
Teriflunomide Inhibits JCPyV Infection and Spread in Glial Cells and Choroid Plexus Epithelial Cells

 ,
,
Abstract
:1. Introduction
2. Results
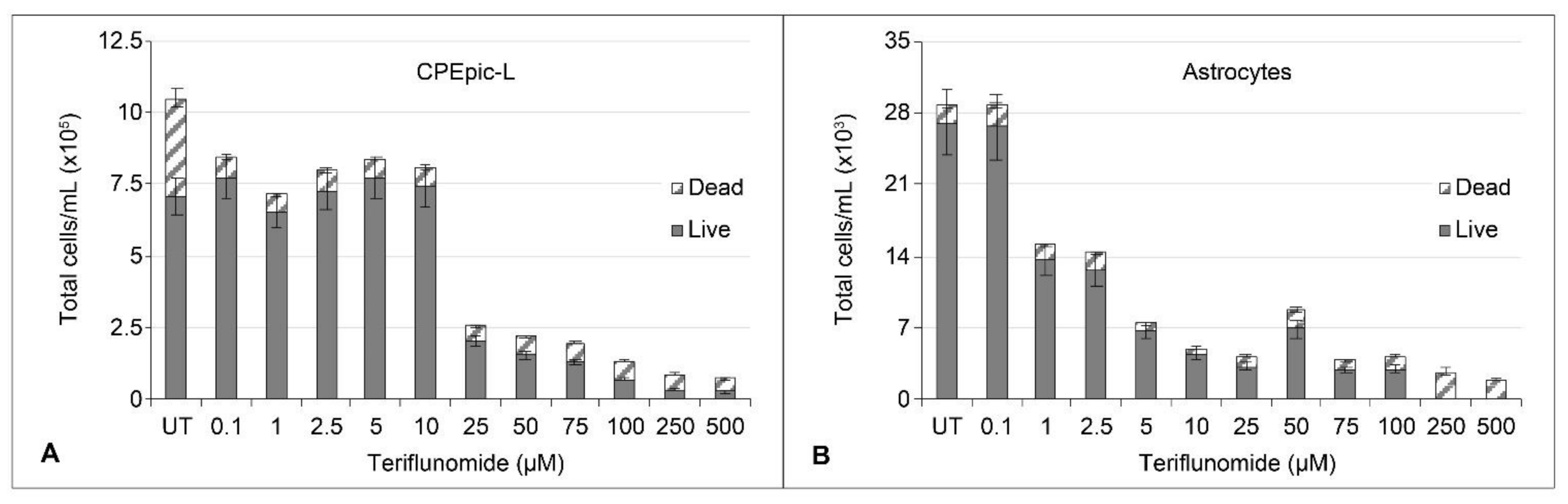
2.1. Cytotoxicity and Cytostatic Effect of Teriflunomide
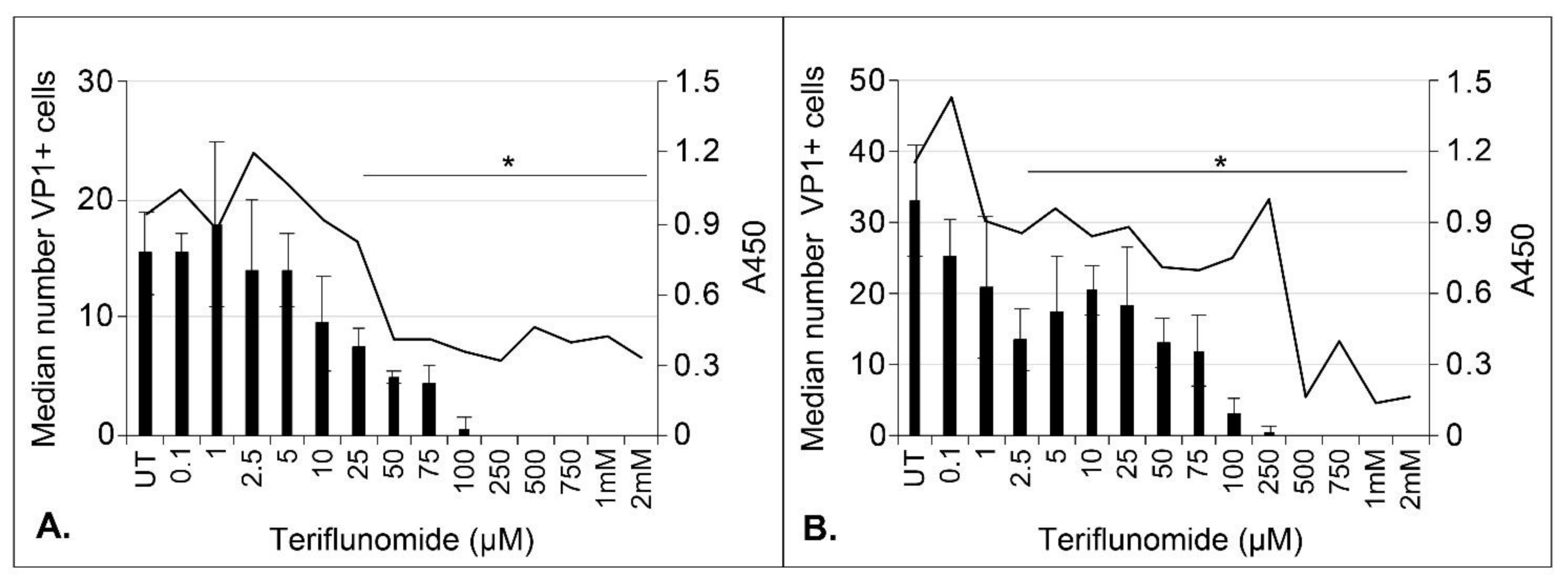
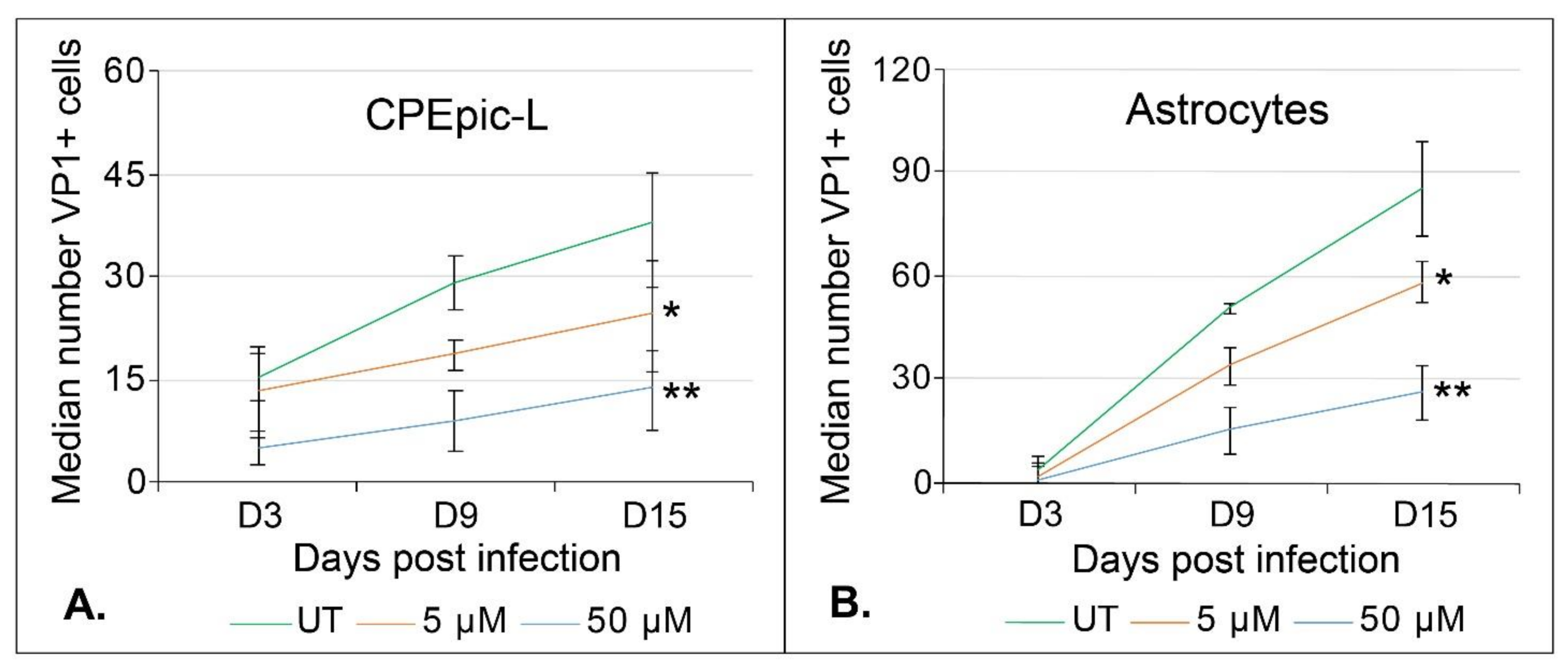
2.2. Teriflunomide Inhibits Initial Infection and Virus Spread over Time
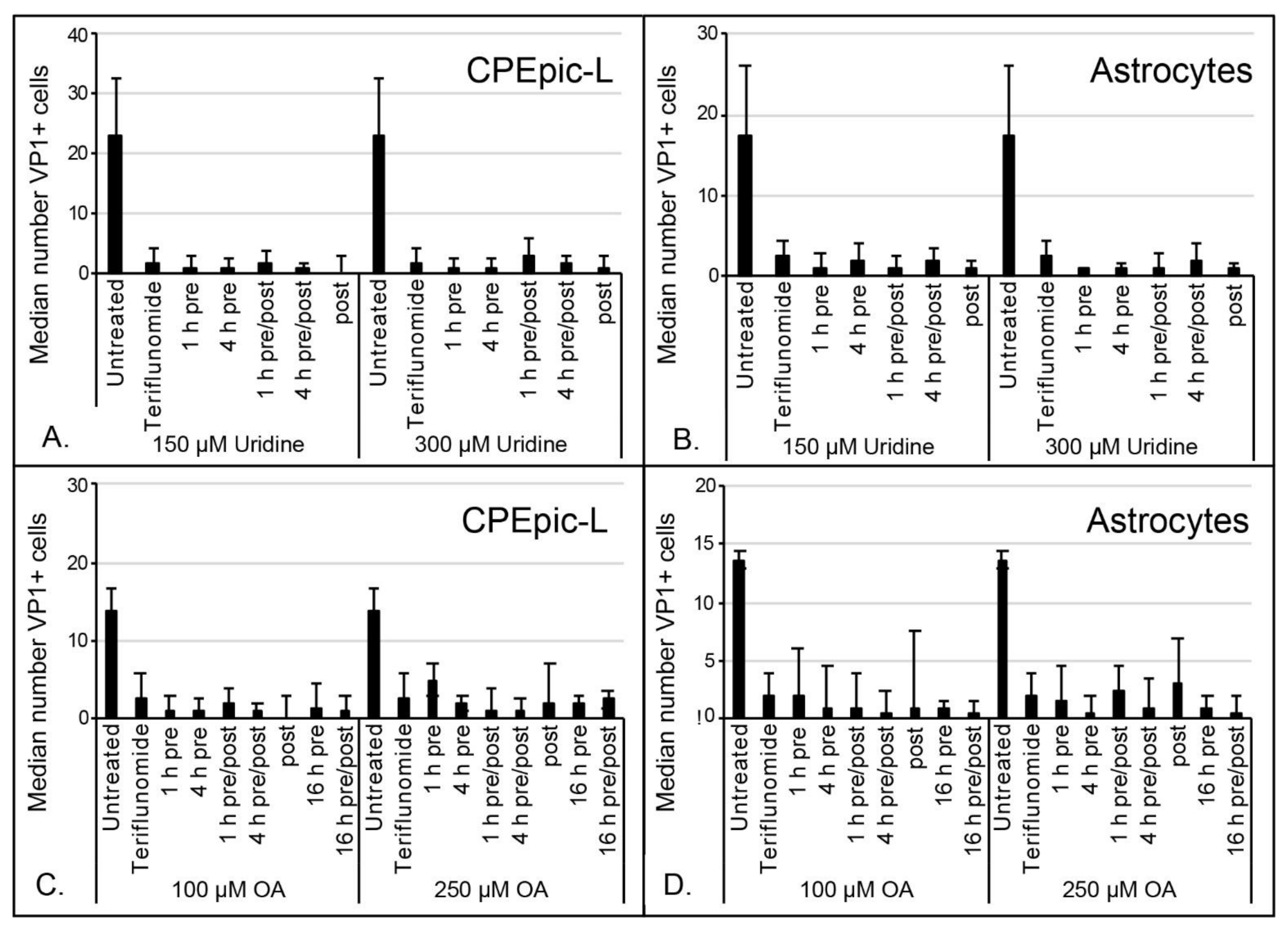
2.3. Inhibitory Effect of Teriflunomide on Viral Infection Cannot Be Rescued by Supplementing Cells with Uridine or Orotic Acid
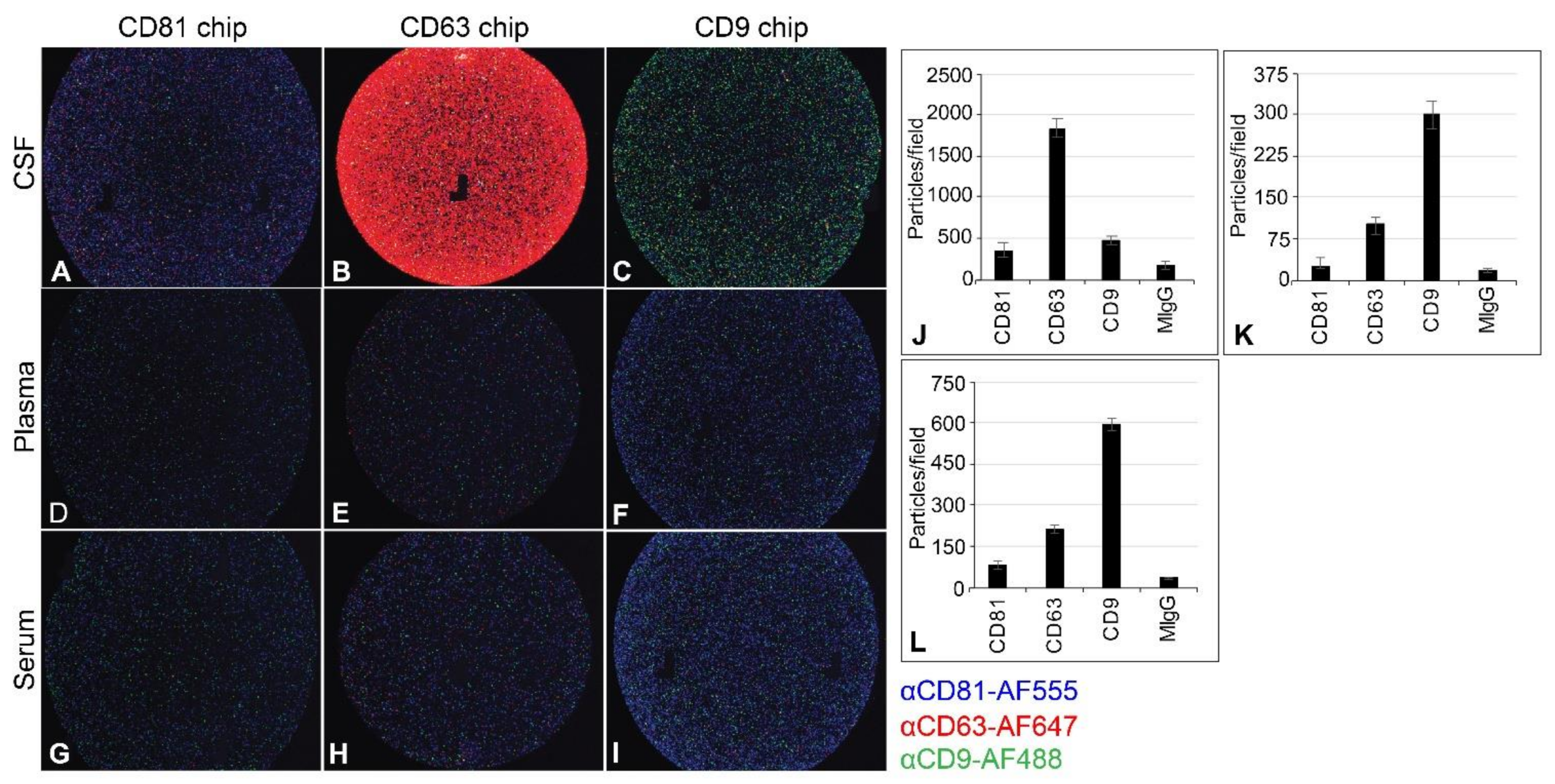
2.4. Capturing EV from CSF, Plasma, and Serum
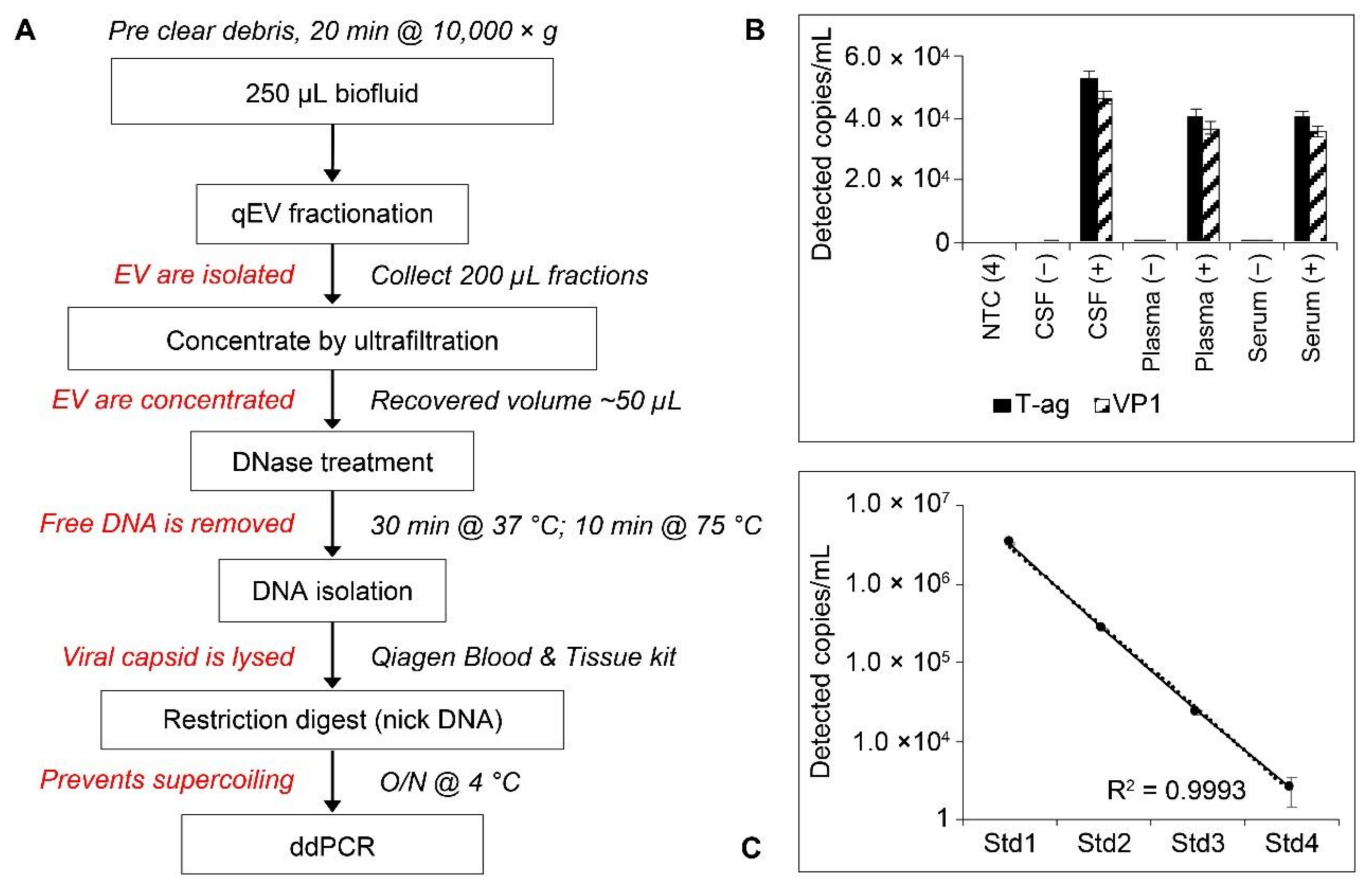
2.5. Workflow and Standard Curves for Droplet Digital PCR (ddPCR) Analysis of EV-Associated JCPyV DNA
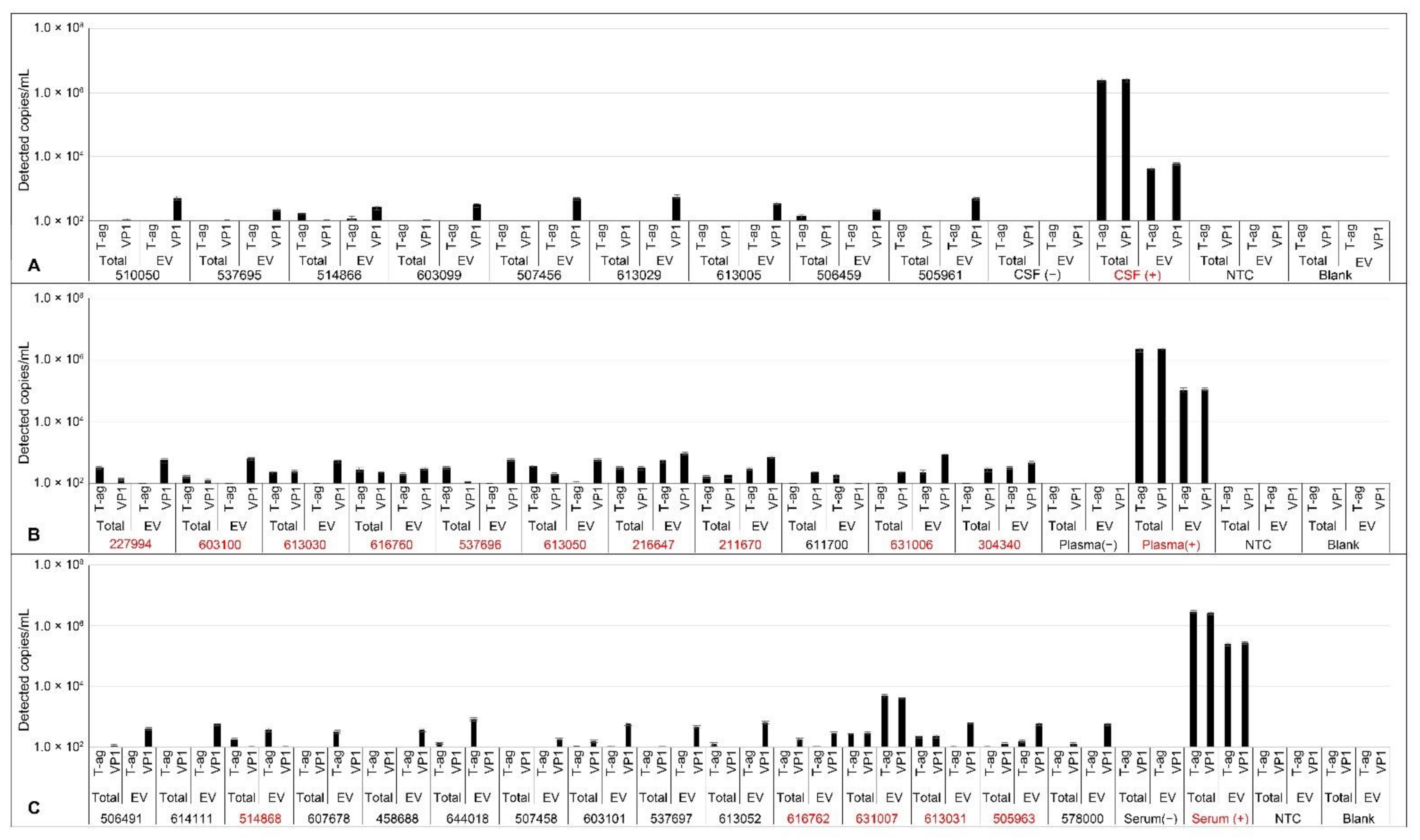
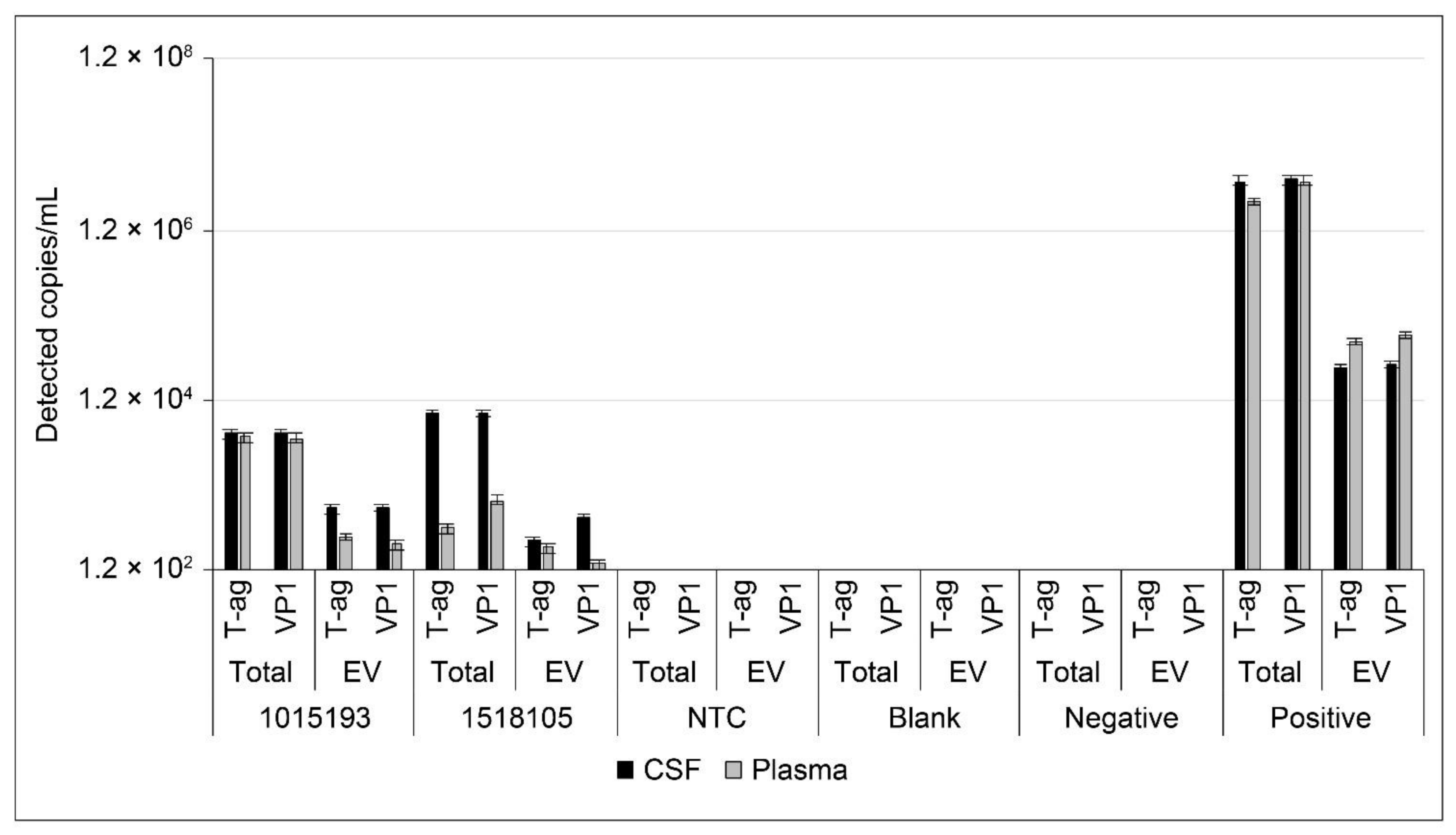
2.6. Analysis of JCPyV DNA in Patient Samples
3. Discussion
4. Materials and Methods
4.1. Cells, Media, and Virus
4.2. Proliferation and Toxicity
4.3. Infections
4.4. Indirect Immunofluorescence
4.5. Patient Samples
4.6. Generation of Control Samples
4.7. Extracellular Vesicles
4.8. DNA Extraction
4.9. Droplet Digital PCR
5. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Previous Presentation
Acknowledgments
Conflicts of Interest
References
- Imperiale, M.J.; Jiang, M. Polyomavirus persistence. Annu. Rev. Virol. 2016, 3, 517–532. [Google Scholar] [CrossRef]
- Haley, S.A.; Atwood, W.J. Progressive multifocal leukoencephalopathy: Endemic viruses and lethal brain disease. Annu. Rev. Virol. 2017, 4, 349–367. [Google Scholar] [CrossRef] [PubMed]
- Assetta, B.; Atwood, W.J. The biology of JC polyomavirus. Biol. Chem. 2017, 398, 839–855. [Google Scholar] [CrossRef]
- Cortese, I.; Reich, D.S.; Nath, A. Progressive multifocal leukoencephalopathy and the spectrum of JC virus-related disease. Nat. Rev. Neurol. 2021, 17, 37–51. [Google Scholar] [CrossRef] [PubMed]
- Berger, J.R. Classifying PML risk with disease modifying therapies. Mult. Scler. Relat. Disord. 2017, 12, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Wuthrich, C.; Popescu, B.F.; Gheuens, S.; Marvi, M.; Ziman, R.; Denq, S.P.; Tham, M.; Norton, E.; Parisi, J.E.; Dang, X.; et al. Natalizumab-associated progressive multifocal leukoencephalopathy in a patient with multiple sclerosis: A postmortem study. J. Neuropathol. Exp. Neurol. 2013, 72, 1043–1051. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Bord, E.; Tompkins, T.; Miller, J.; Tan, C.S.; Kinkel, R.P.; Stein, M.C.; Viscidi, R.P.; Ngo, L.H.; Koralnik, I.J. Asymptomatic reactivation of JC virus in patients treated with natalizumab. N. Engl. J. Med. 2009, 361, 1067–1074. [Google Scholar] [CrossRef] [PubMed]
- Berger, J.R.; Koralnik, I.J. Progressive multifocal leukoencephalopathy and natalizumab--unforeseen consequences. N. Engl. J. Med. 2005, 353, 414–416. [Google Scholar] [CrossRef] [Green Version]
- Miller, A.E.; Olsson, T.P.; Wolinsky, J.S.; Comi, G.; Kappos, L.; Hu, X.; Xu, X.; Lublin, A.L.; Truffinet, P.; Chavin, J.; et al. Long-term safety and efficacy of teriflunomide in patients with relapsing multiple sclerosis: Results from the TOWER extension study. Mult. Scler. Relat. Disord. 2020, 46, 102438. [Google Scholar] [CrossRef] [PubMed]
- Josephson, M.A.; Gillen, D.; Javaid, B.; Kadambi, P.; Meehan, S.; Foster, P.; Harland, R.; Thistlethwaite, R.J.; Garfinkel, M.; Atwood, W.; et al. Treatment of renal allograft polyoma BK virus infection with leflunomide. Transplantation 2006, 81, 704–710. [Google Scholar] [CrossRef]
- Williams, J.W.; Javaid, B.; Kadambi, P.V.; Gillen, D.; Harland, R.; Thistlewaite, J.R.; Garfinkel, M.; Foster, P.; Atwood, W.; Millis, J.M.; et al. Leflunomide for polyomavirus type BK nephropathy. N. Engl. J. Med. 2005, 352, 1157–1158. [Google Scholar] [CrossRef]
- Li, L.; Liu, J.; Delohery, T.; Zhang, D.; Arendt, C.; Jones, C. The effects of teriflunomide on lymphocyte subpopulations in human peripheral blood mononuclear cells in vitro. J. Neuroimmunol. 2013, 265, 82–90. [Google Scholar] [CrossRef] [PubMed]
- O’Hara, B.A.; Morris-Love, J.; Gee, G.V.; Haley, S.A.; Atwood, W.J. JC Virus infected choroid plexus epithelial cells produce extracellular vesicles that infect glial cells independently of the virus attachment receptor. PLoS Pathog. 2020, 16, e1008371. [Google Scholar] [CrossRef] [PubMed]
- O’Hara, B.A.; Gee, G.V.; Atwood, W.J.; Haley, S.A. Susceptibility of primary human choroid plexus epithelial cells and meningeal cells to infection by JC virus. J. Virol. 2018, 92, e00105-18. [Google Scholar] [CrossRef] [Green Version]
- Astrom, K.; Mancall, E.; Richardson, E.P., Jr. Progressive multifocal leukoencephalopathy. Brain 1958, 81, 93–127. [Google Scholar] [CrossRef]
- Richardson, E.P., Jr. Progressive multifocal leukoencephalopathy. N. Engl. J. Med. 1961, 265, 815–823. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.R.; Barrett, R.E.; Britton, C.B.; Tapper, M.L.; Bahr, G.S.; Bruno, P.J.; Marquardt, M.D.; Hays, A.P.; McMurtry, J.G., 3rd; Weissman, J.B.; et al. Progressive multifocal leukoencephalopathy in a male homosexual with T-cell immune deficiency. N. Engl. J. Med. 1982, 307, 1436–1438. [Google Scholar] [CrossRef] [PubMed]
- Bernick, C.; Gregorios, J.B. Progressive multifocal leukoencephalopathy in a patient with acquired immune deficiency syndrome. Arch. Neurol. 1984, 41, 780–782. [Google Scholar] [CrossRef]
- Brooks, B.R.; Walker, D.L. Progressive multifocal leukoencephalopathy. Neurol. Clin. 1984, 2, 299–313. [Google Scholar] [CrossRef]
- Adelman, B.; Sandrock, A.; Panzara, M.A. Natalizumab and progressive multifocal leukoencephalopathy. N. Engl. J. Med. 2005, 353, 432–433. [Google Scholar] [CrossRef] [Green Version]
- Williamson, E.M.; Berger, J.R. Central nervous system infections with immunomodulatory therapies. Continuum. Minneap. Minn. 2015, 21, 1577–1598. [Google Scholar] [CrossRef] [PubMed]
- Major, E.O.; Nath, A. A link between long-term natalizumab dosing in MS and PML: Putting the puzzle together. Neurol. Neuroimmunol. Neuroinflamm. 2016, 3, e235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, J.R.; Fox, R.J. Reassessing the risk of natalizumab-associated PML. J. Neurovirol. 2016, 22, 533–535. [Google Scholar] [CrossRef]
- Zhovtis Ryerson, L.; Frohman, T.C.; Foley, J.; Kister, I.; Weinstock-Guttman, B.; Tornatore, C.; Pandey, K.; Donnelly, S.; Pawate, S.; Bomprezzi, R.; et al. Extended interval dosing of natalizumab in multiple sclerosis. J. Neurol. Neurosurg. Psychiatry 2016, 87, 885–889. [Google Scholar] [CrossRef]
- Chang, I.; Muralidharan, K.K.; Campbell, N.; Ho, P.R. Modeling the efficacy of natalizumab in multiple sclerosis patients who switch from every-4-week dosing to extended-interval dosing. J. Clin. Pharmacol. 2021, 61, 339–348. [Google Scholar] [CrossRef]
- Ryerson, L.Z.; Foley, J.; Chang, I.; Kister, I.; Cutter, G.; Metzger, R.R.; Goldberg, J.D.; Li, X.; Riddle, E.; Smirnakis, K.; et al. Risk of natalizumab-associated PML in patients with MS is reduced with extended interval dosing. Neurology 2019, 93, e1452–e1462. [Google Scholar] [CrossRef] [Green Version]
- Scarpazza, C.; De Rossi, N.; Tabiadon, G.; Turrini, M.V.; Gerevini, S.; Capra, R. Four cases of natalizumab-related PML: A less severe course in extended interval dosing? Neurol. Sci. 2019, 40, 2119–2124. [Google Scholar] [CrossRef]
- Corbridge, S.M.; Rice, R.C.; Bean, L.A.; Wuthrich, C.; Dang, X.; Nicholson, D.A.; Koralnik, I.J. JC virus infection of meningeal and choroid plexus cells in patients with progressive multifocal leukoencephalopathy. J. Neurovirol. 2019, 25, 520–524. [Google Scholar] [CrossRef] [PubMed]
- Grapp, M.; Wrede, A.; Schweizer, M.; Huwel, S.; Galla, H.J.; Snaidero, N.; Simons, M.; Buckers, J.; Low, P.S.; Urlaub, H.; et al. Choroid plexus transcytosis and exosome shuttling deliver folate into brain parenchyma. Nat. Commun. 2013, 4, 2123. [Google Scholar] [CrossRef] [Green Version]
- Scribano, S.; Guerrini, M.; Arvia, R.; Guasti, D.; Nardini, P.; Romagnoli, P.; Giannecchini, S. Archetype JC polyomavirus DNA associated with extracellular vesicles circulates in human plasma samples. J. Clin. Virol. 2020, 128, 104435. [Google Scholar] [CrossRef]
- Prezioso, C.; Zingaropoli, M.A.; Iannetta, M.; Rodio, D.M.; Altieri, M.; Conte, A.; Vullo, V.; Ciardi, M.R.; Palamara, A.T.; Pietropaolo, V. Which is the best PML risk stratification strategy in natalizumab-treated patients affected by multiple sclerosis? Mult. Scler. Relat. Disord. 2020, 41, 102008. [Google Scholar] [CrossRef] [Green Version]
- Morris-Love, J.; Gee, G.V.; O’Hara, B.A.; Assetta, B.; Atkinson, A.L.; Dugan, A.S.; Haley, S.A.; Atwood, W.J. JC polyomavirus uses extracellular vesicles to infect target cells. mBio 2019, 10, e00379-19. [Google Scholar] [CrossRef] [Green Version]
- Tugemann, B.; Berger, J.R. Improving risk-stratification of natalizumab-associated PML. Ann. Clin. Transl. Neurol. 2021, 8, 696–703. [Google Scholar] [CrossRef] [PubMed]
- Toboso, I.; Tejeda-Velarde, A.; Alvarez-Lafuente, R.; Arroyo, R.; Hegen, H.; Deisenhammer, F.; Sainz de la Maza, S.; Alvarez-Cermeno, J.C.; Izquierdo, G.; Paramo, D.; et al. New algorithms improving PML risk stratification in MS patients treated with natalizumab. Front. Neurol. 2020, 11, 579438. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.K.; Atwood, W.J. Propagation and assay of the JC virus. Methods Mol. Biol. 2001, 165, 9–17. [Google Scholar] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| JCPyV Status | ||||||
|---|---|---|---|---|---|---|
| CSF (+/−) | Plasma (+/−) | Serum (+/−) | ||||
| Patient Group | Total | EV | Total | EV | Total | EV |
| RRMS, teriflunomide | 0/7 | 0/7 | 1/1 | 0/2 | 1/5 | 0/6 |
| RRMS, natalizumab | N/A | N/A | 6/7 | 3/4 | 0/7 | 0/7 |
| Nonimmunomodulated | 0/2 | 0/2 | 1/1 | 1/1 | 1/1 | 1/1 |
| HIV-PML | 2/0 | 2/0 | 2/0 | 2/0 | N/A | N/A |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
O’Hara, B.A.; Gee, G.V.; Haley, S.A.; Morris-Love, J.; Nyblade, C.; Nieves, C.; Hanson, B.A.; Dang, X.; Turner, T.J.; Chavin, J.M.; et al. Teriflunomide Inhibits JCPyV Infection and Spread in Glial Cells and Choroid Plexus Epithelial Cells. Int. J. Mol. Sci. 2021, 22, 9809. https://doi.org/10.3390/ijms22189809
O’Hara BA, Gee GV, Haley SA, Morris-Love J, Nyblade C, Nieves C, Hanson BA, Dang X, Turner TJ, Chavin JM, et al. Teriflunomide Inhibits JCPyV Infection and Spread in Glial Cells and Choroid Plexus Epithelial Cells. International Journal of Molecular Sciences. 2021; 22(18):9809. https://doi.org/10.3390/ijms22189809
Chicago/Turabian StyleO’Hara, Bethany A., Gretchen V. Gee, Sheila A. Haley, Jenna Morris-Love, Charlotte Nyblade, Chris Nieves, Barbara A. Hanson, Xin Dang, Timothy J. Turner, Jeffrey M. Chavin, and et al. 2021. "Teriflunomide Inhibits JCPyV Infection and Spread in Glial Cells and Choroid Plexus Epithelial Cells" International Journal of Molecular Sciences 22, no. 18: 9809. https://doi.org/10.3390/ijms22189809