.; Guo, W.; Zhong, L.; Cai, X.; Dai, A.; Jang, W.; I Shakhnovich, E.; et al. Common activation mechanism of class A GPCRs. eLife 2019, 8, e50279. [Google Scholar] [CrossRef] [PubMed]
- Filipek, S. Molecular switches in GPCRs. Curr. Opin. Struct. Biol. 2019, 55, 114–120. [Google Scholar] [CrossRef] [PubMed]
- Mitra, A.; Sarkar, A.; Szabó, M.; Borics, A. Correlated Motions of Conserved Polar Motifs Lay out a Plausible Mechanism of G Protein-Coupled Receptor Activation. Biomolecules 2021, 11, 670. [Google Scholar] [CrossRef]
- Gregorio, G.G.; Masureel, M.; Hilger, D.; Terry, D.S.; Juette, M.; Zhao, H.; Zhou, Z.; Perez-Aguilar, J.M.; Hauge, M.; Mathiasen, S.; et al. Single-molecule analysis of ligand efficacy in β2AR-G-protein activation. Nature 2017, 547, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Fleetwood, O.; Matricon, P.; Carlsson, J.; Delemotte, L. Energy Landscapes Reveal Agonist Control of G Protein-Coupled Receptor Activation via Microswitches. Biochemistry 2020, 59, 880–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, S.; Vogel, H.; Filipek, S. The Role of Water and Sodium Ions in the Activation of the μ-Opioid Receptor. Angew. Chem. Int. Ed. 2013, 52, 10112–10115. [Google Scholar] [CrossRef]
- Shang, Y.; Lerouzic, V.; Schneider, S.; Bisignano, P.; Pasternak, G.W.; Filizola, M. Mechanistic Insights into the Allosteric Modulation of Opioid Receptors by Sodium Ions. Biochemistry 2014, 53, 5140–5149. [Google Scholar] [CrossRef]
- Selent, J.; Sanz, F.; Pastor, M.; De Fabritiis, G. Induced Effects of Sodium Ions on Dopaminergic G-Protein Coupled Receptors. PLoS Comput. Biol. 2010, 6, e1000884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, X.; Wang, Y.; Hunkele, A.; Provasi, D.; Pasternak, G.W.; Filizola, M. Kinetic and thermodynamic insights into sodium ion translocation through the μ-opioid receptor from molecular dynamics and machine learning analysis. PLoS Comput. Biol. 2019, 15, e1006689. [Google Scholar] [CrossRef] [PubMed]
- Hol, W.G. Effects of the α-helix dipole upon the functioning and structure of proteins and peptides. Adv. Biophys. 1985, 19, 133–165. [Google Scholar] [CrossRef]
- Ma, X.; Hu, Y.; Batebi, H.; Heng, J.; Xu, J.; Liu, X.; Niu, X.; Li, H.; Hildebrand, P.W.; Jin, C.; et al. Analysis of β2AR-Gs and β2AR-Gi complex formation by NMR spectroscopy. Proc. Natl. Acad. Sci. USA 2020, 117, 23096–23105. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yang, F.; Ling, S.; Lv, P.; Zhou, Y.; Fang, W.; Sun, W.; Zhang, L.; Shi, P.; Tian, C. Single-particle cryo-EM structural studies of the β2AR–Gs complex bound with a full agonist formoterol. Cell Discov. 2020, 6, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Ling, S.; Zhou, Y.; Zhang, Y.; Lv, P.; Liu, S.; Fang, W.; Sun, W.; Hu, L.A.; Zhang, L.; et al. Different conformational responses of the β2-adrenergic receptor-Gs complex upon binding of the partial agonist salbutamol or the full agonist isoprenaline. Natl. Sci. Rev. 2020, 8, nwaa284. [Google Scholar] [CrossRef]
- Liu, R.; Nahon, D.; le Roy, B.; Lenselink, E.B.; Ijzerman, A.P. Scanning Mutagenesis in a Yeast System Delineates the Role of the NPxxY(x)5,6 F Motif and Helix 8 of the Adenosine A2B Receptor in G Protein Coupling. Biochem. Pharmacol. 2015, 95, 290–300. [Google Scholar] [CrossRef]
- Ballesteros, J.A.; Jensen, A.D.; Liapakis, G.; Rasmussen, S.; Shi, L.; Gether, U.; Javitch, J. Activation of the β2-Adrenergic Receptor Involves Disruption of an Ionic Lock between the Cytoplasmic Ends of Transmembrane Segments 3 and 6. J. Biol. Chem. 2001, 276, 29171–29177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, W.; Manglik, A.; Venkatakrishnan, A.J.; Laeremans, T.; Feinberg, E.; Sanborn, A.L.; Kato, H.; Livingston, K.E.; Thorsen, T.S.; Kling, R.C.; et al. Structural insights into μ-opioid receptor activation. Nature 2015, 524, 315–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jongejan, A.; Bruysters, M.; Ballesteros, J.A.; Haaksma, E.; Bakker, R.A.; Pardo, L.; Leurs, R. Linking Agonist Binding to Histamine H1 Receptor Activation. Nat. Chem. Biol. 2005, 1, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Warne, T.; Nehmé, R.; Pandey, S.; Dwivedi-Agnihotri, H.; Chaturvedi, M.; Edwards, P.C.; García-Nafría, J.; Leslie, A.G.W.; Shukla, A.K.; et al. Molecular basis of β-arrestin coupling to formoterol-bound β1-adrenoceptor. Nature 2020, 583, 862–866. [Google Scholar] [CrossRef] [PubMed]
- Yin, W.; Li, Z.; Jin, M.; Yin, Y.-L.; de Waal, P.W.; Pal, K.; Yin, Y.; Gao, X.; He, Y.; Gao, J.; et al. A complex structure of arrestin-2 bound to a G protein-coupled receptor. Cell Res. 2019, 29, 971–983. [Google Scholar] [CrossRef]
- Zhan, X.; Gimenez, L.E.D.; Gurevich, V.V.; Spiller, B.W. Crystal Structure of Arrestin-3 Reveals the Basis of the Difference in Receptor Binding Between Two Non-visual Subtypes. J. Mol. Biol. 2011, 406, 467–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.; Hu, H.; Ramachandran, S.; Erickson, J.W.; Cerione, R.A.; Skiniotis, G. Structures of the Rhodopsin-Transducin Complex: Insights into G-Protein Activation. Mol. Cell 2019, 75, 781–790. [Google Scholar] [CrossRef]
- Sunahara, R.K.; Tesmer, J.J.G.; Gilman, A.G.; Sprang, S.R. Crystal Structure of the Adenylyl Cyclase Activator Gs. Science 1997, 278, 1943–1947. [Google Scholar] [CrossRef] [Green Version]
- Fiser, A.; Do, R.K.G.; Šali, A. Modeling of loops in protein structures. Protein Sci. 2000, 9, 1753–1773. [Google Scholar] [CrossRef] [Green Version]
- Johansson, M.U.; Zoete, V.; Michielin, O.; Guex, N. Defining and searching for structural motifs using DeepView/Swiss-PdbViewer. BMC Bioinform. 2012, 13, 173. [Google Scholar] [CrossRef] [Green Version]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Mialet-Perez, J.; Green, S.A.; Miller, W.E.; Liggett, S.B. A primate-dominant third glycosylation site of the β2-adrenergic receptor routes receptors to degradation during agonist regulation. J. Biol. Chem. 2004, 279, 38603–38607. [Google Scholar] [CrossRef] [Green Version]
- O’Dowd, B.F.; Hnatowich, M.; Caron, M.G.; Lefkowitz, R.J.; Bouvier, M. Palmitoylation of the human beta 2-adrenergic receptor. Mutation of Cys341 in the carboxyl tail leads to an uncoupled nonpalmitoylated form of the receptor. J. Biol. Chem. 1989, 264, 7564–7569. [Google Scholar] [CrossRef]
- Zamah, A.M.; Delahunty, M.; Luttrell, L.; Lefkowitz, R.J. Protein Kinase A-mediated Phosphorylation of the β2-Adrenergic Receptor Regulates Its Coupling to Gs and Gi. J. Biol. Chem. 2002, 277, 31249–31256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hausdorff, W.P.; Bouvier, M.; O’Dowd, B.F.; Irons, G.P.; Caron, M.G.; Lefkowitz, R.J. Phosphorylation Sites on Two Domains of the β2-Adrenergic Receptor Are Involved in Distinct Pathways of Receptor Desensitization. J. Biol. Chem. 1989, 264, 12657–12665. [Google Scholar] [CrossRef]
- Pike, L.J.; Han, X.; Chung, K.N.; Gross, R.W. Lipid rafts are enriched in arachidonic acid and plasmenylethanolamine and their composition is independent of caveolin-1 expression: A quantitative electrospray ionization/mass spectrometric analysis. Biochemistry 2002, 41, 2075–2088. [Google Scholar] [CrossRef] [PubMed]
- Ingólfsson, H.I.; Melo, M.N.; van Eerden, F.J.; Arnarez, C.; Lopez, C.A.; Wassenaar, T.A.; Periole, X.; de Vries, A.H.; Tieleman, D.P.; Marrink, S.J. Lipid Organization of the Plasma Membrane. J. Am. Chem. Soc. 2014, 136, 14554–14559. [Google Scholar] [CrossRef]
- Huang, J.; MacKerell, A.D., Jr. CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data. J. Comput. Chem. 2013, 34, 2135–2145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [Green Version]
- Berendsen, H.J.C.; Postma, J.P.M.; Van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef]
- Lange, O.F.; Grubmüller, H. Generalized correlation for biomolecular dynamics. Proteins Struct. Funct. Bioinform. 2005, 62, 1053–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; López, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.M.; Procter, J.; Martin, D.; Clamp, M.; Barton, G.J. Jalview Version 2—A multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Residue | Frequency of Contact/% | |||||||
|---|---|---|---|---|---|---|---|---|
| Epinephrine-Bound β2AR | Ligand-Free β2AR | |||||||
| Gs Protein Complex | β-Arrestin-2 Complex | Gs Protein Complex | β-Arrestin-2 Complex | |||||
| Active | Inactive | Active | Inactive | Active | Inactive | Active | Inactive | |
| D792.50 | 0.0 | 0.0 | 0.0 | 0.0 | 52.15 | 93.41 | 25.91 | 26.04 |
| D1133.32 | <0.1 | 32.33 | 0.0 | 0.0 | 9.22 | 45.06 | 5.02 | 72.03 |
| S1203.39 | 0.0 | 0.0 | 0.0 | 0.0 | 44.95 | 96.36 | 5.55 | 25.86 |
| W2866.48 | 0.0 | 6.88 | 0.0 | 0.0 | 3.36 | 19.51 | 29.16 | 10.92 |
| N3187.45 | 0.0 | 0.0 | 0.0 | 0.0 | 2.78 | 0.46 | 46.14 | 0.22 |
| S3197.46 | 0.0 | 0.0 | 0.0 | 0.0 | 30.52 | 14.39 | 79.75 | 24.69 |
| N3227.49 | 0.0 | 0.0 | 0.0 | 0.0 | 45.87 | 51.12 | 8.17 | 20.06 |
| Y3267.53 | 0.0 | 0.0 | 0.0 | 0.0 | 0.12 | 0.60 | 0.0 | 0.0 |
| Interactions | Residues Involved | Epinephrine-Bound | Ligand-Free | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Active State | Inactive State | Active State | Inactive State | |||||||||
| Gs Protein Complex | β-Arrestin-2 | Gs Protein Complex | β-Arrestin-2 | Gs Protein Complex | β-Arrestin-2 | Gs Protein Complex | β-Arrestin-2 | |||||
| 1 | 2 | 3 | Restrained | |||||||||
| Salt Bridge | ||||||||||||
| Intra-DRY | D1303.49; R1313.50 | 26.5 | 5.7 | 6.0 | 0 | 65.1 | 40.4 | 35.5 | 40.5 | 10.7 | 51.2 | 41.3 |
| H-bonds | ||||||||||||
| DRY-H8 | R1313.50; S3298.47 | 0.1 | 0 | 0.3 | 0 | 0.1 | 0 | 0 | 0.1 | 0.1 | 0.0 | 0.0 |
| BP | D1133.32; Y3167.43 | 14.0 | 80.6 | 82.1 | 95.3 | 28.3 | 65.9 | 69.2 | 96.1 | 9.9 | 0.1 | 14.6 |
| intra-DRY | D1303.49; R1313.50 | 65.9 | 64.5 | 17.9 | 16.8 | 35.7 | 80.0 | 72.9 | 82.3 | 30.5 | 99.8 | 83.5 |
| DRY-ICL2 | D1303.49; Y141ICL2 | 99.3 | 97.5 | 92.4 | 98.0 | 99.8 | 0 | 0 | 98.1 | 86.2 | 0 | 0 |
| D1303.49; S143ICL2; L144ICL2 | 0 | 0 | 0 | 0 | 0 | 99.5 | 90.8 | 0 | 0 | 0 | 37.3 | |
| DRY-TM5 | R1313.50; Y2195.58 | 3.9 | 10.5 | 4.6 | 45.2 | 30.9 | 0 | 0 | 5.7 | 3.3 | 0 | 0 |
| DRY-TM6 | R1313.50; E2686.30 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| CWxP-TM7 | C2856.47; W2866.48; N3187.45 | 1.8 | 3.4 | 45.1 | 4.1 | 5.3 | 11.7 | 4.3 | 17.6 | 0.5 | 53.7 | 3.8 |
| Interactions | Residues Involved, Respectively | Epinephrine-Bound | Ligand-Free | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Active State | Inactive State | Active State | Inactive State | |||||||||
| Gs Protein Complex | β-Arrestin-2 | Gs Protein Complex | β-Arrestin-2 | Gs Protein Complex | β-Arrestin-2 | Gs Protein Complex | β-Arrestin-2 | |||||
| 1 | 2 | 3 | Restrained | |||||||||
| Salt Bridge | ||||||||||||
| BP-epi | D1133.32; epi | 67.5 | 51.0 | 4.8 | 52.7 | 36.0 | 29.3 | 60.1 | - | - | - | - |
| H-bonds | ||||||||||||
| BP-epi | D1133.32; epi | 96.7 | 96.0 | 8.3 | 99.3 | 55.5 | 53.8 | 93.3 | - | - | - | - |
| H8-H5Gα | S329-L340; T369-L394 | 0.0 | 7.3 | 4.0 | 0.0 | - | 0.0 | - | 0.0 | - | 0.0 | - |
| ICl1-H5Gα | F61-T66; T369-L394 | 0.2 | 0.1 | 0.0 | 0.2 | - | 0.6 | - | 30.3 | - | 40.0 | - |
| ICl2-H5Gα | S137-T146; T369-L394 | 0.1 | 0.1 | 17.9 | 0.5 | - | 9.9 | - | 5.1 | - | 31.5 | - |
| ICL3-H5Gα | E237-K267; T369-L394 | 90.5 | 92.5 | 97.5 | 96.3 | - | 77.1 | - | 76.2 | - | 76.7 | - |
| H8-FL | S329-L340; G65-K78 | - | - | - | - | 33.4 | - | 0.0 | - | 0.0 | - | 0.0 |
| ICl1-FL | F61-T66; G65-K78 | - | - | - | - | 25.6 | - | 58.8 | - | 22.9 | - | 49.3 |
| ICL2-FL | S137-T146; G65-K78 | - | - | - | - | 8.7 | - | 30.4 | - | 77.8 | - | 35.6 |
| ICL3-FL | E237-K267; G65-K78 | - | - | - | - | 85.5 | - | 71.7 | - | 14.8 | - | 14.0 |
| H8-ML | S329-L340; P132-A140 | - | - | - | - | 0.0 | - | 0.0 | - | 0.0 | - | 0.0 |
| ICL1-ML | F61-T66; P132-A140 | - | - | - | - | 0.9 | - | 0.1 | - | 0.0 | - | 0.0 |
| ICL2-ML | S137-T146; P132-A140 | - | - | - | - | 7.1 | - | 4.2 | - | 9.3 | - | 30.3 |
| ICL3-ML | E237-K267; P132-A140 | - | - | - | - | 0.0 | - | 0.0 | - | 0 | - | 0 |
| H8-CL | S329-L340; V307-G317 | - | - | - | - | 0.0 | - | 0.0 | - | 0.0 | - | 0.0 |
| ICl1-CL | F61-T66; V307-G317 | - | - | - | - | 0.0 | - | 0.0 | - | 0.0 | - | 0.0 |
| ICL2-CL | S137-T146; V307-G317 | - | - | - | - | 4.6 | - | 58.4 | - | 0.2 | - | 8.9 |
| ICL3-CL | E237-K267; V307-G317 | - | - | - | - | 0.0 | - | 0.0 | - | 4.0 | - | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mitra, A.; Sarkar, A.; Borics, A. Universal Properties and Specificities of the β2-Adrenergic Receptor-Gs Protein Complex Activation Mechanism Revealed by All-Atom Molecular Dynamics Simulations. Int. J. Mol. Sci. 2021, 22, 10423. https://doi.org/10.3390/ijms221910423
Mitra A, Sarkar A, Borics A. Universal Properties and Specificities of the β2-Adrenergic Receptor-Gs Protein Complex Activation Mechanism Revealed by All-Atom Molecular Dynamics Simulations. International Journal of Molecular Sciences. 2021; 22(19):10423. https://doi.org/10.3390/ijms221910423
Chicago/Turabian StyleMitra, Argha, Arijit Sarkar, and Attila Borics. 2021. "Universal Properties and Specificities of the β2-Adrenergic Receptor-Gs Protein Complex Activation Mechanism Revealed by All-Atom Molecular Dynamics Simulations" International Journal of Molecular Sciences 22, no. 19: 10423. https://doi.org/10.3390/ijms221910423