
Regulation of Survival Motor Neuron Gene Expression by Calcium Signaling

 , , , , , and
, , , , , and 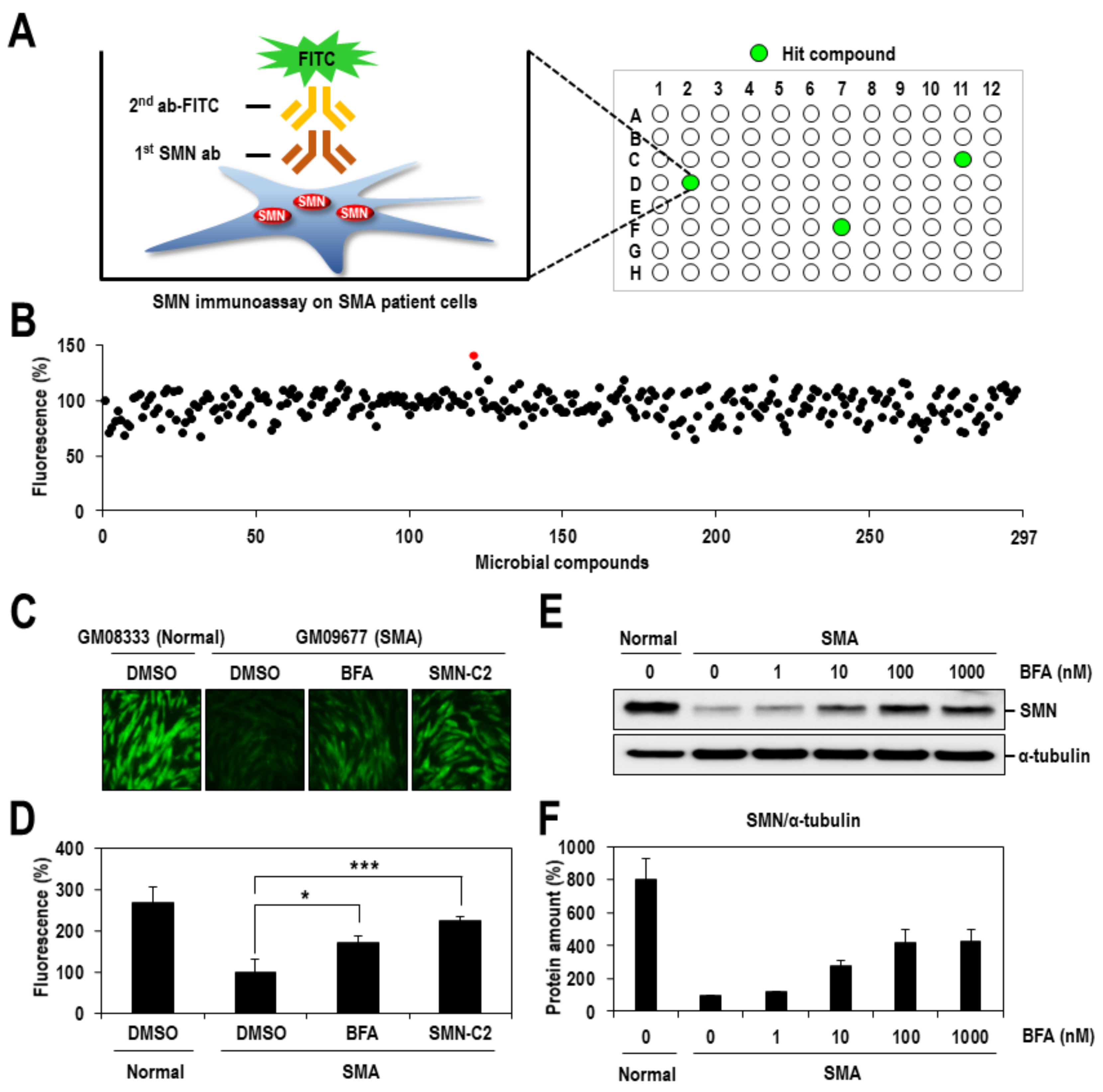{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Identification of BFA as a Chemical Inducer of SMN Protein
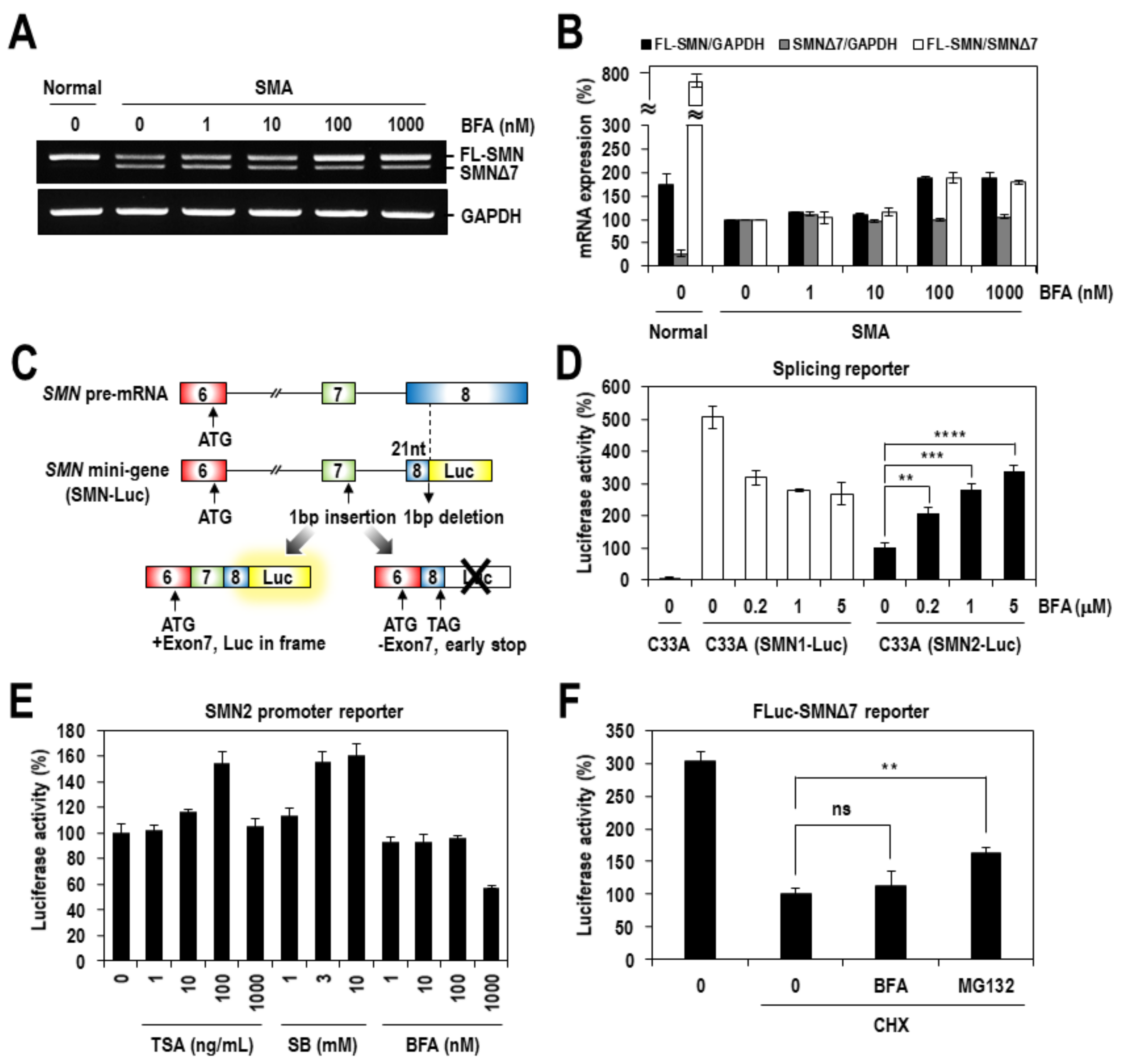
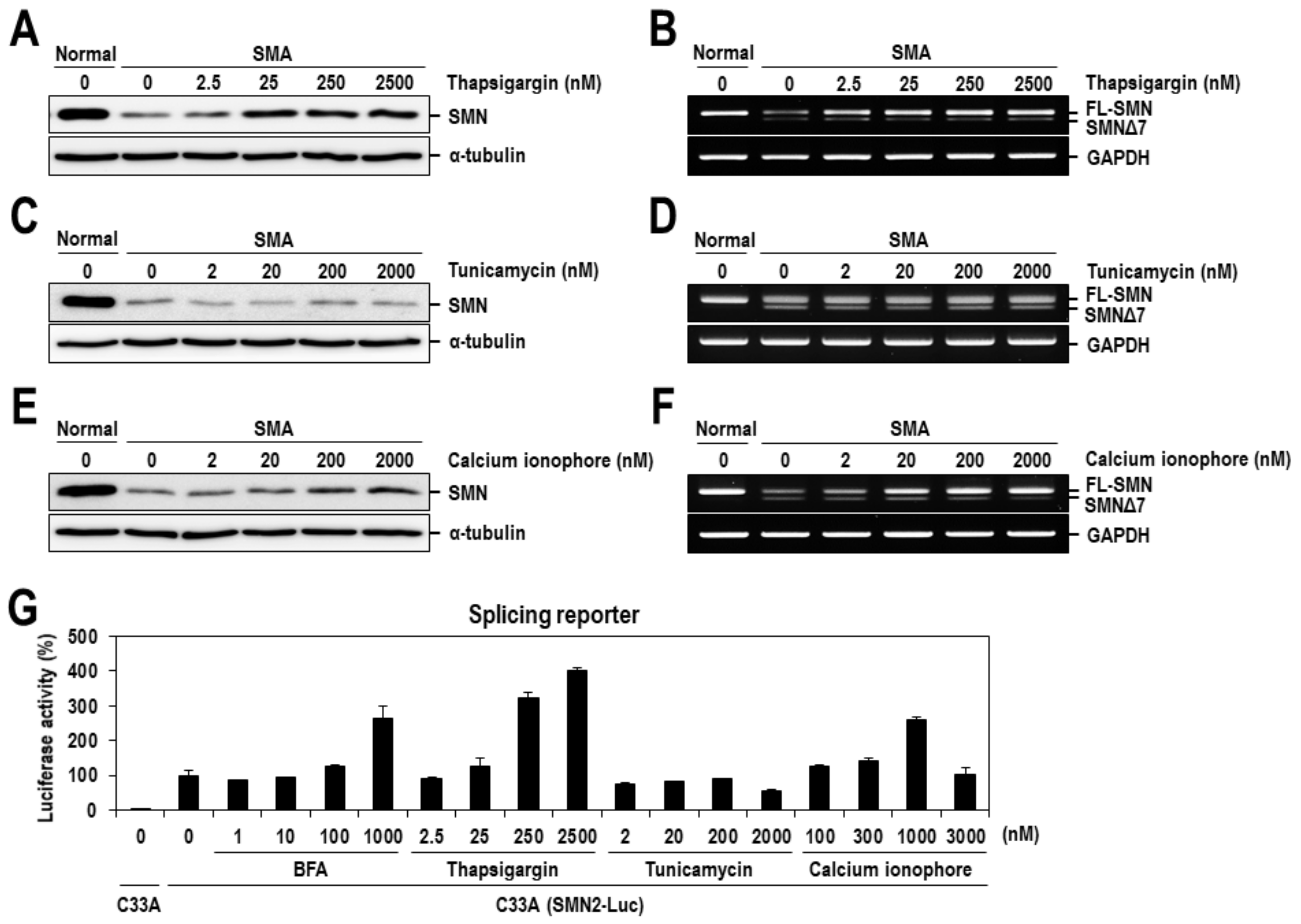
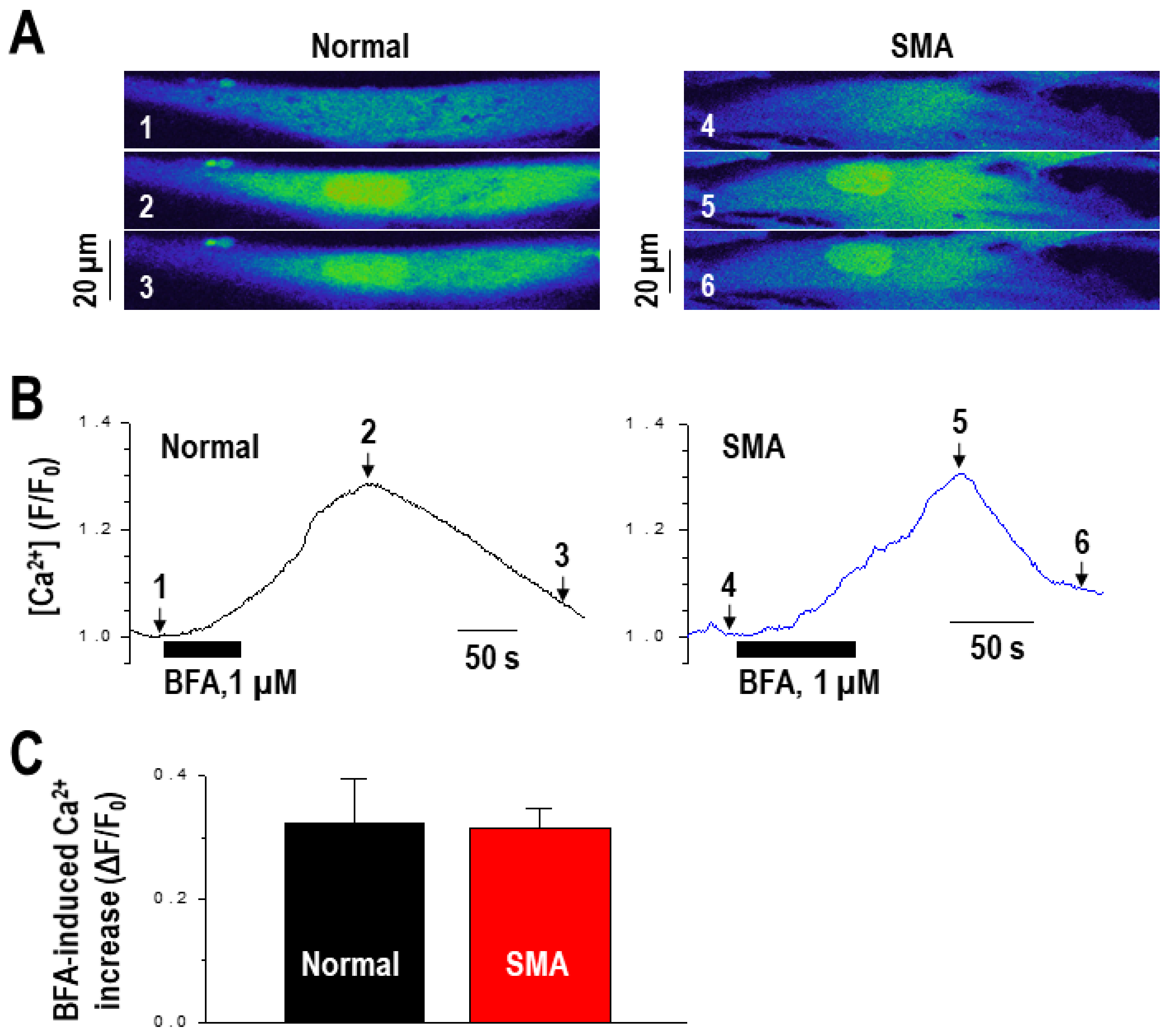
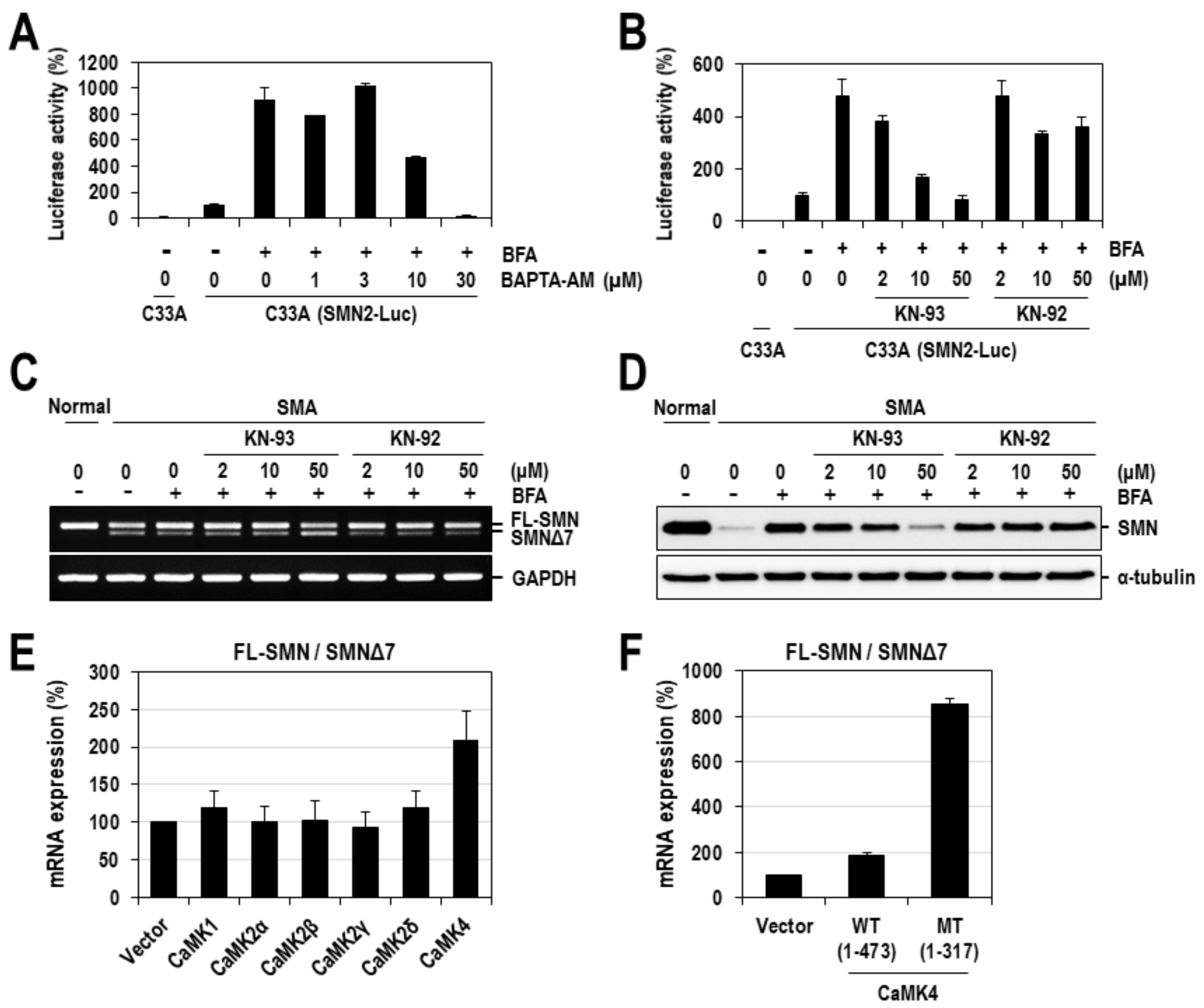
2.2. Calcium Mediates BFA-Induced SMN Expression Partly by Modulating the Alternative Splicing of SMN2 Exon 7
2.3. CaMK4 Regulates the Alternative Splicing of SMN2 Exon 7
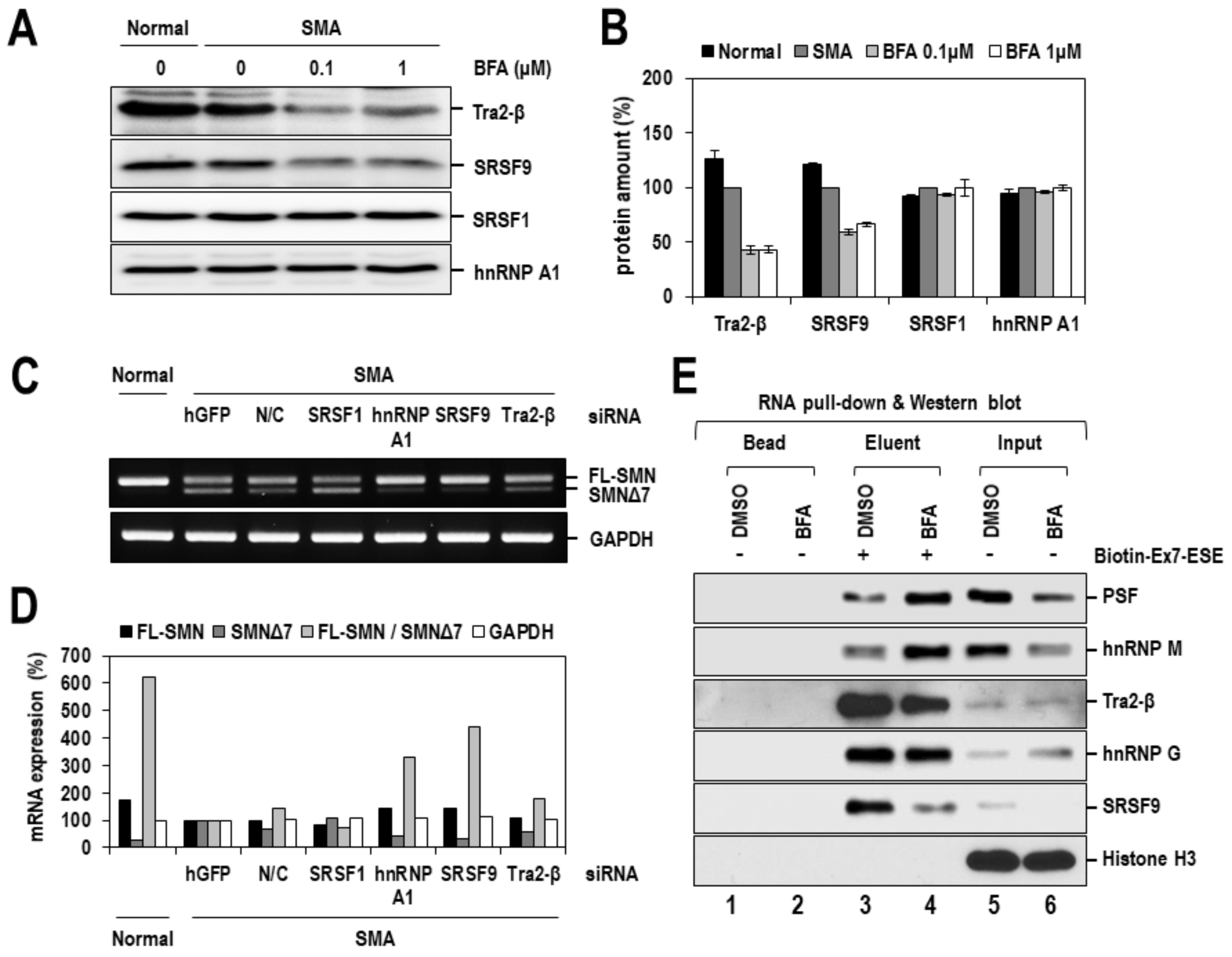
2.4. BFA Altered the Expression or RNA-Binding Activity of Splicing Factors
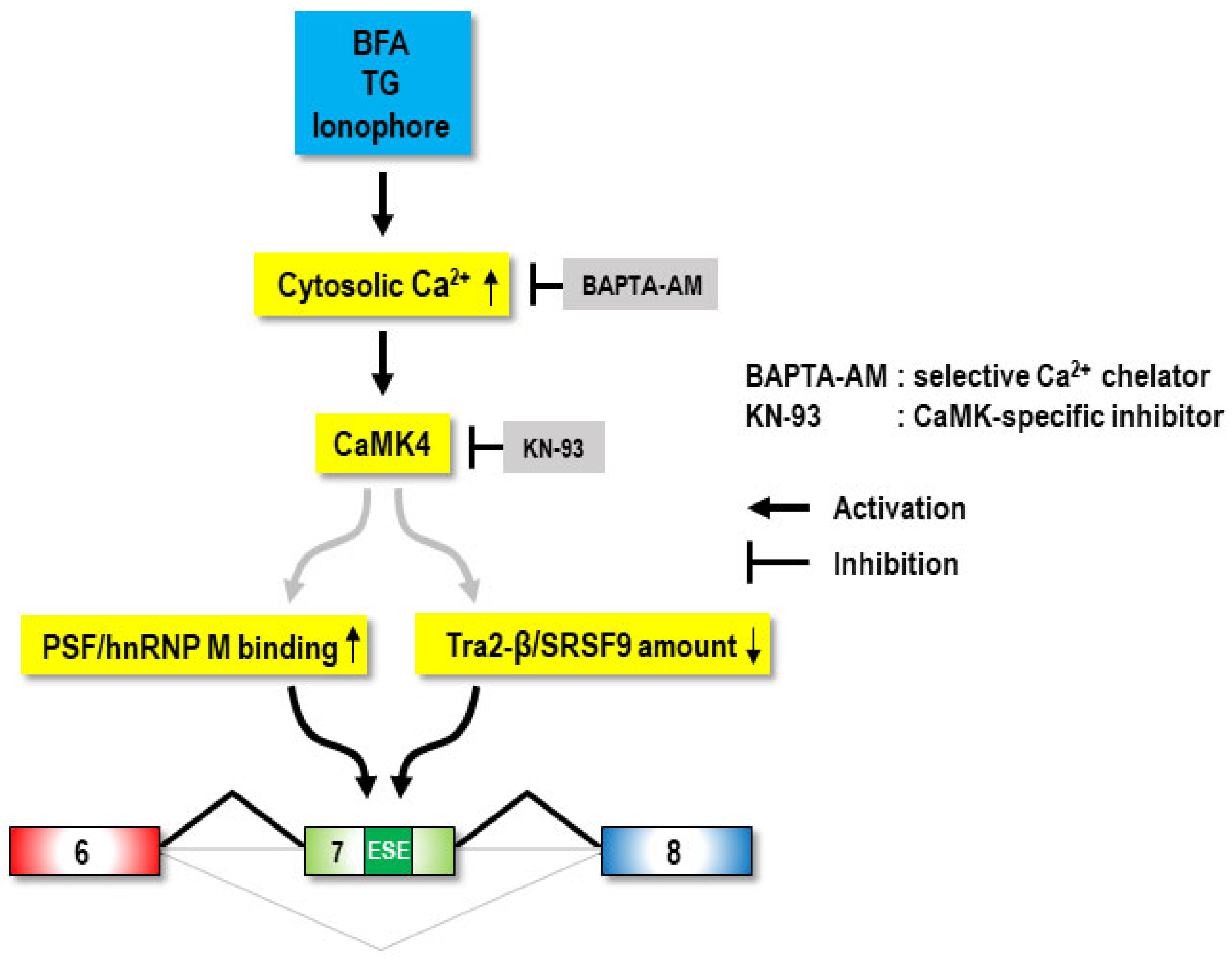
3. Discussion
4. Materials and Methods
4.1. Cell Cultures
4.2. Plasmid Construction and DNA Transfection
4.3. Antibodies and Chemicals
4.4. Immunoassay-Based Screening
4.5. Quantitative Western Blot Analysis
4.6. Analysis of the Alternative Splicing of SMN2 Exon 7
4.7. RT-PCR Analysis
4.8. Real-Time PCR Analysis
4.9. Calcium Imaging
4.10. 5′-Biotinylated RNA Oligomer-Capture Pull-Down Assay
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Talbot, K.; Davies, K.E. Spinal muscular atrophy. Semin. Neurol. 2001, 21, 189–197. [Google Scholar] [CrossRef] [Green Version]
- Wirth, B.; Brichta, L.; Hahnen, E. Spinal muscular atrophy: From gene to therapy. Semin. Pediatr. Neurol. 2006, 13, 121–131. [Google Scholar] [CrossRef]
- Burnett, B.G.; Crawford, T.O.; Sumner, C.J. Emerging treatment options for spinal muscular atrophy. Curr. Treat. Options Neurol. 2009, 11, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Oskoui, M.; Darras, B.; De Vivo, D. Spinal muscular atrophy: 125 years later and on the verge of a cure. In Spinal Muscular Atrophy; Elsevier: Amsterdam, The Netherlands, 2017; pp. 3–19. [Google Scholar]
- Munsat, T.L.; Davies, K.E. International SMA consortium meeting. (26–28 June 1992, Bonn, Germany). Neuromuscul. Disord. 1992, 2, 423–428. [Google Scholar] [CrossRef]
- Yong, J.; Wan, L.; Dreyfuss, G. Why do cells need an assembly machine for RNA-protein complexes? Trends Cell Biol. 2004, 14, 226–232. [Google Scholar] [CrossRef]
- Neuenkirchen, N.; Chari, A.; Fischer, U. Deciphering the assembly pathway of Sm-class U snRNPs. FEBS Lett. 2008, 582, 1997–2003. [Google Scholar] [CrossRef] [Green Version]
- Fischer, U.; Liu, Q.; Dreyfuss, G. The SMN-SIP1 complex has an essential role in spliceosomal snRNP biogenesis. Cell 1997, 9, 1023–1029. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Fischer, U.; Wang, F.; Dreyfuss, G. The spinal muscular atrophy disease gene product, SMN, and its associated protein SIP1 are in a complex with spliceosomal snRNP proteins. Cell 1997, 90, 1013–1021. [Google Scholar] [CrossRef] [Green Version]
- Meister, G.; Buhler, D.; Pillai, R.; Lottspeich, F.; Fischer, U. A multiprotein complex mediates the ATP-dependent assembly of spliceosomal U snRNPs. Nat. Cell Biol. 2001, 3, 945–949. [Google Scholar] [CrossRef] [PubMed]
- Pellizzoni, L.; Charroux, B.; Dreyfuss, G. SMN mutants of spinal muscular atrophy patients are defective in binding to snRNP proteins. Proc. Natl. Acad Sci. USA 1999, 96, 11167–11172. [Google Scholar] [CrossRef] [Green Version]
- Gabanella, F.; Butchbach, M.E.; Saieva, L.; Carissimi, C.; Burghes, A.H.; Pellizzoni, L. Ribonucleoprotein assembly defects correlate with spinal muscular atrophy severity and preferentially affect a subset of spliceosomal snRNPs. PLoS ONE 2007, 2, e921. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Lotti, F.; Dittmar, K.; Younis, I.; Wan, L.; Kasim, M.; Dreyfuss, G. SMN deficiency causes tissue-specific perturbations in the repertoire of snRNAs and widespread defects in splicing. Cell 2008, 133, 585–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, S.; Dreyfuss, G. A degron created by SMN2 exon 7 skipping is a principal contributor to spinal muscular atrophy severity. Genes Dev. 2010, 24, 438–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefebvre, S.; Burglen, L.; Reboullet, S.; Clermont, O.; Burlet, P.; Viollet, L.; Benichou, B.; Cruaud, C.; Millasseau, P.; Zeviani, M.; et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 1995, 80, 155–165. [Google Scholar] [CrossRef] [Green Version]
- Mendell, J.R.; Al-Zaidy, S.; Shell, R.; Arnold, W.D.; Rodino-Klapac, L.R.; Prior, T.W.; Lowes, L.; Alfano, L.; Berry, K.; Church, K.; et al. Single-Dose Gene-Replacement Therapy for Spinal Muscular Atrophy. N. Engl. J. Med. 2017, 377, 1713–1722. [Google Scholar] [CrossRef]
- Ratni, H.; Ebeling, M.; Baird, J.; Bendels, S.; Bylund, J.; Chen, K.S.; Denk, N.; Feng, Z.; Green, L.; Guerard, M.; et al. Discovery of Risdiplam, a Selective Survival of Motor Neuron-2 (SMN2) Gene Splicing Modifier for the Treatment of Spinal Muscular Atrophy (SMA). J. Med. Chem. 2018, 61, 6501–6517. [Google Scholar] [CrossRef] [Green Version]
- Sakla, M.S.; Lorson, C.L. Induction of full-length survival motor neuron by polyphenol botanical compounds. Hum. Genet. 2008, 122, 635–643. [Google Scholar] [CrossRef]
- Markus, M.A.; Marques, F.Z.; Morris, B.J. Resveratrol, by modulating RNA processing factor levels, can influence the alternative splicing of pre-mRNAs. PLoS ONE 2011, 6, e28926. [Google Scholar] [CrossRef] [Green Version]
- Baek, J.; Jeong, H.; Ham, Y.; Jo, Y.H.; Choi, M.; Kang, M.; Son, B.; Choi, S.; Ryu, H.W.; Kim, J.; et al. Improvement of spinal muscular atrophy via correction of the SMN2 splicing defect by Brucea javanica (L.) Merr. extract and Bruceine, D. Phytomedicine 2019, 65, 153089. [Google Scholar] [CrossRef]
- Williams, P.G.; Buchanan, G.O.; Feling, R.H.; Kauffman, C.A.; Jensen, P.R.; Fenical, W. New cytotoxic salinosporamides from the marine Actinomycete Salinispora tropica. J. Org. Chem. 2005, 70, 6196–6203. [Google Scholar] [CrossRef]
- Lee, U.; Kim, S.O.; Hwang, J.A.; Jang, J.H.; Son, S.; Ryoo, I.J.; Ahn, J.S.; Kim, B.Y.; Lee, K.H. The Fungal Metabolite Brefeldin A Inhibits Dvl2-Plk1-Dependent Primary Cilium Disassembly. Mol. Cells 2017, 40, 401–409. [Google Scholar] [CrossRef] [Green Version]
- Naryshkin, N.A.; Weetall, M.; Dakka, A.; Narasimhan, J.; Zhao, X.; Feng, Z.; Ling, K.K.; Karp, G.M.; Qi, H.; Woll, M.G.; et al. Motor neuron disease. SMN2 splicing modifiers improve motor function and longevity in mice with spinal muscular atrophy. Science 2014, 345, 688–693. [Google Scholar] [CrossRef]
- Zhang, M.L.; Lorson, C.L.; Androphy, E.J.; Zhou, J. An in vivo reporter system for measuring increased inclusion of exon 7 in SMN2 mRNA: Potential therapy of SMA. Gene. Ther. 2001, 8, 1532–1538. [Google Scholar] [CrossRef] [Green Version]
- Jarecki, J.; Chen, X.; Bernardino, A.; Coovert, D.D.; Whitney, M.; Burghes, A.; Stack, J.; Pollok, B.A. Diverse small-molecule modulators of SMN expression found by high-throughput compound screening: Early leads towards a therapeutic for spinal muscular atrophy. Hum. Mol. Genet. 2005, 14, 2003–2018. [Google Scholar] [CrossRef] [Green Version]
- Chang, J.G.; Hsieh-Li, H.M.; Jong, Y.J.; Wang, N.M.; Tsai, C.H.; Li, H. Treatment of spinal muscular atrophy by sodium butyrate. Proc. Natl. Acad Sci. USA 2001, 98, 9808–9813. [Google Scholar] [CrossRef] [Green Version]
- Kawamoto, K.; Yoshida, Y.; Tamaki, H.; Torii, S.; Shinotsuka, C.; Yamashina, S.; Nakayama, K. GBF1, a guanine nucleotide exchange factor for ADP-ribosylation factors, is localized to the cis-Golgi and involved in membrane association of the COPI coat. Traffic 2002, 3, 483–495. [Google Scholar] [CrossRef]
- Nebenfuhr, A.; Ritzenthaler, C.; Robinson, D.G. Brefeldin A: Deciphering an enigmatic inhibitor of secretion. Plant Physiol. 2002, 130, 1102–1108. [Google Scholar] [CrossRef] [Green Version]
- Brandizzi, F.; Barlowe, C. Organization of the ER-Golgi interface for membrane traffic control. Nat. Rev. Mol. Cell Biol. 2013, 14, 382–392. [Google Scholar] [CrossRef] [Green Version]
- Mekahli, D.; Bultynck, G.; Parys, J.B.; De Smedt, H.; Missiaen, L. Endoplasmic-reticulum calcium depletion and disease. Cold Spring Harb. Perspect Biol. 2011, 3, a004317. [Google Scholar] [CrossRef]
- Tordai, A.; Brass, L.F.; Gelfand, E.W. Tunicamycin inhibits the expression of functional thrombin receptors on human T-lymphoblastoid cells. Biochem. Biophys. Res. Commun. 1995, 206, 857–862. [Google Scholar] [CrossRef]
- Ziomek, G.; van Breemen, C.; Esfandiarei, M. Drop in endo/sarcoplasmic calcium precedes the unfolded protein response in Brefeldin A-treated vascular smooth muscle cells. Eur. J. Pharmacol. 2015, 764, 328–339. [Google Scholar] [CrossRef]
- Biondi, O.; Branchu, J.; Sanchez, G.; Lancelin, C.; Deforges, S.; Lopes, P.; Pariset, C.; Lecolle, S.; Cote, J.; Chanoine, C.; et al. In vivo NMDA receptor activation accelerates motor unit maturation, protects spinal motor neurons, and enhances SMN2 gene expression in severe spinal muscular atrophy mice. J. Neurosci. 2010, 30, 11288–11299. [Google Scholar] [CrossRef] [Green Version]
- Branchu, J.; Biondi, O.; Chali, F.; Collin, T.; Leroy, F.; Mamchaoui, K.; Makoukji, J.; Pariset, C.; Lopes, P.; Massaad, C.; et al. Shift from extracellular signal-regulated kinase to AKT/cAMP response element-binding protein pathway increases survival-motor-neuron expression in spinal-muscular-atrophy-like mice and patient cells. J. Neurosci. 2013, 33, 4280–4294. [Google Scholar] [CrossRef] [Green Version]
- Pellicena, P.; Schulman, H. CaMKII inhibitors: From research tools to therapeutic agents. Front. Pharmacol. 2014, 5, 21. [Google Scholar] [CrossRef] [Green Version]
- Kashima, T.; Manley, J.L. A negative element in SMN2 exon 7 inhibits splicing in spinal muscular atrophy. Nat. Genet. 2003, 34, 460–463. [Google Scholar] [CrossRef]
- Long, J.C.; Caceres, J.F. The SR protein family of splicing factors: Master regulators of gene expression. Biochem. J. 2009, 417, 15–27. [Google Scholar] [CrossRef] [Green Version]
- Sanford, J.R.; Bruzik, J.P. Regulation of SR protein localization during development. Proc. Natl. Acad Sci. USA 2001, 98, 10184–10189. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.N.; Singh, N.N. Mechanism of Splicing Regulation of Spinal Muscular Atrophy Genes. Adv. Neurobiol. 2018, 20, 31–61. [Google Scholar]
- Claude, A.; Zhao, B.P.; Kuziemsky, C.E.; Dahan, S.; Berger, S.J.; Yan, J.P.; Armold, A.D.; Sullivan, E.M.; Melancon, P. GBF1: A novel Golgi-associated BFA-resistant guanine nucleotide exchange factor that displays specificity for ADP-ribosylation factor 5. J. Cell Biol. 1999, 146, 71–84. [Google Scholar] [CrossRef] [Green Version]
- Niu, T.K.; Pfeifer, A.C.; Lippincott-Schwartz, J.; Jackson, C.L. Dynamics of GBF1, a Brefeldin A-sensitive Arf1 exchange factor at the Golgi. Mol. Biol. Cell 2005, 16, 1213–1222. [Google Scholar] [CrossRef] [Green Version]
- Breckenridge, D.G.; Germain, M.; Mathai, J.P.; Nguyen, M.; Shore, G.C. Regulation of apoptosis by endoplasmic reticulum pathways. Oncogene 2003, 22, 8608–8618. [Google Scholar] [CrossRef] [Green Version]
- Wlodkowic, D.; Skommer, J.; McGuinness, D.; Hillier, C.; Darzynkiewicz, Z. ER-Golgi network—A future target for anti-cancer therapy. Leuk. Res. 2009, 33, 1440–1447. [Google Scholar] [CrossRef] [Green Version]
- Makhortova, N.R.; Hayhurst, M.; Cerqueira, A.; Sinor-Anderson, A.D.; Zhao, W.N.; Heiser, P.W.; Arvanites, A.C.; Davidow, L.S.; Waldon, Z.O.; Steen, J.A.; et al. A screen for regulators of survival of motor neuron protein levels. Nat. Chem. Biol. 2011, 7, 544–552. [Google Scholar] [CrossRef]
- Hsieh-Li, H.M.; Chang, J.G.; Jong, Y.J.; Wu, M.H.; Wang, N.M.; Tsai, C.H.; Li, H. A mouse model for spinal muscular atrophy. Nat. Genet. 2000, 24, 66–70. [Google Scholar] [CrossRef]
- Monani, U.R.; Sendtner, M.; Coovert, D.D.; Parsons, D.W.; Andreassi, C.; Le, T.T.; Jablonka, S.; Schrank, B.; Rossoll, W.; Prior, T.W.; et al. The human centromeric survival motor neuron gene (SMN2) rescues embryonic lethality in Smn(-/-) mice and results in a mouse with spinal muscular atrophy. Hum. Mol. Genet. 2000, 9, 333–339. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Razanau, A.; Hai, Y.; Yu, J.; Sohail, M.; Lobo, V.G.; Chu, J.; Kung, S.K.; Xie, J. A conserved serine of heterogeneous nuclear ribonucleoprotein L (hnRNP L) mediates depolarization-regulated alternative splicing of potassium channels. J. Biol. Chem. 2012, 287, 22709–22716. [Google Scholar] [CrossRef] [Green Version]
- Xiao, R.; Tang, P.; Yang, B.; Huang, J.; Zhou, Y.; Shao, C.; Li, H.; Sun, H.; Zhang, Y.; Fu, X.D. Nuclear matrix factor hnRNP U/SAF-A exerts a global control of alternative splicing by regulating U2 snRNP maturation. Mol. Cell 2012, 45, 656–668. [Google Scholar] [CrossRef] [Green Version]
- Cho, S.; Moon, H.; Loh, T.J.; Oh, H.K.; Cho, S.; Choy, H.E.; Song, W.K.; Chun, J.S.; Zheng, X.; Shen, H. hnRNP M facilitates exon 7 inclusion of SMN2 pre-mRNA in spinal muscular atrophy by targeting an enhancer on exon 7. Biochim. Biophys. Acta 2014, 1839, 306–315. [Google Scholar] [CrossRef]
- Cho, S.; Moon, H.; Loh, T.J.; Oh, H.K.; Williams, D.R.; Liao, D.J.; Zhou, J.; Green, M.R.; Zheng, X.; Shen, H. PSF contacts exon 7 of SMN2 pre-mRNA to promote exon 7 inclusion. Biochim. Biophys. Acta 2014, 1839, 517–525. [Google Scholar] [CrossRef] [Green Version]
- Shav-Tal, Y.; Cohen, M.; Lapter, S.; Dye, B.; Patton, J.G.; Vandekerckhove, J.; Zipori, D. Nuclear relocalization of the pre-mRNA splicing factor PSF during apoptosis involves hyperphosphorylation, masking of antigenic epitopes, and changes in protein interactions. Mol. Biol. Cell 2001, 12, 2328–2340. [Google Scholar] [CrossRef] [Green Version]
- Buxade, M.; Morrice, N.; Krebs, D.L.; Proud, C.G. The PSF.p54nrb complex is a novel Mnk substrate that binds the mRNA for tumor necrosis factor alpha. J. Biol. Chem. 2008, 283, 57–65. [Google Scholar] [CrossRef] [Green Version]
- Galietta, A.; Gunby, R.H.; Redaelli, S.; Stano, P.; Carniti, C.; Bachi, A.; Tucker, P.W.; Tartari, C.J.; Huang, C.J.; Colombo, E.; et al. NPM/ALK binds and phosphorylates the RNA/DNA-binding protein PSF in anaplastic large-cell lymphoma. Blood 2007, 110, 2600–2609. [Google Scholar] [CrossRef] [Green Version]
- Lukong, K.E.; Huot, M.E.; Richard, S. BRK phosphorylates PSF promoting its cytoplasmic localization and cell cycle arrest. Cell Signal. 2009, 21, 1415–1422. [Google Scholar] [CrossRef]
- Ebert, A.D.; Yu, J.; Rose, F.F., Jr.; Mattis, V.B.; Lorson, C.L.; Thomson, J.A.; Svendsen, C.N. Induced pluripotent stem cells from a spinal muscular atrophy patient. Nature 2009, 457, 277–280. [Google Scholar] [CrossRef]
- Frattini, E.; Ruggieri, M.; Salani, S.; Faravelli, I.; Zanetta, C.; Nizzardo, M.; Simone, C.; Magri, F.; Corti, S. Pluripotent stem cell-based models of spinal muscular atrophy. Mol. Cell Neurosci. 2015, 64, 44–50. [Google Scholar] [CrossRef]
- Valetdinova, K.R.; Medvedev, S.P.; Zakian, S.M. Model systems of motor neuron diseases as a platform for studying pathogenic mechanisms and searching for therapeutic agents. Acta. Naturae 2015, 7, 19–36. [Google Scholar] [CrossRef] [Green Version]
- Ando, S.; Suzuki, S.; Okubo, S.; Ohuchi, K.; Takahashi, K.; Nakamura, S.; Shimazawa, M.; Fuji, K.; Hara, H. Discovery of a CNS penetrant small molecule SMN2 splicing modulator with improved tolerability for spinal muscular atrophy. Sci. Rep. 2020, 10, 17472. [Google Scholar] [CrossRef]
- Son, Y.S.; Choi, K.; Lee, H.; Kwon, O.; Jung, K.B.; Cho, S.; Baek, J.; Son, B.; Kang, S.M.; Kang, M.; et al. A SMN2 Splicing Modifier Rescues the Disease Phenotypes in an In Vitro Human Spinal Muscular Atrophy Model. Stem. Cells Dev. 2019, 28, 438–453. [Google Scholar] [CrossRef]
- Kim, J.C.; Woo, S.H. Shear stress induces a longitudinal Ca(2+) wave via autocrine activation of P2Y1 purinergic signalling in rat atrial myocytes. J. Physiol. 2015, 593, 5091–5109. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, K.; Yang, A.; Baek, J.; Jeong, H.; Kang, Y.; Baek, W.; Kim, J.-C.; Kang, M.; Choi, M.; Ham, Y.; et al. Regulation of Survival Motor Neuron Gene Expression by Calcium Signaling. Int. J. Mol. Sci. 2021, 22, 10234. https://doi.org/10.3390/ijms221910234
Choi K, Yang A, Baek J, Jeong H, Kang Y, Baek W, Kim J-C, Kang M, Choi M, Ham Y, et al. Regulation of Survival Motor Neuron Gene Expression by Calcium Signaling. International Journal of Molecular Sciences. 2021; 22(19):10234. https://doi.org/10.3390/ijms221910234
Chicago/Turabian StyleChoi, Kwangman, Ansook Yang, Jiyeon Baek, Hyejeong Jeong, Yura Kang, Woosun Baek, Joon-Chul Kim, Mingu Kang, Miri Choi, Youngwook Ham, and et al. 2021. "Regulation of Survival Motor Neuron Gene Expression by Calcium Signaling" International Journal of Molecular Sciences 22, no. 19: 10234. https://doi.org/10.3390/ijms221910234