
Expression Patterns of DNA Methylation and Demethylation Genes during Plant Development and in Response to Phytohormones

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Generation of GUS Reporter Lines of 15 DNA Methylation and Demethylation-Related Genes
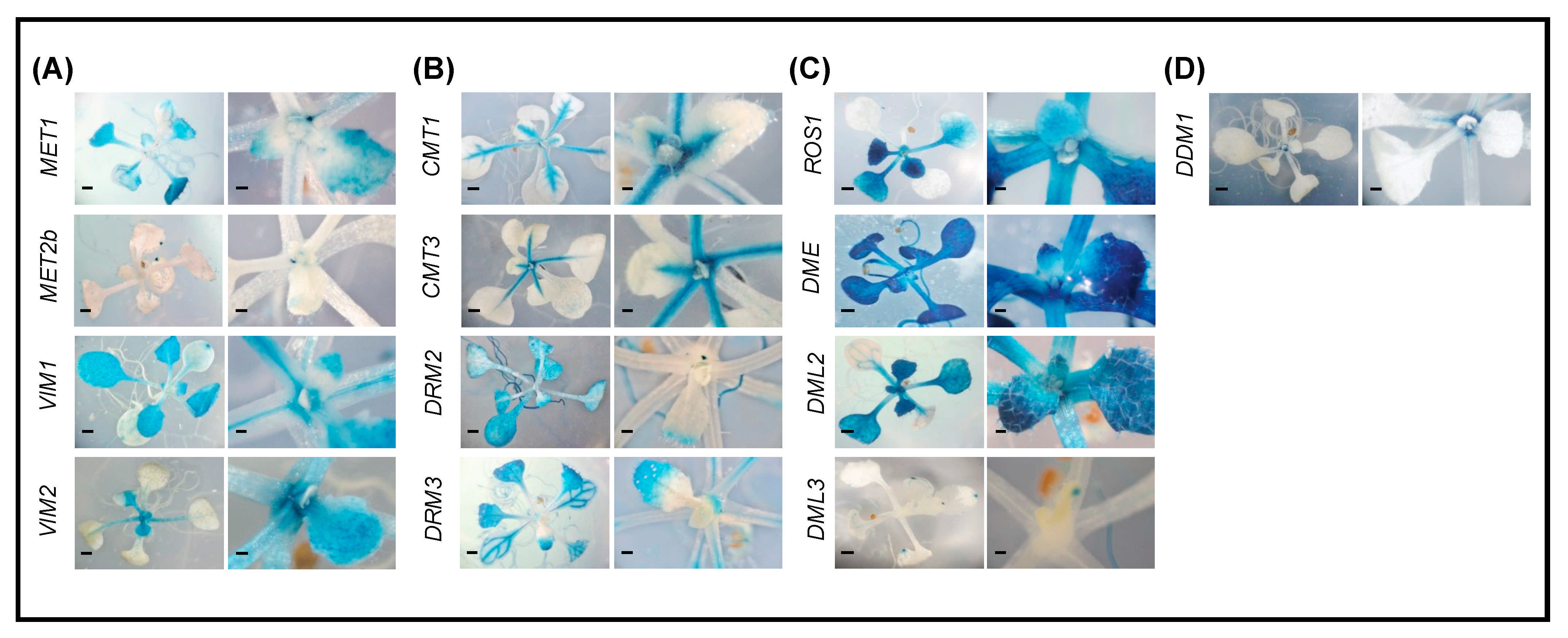
2.2. Promoter Activity of DNA Methylation and Demethylation-Related Genes in Roots
2.3. Promoter Activity of DNA Methylation and Demethylation-Related Genes in Shoots
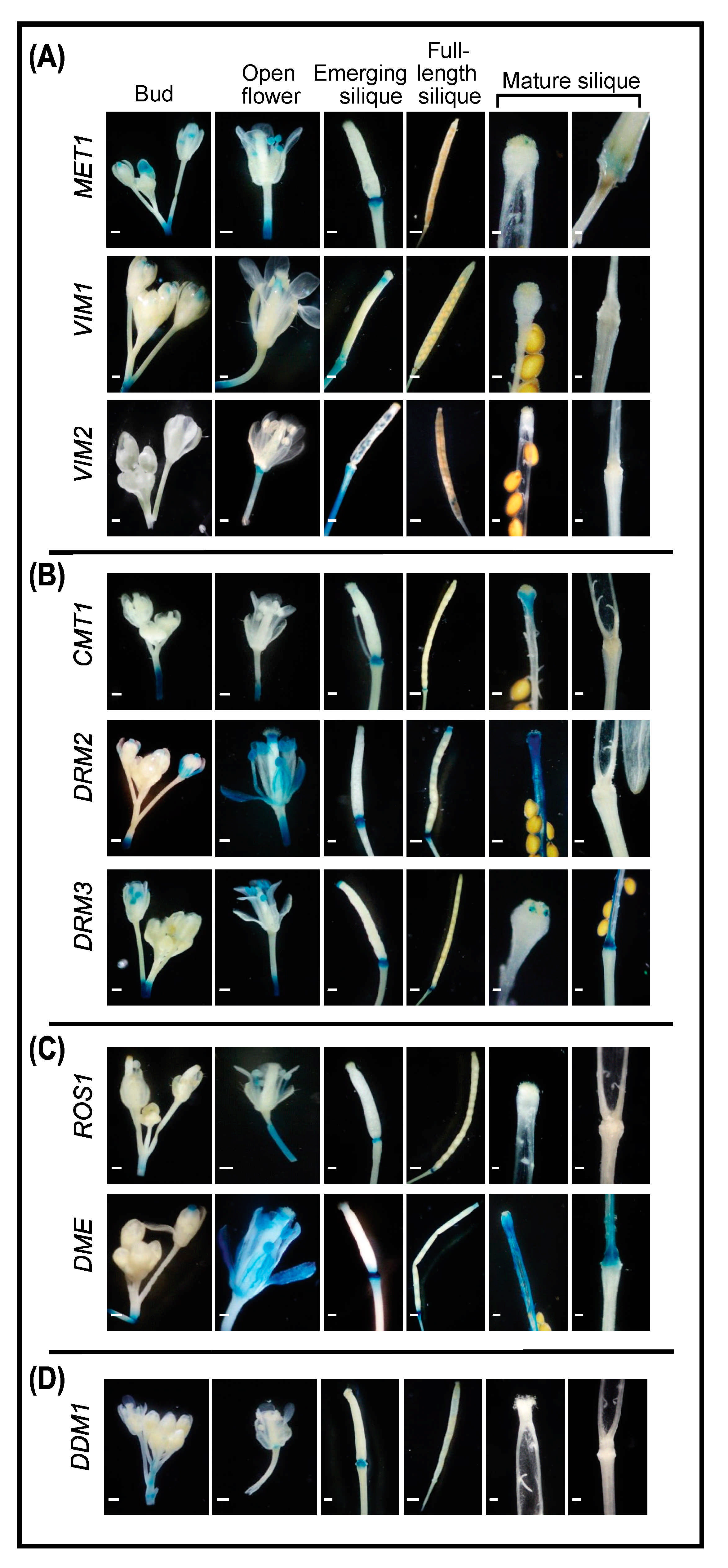
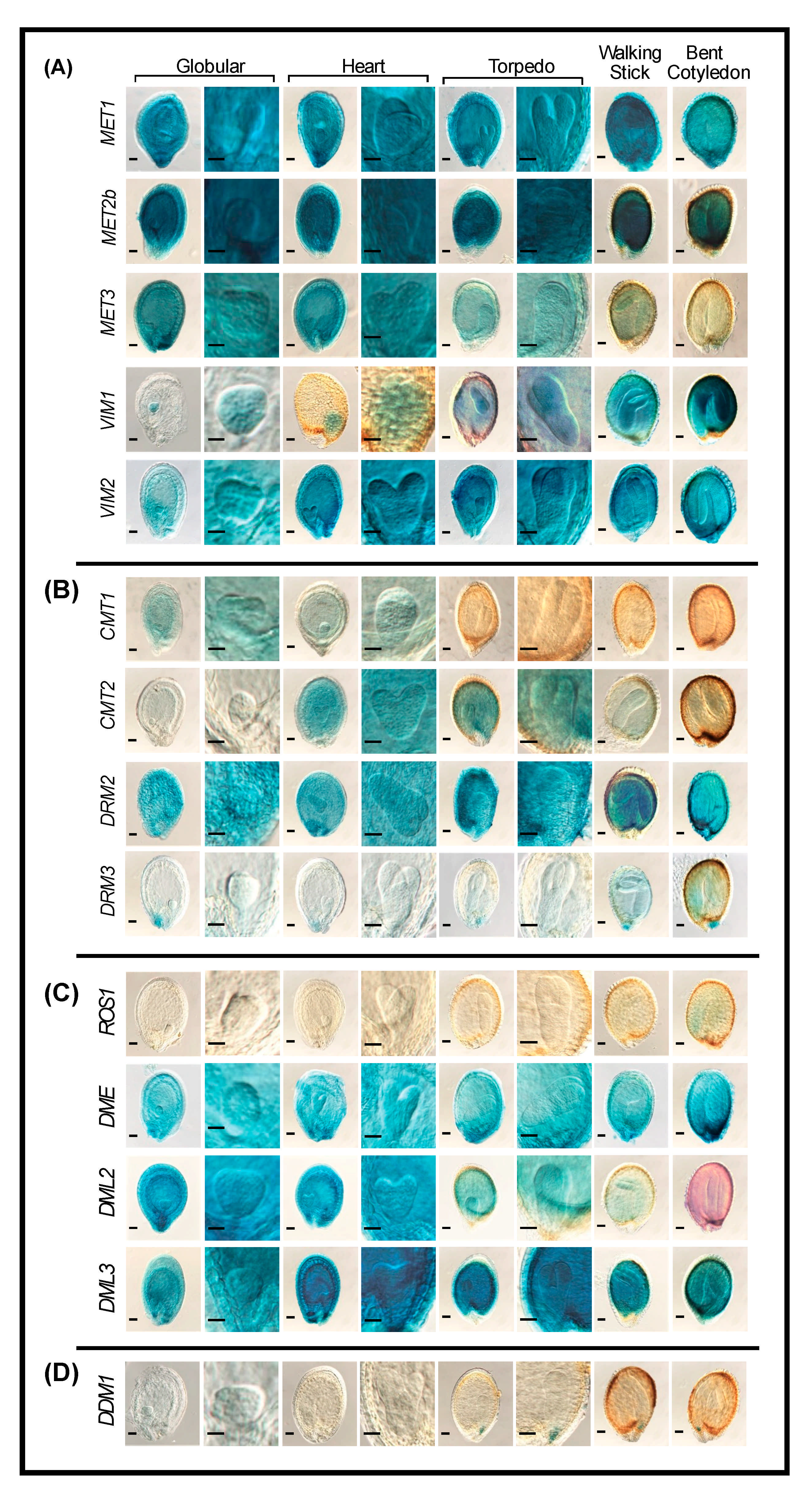
2.4. Promoter Activity of DNA Methylation and Demethylation in Reproductive Tissues
2.4.1. Buds and Open Flowers
2.4.2. Siliques
2.4.3. Seeds
2.5. RNA-seq and RT-qPCR Data Support Promoter Activity of DNA Methylation and Demethylation-Related Genes
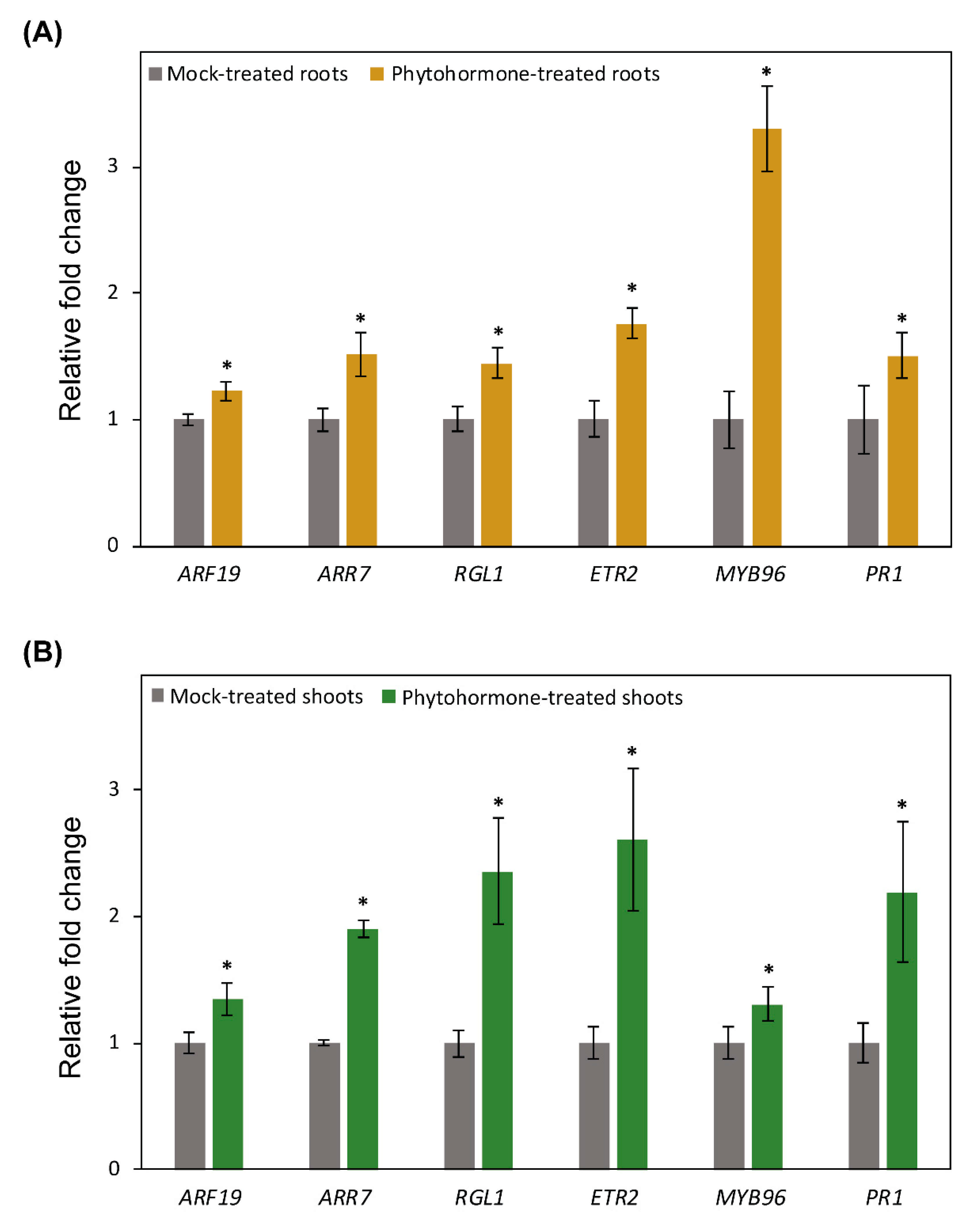
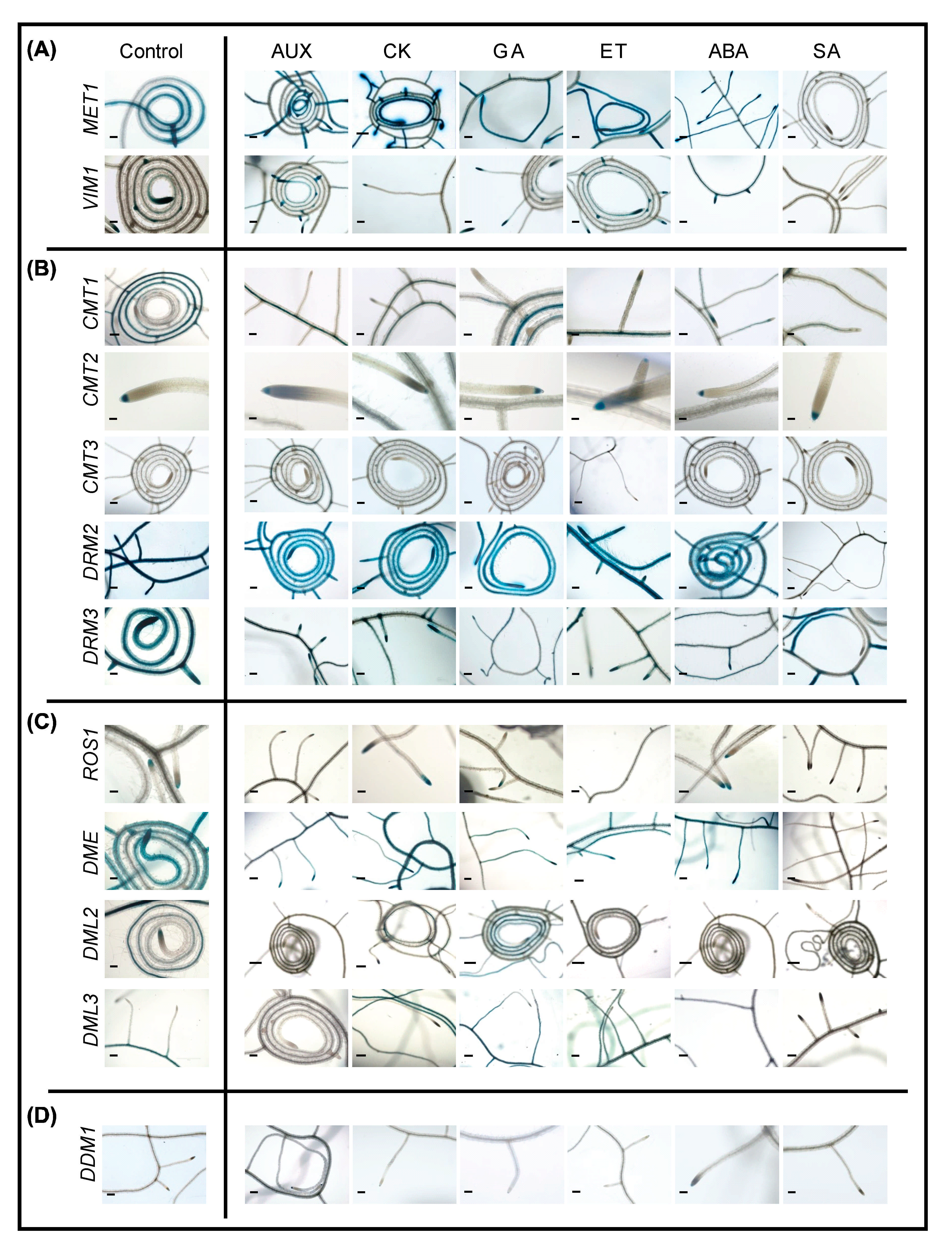
2.6. Promoter Activity of DNA Methylation and Demethylation-Related Genes in Roots following Phytohormone Treatments
2.7. Promoter Activity of DNA Methylation and Demethylation-Related Gene in Shoots following Phytohormone Treatments
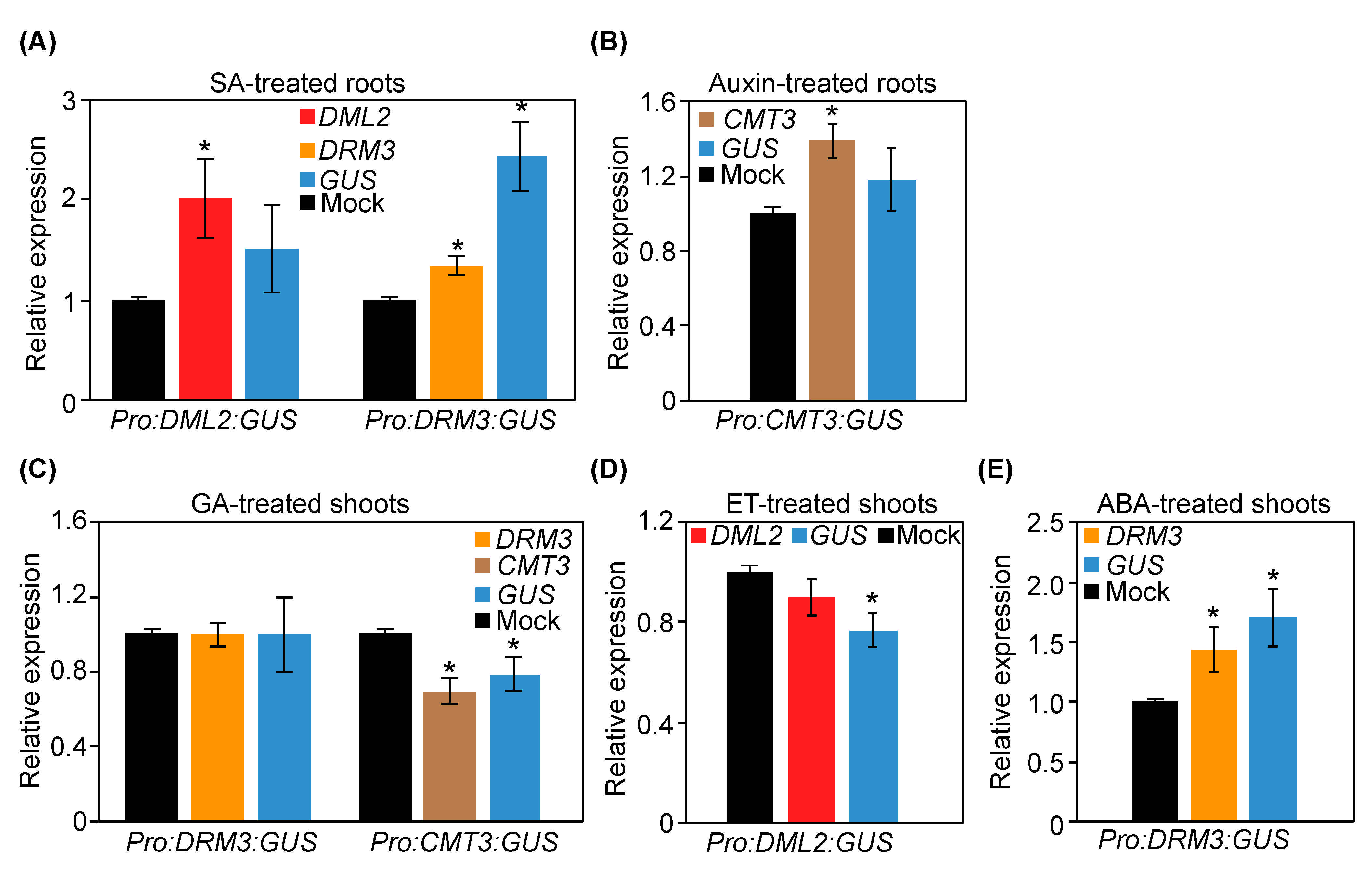
2.8. RT-qPCR Data Validate Gene Expression Changes Induced by Phytohormone Treatments
3. Discussion
3.1. Promoter Activity of DNA Methylation and Demethylation-Related Genes in Various Plant Tissues and Organs
3.2. Phytohormone-Induced Changes in the Promoter Activity of DNA Methylation and Demethylation-Related Genes
4. Materials and Methods
4.1. Plant Materials and Growth Conditions
4.2. Generation of GUS Reporter Lines
4.3. Histochemical Analysis of GUS Activity
4.4. Phytohormone Treatments
4.5. RT-qPCR Analysis
4.6. Light Microscopy and Imaging
4.7. RNA-Seq Data Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ABA | Abscisic acid |
| AUX | Auxin |
| CK | Cytokinin |
| ET | Ethylene |
| GA | Gibberellic acid |
| RdDM | RNA-directed DNA methylation |
| SA | Salicylic acid |
| RAM | Root apical meristem |
References
- Henderson, I.R.; Jacobsen, S.E. Epigenetic inheritance in plants. Nature 2007, 447, 418–424. [Google Scholar] [CrossRef]
- Fransz, P.F.; De Jong, J.H. Chromatin dynamics in plants. Curr. Opin. Plant Biol. 2002, 5, 560–567. [Google Scholar] [CrossRef]
- Zilberman, D.; Gehring, M.; Tran, R.K.; Ballinger, T.; Henikoff, S. Genome-wide analysis of Arabidopsis thaliana DNA methylation uncovers an interdependence between methylation and transcription. Nat. Genet. 2007, 39, 61–69. [Google Scholar] [CrossRef]
- Zhang, M.; Kimatu, J.N.; Xu, K.; Liu, B. DNA cytosine methylation in plant development. J Genet. Genom. 2010, 37, 1–12. [Google Scholar] [CrossRef]
- Matzke, M.A.; Mosher, R.A. RNA-directed DNA methylation: An epigenetic pathway of increasing complexity. Nat. Genet. 2014, 15, 394–408. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Johnson, L.M.; Jacobsen, S.E.; Patel, D.J. DNA methylation pathways and their crosstalk with histone methylation. Nat. Rev. Mol. Cell Biol. 2015, 16, 519–532. [Google Scholar] [CrossRef] [Green Version]
- López Sánchez, A.; Stassen, J.H.; Furci, L.; Smith, L.M.; Ton, J. The role of DNA (de) methylation in immune responsiveness of Arabidopsis. Plant J. 2016, 88, 361–374. [Google Scholar] [CrossRef]
- Hewezi, T.; Pantalone, V.; Bennett, M.; Stewart, C.N.; Burch-Smith, T.M. Phytopathogen-induced changes to plant methylomes. Plant Cell Rep. 2018, 37, 17–23. [Google Scholar] [CrossRef]
- Hewezi, T. Epigenetic Mechanisms in Nematode–Plant Interactions. Annu. Rev. Phytopathol. 2020, 58, 119–138. [Google Scholar] [CrossRef]
- Hewezi, T.; Lane, T.; Piya, S.; Rambani, A.; Rice, J.H.; Staton, M. Cyst nematode parasitism induces dynamic changes in the root epigenome. Plant Physiol. 2017, 174, 405–420. [Google Scholar] [CrossRef] [Green Version]
- Rambani, A.; Pantalone, V.; Yang, S.; Rice, J.H.; Song, Q.; Mazarei, M.; Arelli, P.R.; Meksem, K.; Stewart, C.N.; Hewezi, T. Identification of introduced and stably inherited DNA methylation variants in soybean associated with soybean cyst nematode parasitism. New Phytol. 2020, 227, 168–184. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Liu, R.; Niu, Q.; Tang, K.; Zhang, B.; Zhang, H.; Chen, K.; Zhu, J.K.; Lang, Z. Global increase in DNA methylation during orange fruit development and ripening. Proc. Natl. Acad. Sci. USA 2019, 116, 1430–1436. [Google Scholar] [CrossRef] [Green Version]
- Rambani, A.; Hu, Y.; Piya, S.; Long, M.; Rice, J.H.; Pantalone, V.; Hewezi, T. Identification of differentially methylated miRNA genes during compatible and incompatible interactions between soybean and soybean cyst nematode. Mol. Plant-Microbe Interact. 2020, 33, 1340–1352. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.T.; Hu, L.J.; Wang, X.F.; Li, N.; Xu, C.M.; Gong, L.; Liu, B. DNA methylation affects gene alternative splicing in plants: An example from rice. Mol. Plant 2016, 9, 305–307. [Google Scholar] [CrossRef] [Green Version]
- Bewick, A.J.; Schmitz, R.J. Gene body DNA methylation in plants. Curr. Opin. Plant Biol. 2017, 36, 103–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niyikiza, D.; Piya, S.; Routray, P.; Miao, L.; Kim, W.S.; Burch-Smith, T.; Gill, T.; Sams, C.; Arelli, P.R.; Pantalone, V.; et al. Interactions of gene expression, alternative splicing, and DNA methylation in determining nodule identity. Plant J. 2020, 103, 1744–1766. [Google Scholar] [CrossRef]
- Lister, R.; O’Malley, R.C.; Tonti-Filippini, J.; Gregory, B.D.; Berry, C.C.; Millar, A.H.; Ecker, J.R. Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell 2008, 133, 523–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, X.; Jacobsen, S.E. Role of the Arabidopsis DRM methyltransferases in de novo DNA methylation and gene silencing. Curr. Biol. 2002, 12, 1138–1144. [Google Scholar] [CrossRef] [Green Version]
- Cao, X.; Aufsatz, W.; Zilberman, D.; Mette, M.F.; Huang, M.S.; Matzke, M.; Jacobsen, S.E. Role of the DRM and CMT3 methyltransferases in RNA-directed DNA methylation. Curr. Biol. 2003, 13, 2212–2217. [Google Scholar] [CrossRef]
- Henderson, I.R.; Deleris, A.; Wong, W.; Zhong, X.; Chin, H.G.; Horwitz, G.A.; Kelly, K.A.; Pradhan, S.; Jacobsen, S.E. The de novo cytosine methyltransferase DRM2 requires intact UBA domains and a catalytically mutated paralog DRM3 during RNA–directed DNA methylation in Arabidopsis thaliana. PLoS Genet. 2010, 6, e1001182. [Google Scholar] [CrossRef] [Green Version]
- Saze, H.; Scheid, O.M.; Paszkowski, J. Maintenance of CpG methylation is essential for epigenetic inheritance during plant gametogenesis. Nat. Genet. 2003, 34, 65–69. [Google Scholar] [CrossRef] [PubMed]
- Kankel, M.W.; Ramsey, D.E.; Stokes, T.L.; Flowers, S.K.; Haag, J.R.; Jeddeloh, J.A.; Riddle, N.C.; Verbsky, M.L.; Richards, E.J. Arabidopsis MET1 cytosine methyltransferase mutants. Genetics 2003, 163, 1109–1122. [Google Scholar] [CrossRef] [PubMed]
- Quadrana, L.; Silveira, A.B.; Mayhew, G.F.; LeBlanc, C.; Martienssen, R.A.; Jeddeloh, J.A.; Colot, V. The Arabidopsis thaliana mobilome and its impact at the species level. eLife 2016, 5, e15716. [Google Scholar] [CrossRef]
- Zemach, A.; Kim, M.Y.; Hsieh, P.H.; Coleman-Derr, D.; Eshed-Williams, L.; Thao, K.; Harmer, S.L.; Zilberman, D. The Arabidopsis nucleosome remodeler DDM1 allows DNA methyltransferases to access H1-containing heterochromatin. Cell 2013, 153, 193–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Kim, J.H.; Richards, E.J.; Chung, K.M.; Woo, H.R. Arabidopsis VIM proteins regulate epigentic silencing by modulating DNA methylation and histone modification in cooperation with MET1. Mol. Plant 2014, 7, 1470–1485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, J.C.; Xia, A.A.; Liu, J.H.; Jing, J.L.; Wang, Y.Z.; Qi, C.Y.; He, Y. Decrease in DNA methylation 1 (DDM1) is required for the formation of mCHH islands in maize. J. Integr. Plant Biol. 2019, 61, 749–764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stroud, H.; Do, T.; Du, J.; Zhong, X.; Feng, S.; Johnson, L.; Patel, D.J.; Jacobsen, S.E. Non-CG methylation patterns shape the epigenetic landscape in Arabidopsis. Nat. Struct. Biol. 2014, 21, 64. [Google Scholar] [CrossRef] [Green Version]
- Stroud, H.; Greenberg, M.V.; Feng, S.; Bernatavichute, Y.V.; Jacobsen, S.E. Comprehensive analysis of silencing mutants reveals complex regulation of the Arabidopsis methylome. Cell 2013, 152, 352–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Law, J.A.; Jacobsen, S.E. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Genet. 2010, 11, 204–220. [Google Scholar] [CrossRef]
- Costa-Nunes, P.; Kim, J.Y.; Hong, E.; Pontes, O. The cytological and molecular role of domains rearranged methyltransferase3 in RNA-dependent DNA methylation of Arabidopsis thaliana. BMC Res. Notes 2014, 7, 721. [Google Scholar] [CrossRef] [Green Version]
- Gong, Z.; Morales-Ruiz, T.; Ariza, R.R.; Roldán-Arjona, T.; David, L.; Zhu, J.K. ROS1, a repressor of transcriptional gene silencing in Arabidopsis, encodes a DNA glycosylase/lyase. Cell 2002, 111, 803–814. [Google Scholar] [CrossRef] [Green Version]
- Penterman, J.; Uzawa, R.; Fischer, R.L. Genetic interactions between DNA demethylation and methylation in Arabidopsis. Plant Physiol. 2007, 145, 1549–1557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penterman, J.; Zilberman, D.; Huh, J.H.; Ballinger, T.; Henikoff, S.; Fischer, R.L. DNA demethylation in the Arabidopsis genome. Proc. Natl. Acad. Sci. USA 2007, 104, 6752–6757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.; Kapoor, A.; Sridhar, V.V.; Agius, F.; Zhu, J.K. The DNA glycosylase/lyase ROS1 functions in pruning DNA methylation patterns in Arabidopsis. Curr. Biol. 2007, 17, 54–59. [Google Scholar] [CrossRef] [Green Version]
- Ortega-Galisteo, A.P.; Morales-Ruiz, T.; Ariza, R.R.; Roldán-Arjona, T. Arabidopsis DEMETER-LIKE proteins DML2 and DML3 are required for appropriate distribution of DNA methylation marks. Plant Mol. Biol. 2008, 67, 671–681. [Google Scholar] [CrossRef]
- Choi, Y.; Gehring, M.; Johnson, L.; Hannon, M.; Harada, J.J.; Goldberg, R.B.; Jacobsen, S.E.; Fischer, R.L. DEMETER, a DNA glycosylase domain protein, is required for endosperm gene imprinting and seed viability in Arabidopsis. Cell 2002, 110, 33–42. [Google Scholar] [CrossRef] [Green Version]
- Agius, F.; Kapoor, A.; Zhu, J.K. Role of the Arabidopsis DNA glycosylase/lyase ROS1 in active DNA demethylation. Proc. Natl. Acad. Sci. USA 2006, 103, 11796–11801. [Google Scholar] [CrossRef] [Green Version]
- Gehring, M.; Huh, J.H.; Hsieh, T.F.; Penterman, J.; Choi, Y.; Harada, J.J.; Goldberg, R.B.; Fischer, R.L. DEMETER DNA glycosylase establishes MEDEA polycomb gene self-imprinting by allele-specific demethylation. Cell 2006, 124, 495–506. [Google Scholar] [CrossRef] [Green Version]
- Morales-Ruiz, T.; Ortega-Galisteo, A.P.; Ponferrada-Marín, M.I.; Martínez-Macías, M.I.; Ariza, R.R.; Roldán-Arjona, T. Demeter and repressor of silencing 1 encode 5-methylcytosine DNA glycosylases. Proc. Natl. Acad. Sci. USA 2006, 103, 6853–6858. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Zhu, J.K. Active DNA demethylation in plants and animals. Cold Spring Harb. Symp. Quant. Biol. 2012, 77, 161–173. [Google Scholar] [CrossRef] [Green Version]
- Lei, M.; Zhang, H.; Julian, R.; Tang, K.; Xie, S.; Zhu, J.K. Regulatory link between DNA methylation and active demethylation in Arabidopsis. Proc. Natl. Acad. Sci. USA 2015, 112, 3553–3557. [Google Scholar] [CrossRef] [Green Version]
- Parrilla-Doblas, J.T.; Roldán-Arjona, T.; Ariza, R.R.; Córdoba-Cañero, D. Active DNA Demethylation in Plants. Int. J. Mol. Sci. 2019, 20, 4683. [Google Scholar] [CrossRef] [Green Version]
- Dowen, R.H.; Pelizzola, M.; Schmitz, R.J.; Lister, R.; Dowen, J.M.; Nery, J.R.; Dixon, J.E.; Ecker, J.R. Widespread dynamic DNA methylation in response to biotic stress. Proc. Natl. Acad. Sci. USA 2012, 109, E2183–E2191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, T.N.; Schumann, U.; Smith, N.A.; Tiwari, S.; Au, P.C.K.; Zhu, Q.H.; Taylor, J.M.; Kazan, K.; Llewellyn, D.J.; Zhang, R.; et al. DNA demethylases target promoter transposable elements to positively regulate stress responsive genes in Arabidopsis. Genome Biol. 2014, 15, 458. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Prado, J.S.; Abulfaraj, A.A.; Rayapuram, N.; Benhamed, M.; Hirt, H. Plant immunity: From signaling to epigenetic control of defense. Trends Plant Sci. 2018, 23, 833–844. [Google Scholar] [CrossRef] [PubMed]
- Secco, D.; Wang, C.; Shou, H.; Schultz, M.D.; Chiarenza, S.; Nussaume, L.; Ecker, J.R.; Whelan, J.; Lister, R. Stress induced gene expression drives transient DNA methylation changes at adjacent repetitive elements. eLife 2015, 4, e09343. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Tieman, D.M.; Jiao, C.; Xu, Y.; Chen, K.; Fei, Z.; Giovannoni, J.J.; Klee, H.J. Chilling-induced tomato flavor loss is associated with altered volatile synthesis and transient changes in DNA methylation. Proc. Natl. Acad. Sci. USA 2016, 113, 12580–12585. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Xie, Q.; Li, B.; Su, H. Molecular characterization and transcription analysis of DNA methyltransferase genes in tomato (Solanum lycopersicum). Genet. Mol. Biol. 2020, 43, e20180295. [Google Scholar] [CrossRef] [Green Version]
- Santner, A.; Calderon-Villalobos, L.I.A.; Estelle, M. Plant hormones are versatile chemical regulators of plant growth. Nat. Chem. Biol. 2009, 5, 301–307. [Google Scholar] [CrossRef]
- Berens, M.L.; Berry, H.M.; Mine, A.; Argueso, C.T.; Tsuda, K. Evolution of hormone signaling networks in plant defense. Annu. Rev. Phytopath. 2017, 55, 401–425. [Google Scholar] [CrossRef]
- Blázquez, M.A.; Nelson, D.C.; Weijers, D. Evolution of plant hormone response pathways. Annu. Rev. Plant Biol. 2020, 71, 327–353. [Google Scholar] [CrossRef] [Green Version]
- Waadt, R. Phytohormone signaling mechanisms and genetic methods for their modulation and detection. Curr. Opin. Plant Biol. 2020, 57, 31–40. [Google Scholar] [CrossRef]
- Zhu, Y. The epigenetic involvement in plant hormone signaling. Chin. Sci. Bull. 2010, 55, 2198–2203. [Google Scholar] [CrossRef]
- Yamamuro, C.; Zhu, J.K.; Yang, Z. Epigenetic modifications and plant hormone action. Mol. Plant 2016, 9, 57–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maury, S.; Sow, M.D.; Le Gac, A.L.; Genitoni, J.; Lafon-Placette, C.; Mozgova, I. Phytohormone and Chromatin Crosstalk: The Missing Link for Developmental Plasticity? Front. Plant Sci. 2019, 10, 395. [Google Scholar] [CrossRef] [PubMed]
- Xiao, K.; Chen, J.; He, Q.; Wang, Y.; Shen, H.; Sun, L. DNA methylation is involved in the regulation of pepper fruit ripening and interacts with phytohormones. J. Exp. Bot. 2020, 71, 1928–1942. [Google Scholar] [CrossRef] [PubMed]
- Vanyushin, B.F. Enzymatic DNA methylation is an epigenetic control for genetic functions of the cell. Biochemistry 2005, 70, 488–499. [Google Scholar] [CrossRef]
- Kooke, R.; Johannes, F.; Wardenaar, R.; Becker, F.; Etcheverry, M.; Colot, V.; Vreugdenhil, D.; Keurentjes, J.J. Epigenetic basis of morphological variation and phenotypic plasticity in Arabidopsis thaliana. Plant Cell 2015, 27, 337–348. [Google Scholar] [CrossRef] [Green Version]
- Herman, J.J.; Sultan, S.E. DNA methylation mediates genetic variation for adaptive transgenerational plasticity. Proc. R. Soc. B 2016, 283, 20160988. [Google Scholar] [CrossRef] [Green Version]
- Baubec, T.; Finke, A.; Scheid, O.M.; Pecinka, A. Meristem-specific expression of epigenetic regulators safeguards transposon silencing in Arabidopsis. EMBO Rep. 2014, 15, 446–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Wang, H.; Liang, W.; Xie, X.; Guo, G. Developmental expression of Arabidopsis methyltransferase genes MET1, DRM2, and CMT3. Mol. Biol. 2014, 48, 681–687. [Google Scholar] [CrossRef]
- Jullien, P.E.; Susaki, D.; Yelagandula, R.; Higashiyama, T.; Berger, F. DNA methylation dynamics during sexual reproduction in Arabidopsis thaliana. Curr. Biol. 2012, 22, 1825–1830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, X.; Hale, C.J.; Nguyen, M.; Ausin, I.; Groth, M.; Hetzel, J.; Vashisht, A.A.; Henderson, I.R.; Wohlschlegel, J.A.; Jacobsen, S.E. Domains rearranged methyltransferase3 controls DNA methylation and regulates RNA polymerase V transcript abundance in Arabidopsis. Proc. Natl. Acad. Sci. USA 2015, 112, 911–916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roeder, A.H.; Yanofsky, M.F. Fruit development in Arabidopsis. In The Arabidopsis Book; Somerville, C., Meyerowitz, E., Eds.; American Society of Plant Biologists: Rockville, MD, USA, 2006. [Google Scholar] [CrossRef] [Green Version]
- Klepikova, A.V.; Kasianov, A.S.; Gerasimov, E.S.; Logacheva, M.D.; Penin, A.A. A high resolution map of the Arabidopsis thaliana developmental transcriptome based on RNA-seq profiling. Plant J. 2016, 88, 1058–1070. [Google Scholar] [CrossRef]
- Wen, C.K.; Chang, C. Arabidopsis RGL1 encodes a negative regulator of gibberellin responses. Plant Cell 2002, 14, 87–100. [Google Scholar] [CrossRef] [Green Version]
- Nemhauser, J.L.; Hong, F.; Chory, J. Different plant hormones regulate similar processes through largely nonoverlapping transcriptional responses. Cell 2006, 126, 467–475. [Google Scholar] [CrossRef] [Green Version]
- Mathur, V.; Tytgat, T.O.; Hordijk, C.A.; Harhangi, H.R.; Jansen, J.J.; Reddy, A.S.; Harvey, J.A.; Vet, L.E.; Van Dam, N.M. An ecogenomic analysis of herbivore-induced plant volatiles in Brassica juncea. Mol. Ecol. 2013, 22, 6179–6196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartels, A.; Han, Q.; Nair, P.; Stacey, L.; Gaynier, H.; Mosley, M.; Huang, Q.Q.; Pearson, J.K.; Hsieh, T.F.; An, Y.Q.C.; et al. Dynamic DNA methylation in plant growth and development. Int. J. Mol. Sci. 2018, 19, 2144. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Chang, F.; You, C.; Cui, J.; Zhu, G.; Wang, L.; Zheng, Y.; Qi, J.; Ma, H. Whole-genome DNA methylation patterns and complex associations with gene structure and expression during flower development in Arabidopsis. Plant J. 2015, 81, 268–281. [Google Scholar] [CrossRef]
- Campos-Rivero, G.; Osorio-Montalvo, P.; Sanchez-Borges, R.; Us-Camas, R.; Duarte-Ake, F.; De-la-Pena, C. Plant hormone signaling in flowering: An epigenetic point of view. Plant Physiol. 2017, 214, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Yang, T.; Dong, X.; Li, D.Z.; Liu, A. Genomic DNA methylation analyses reveal the distinct profiles in castor bean seeds with persistent endosperms. Plant Physiol. 2016, 171, 1242–1258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsieh, T.F.; Ibarra, C.A.; Silva, P.; Zemach, A.; Eshed-Williams, L.; Fischer, R.L.; Zilberman, D. Genome-wide demethylation of Arabidopsis endosperm. Science 2009, 324, 1451–1454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zemach, A.; Kim, M.Y.; Silva, P.; Rodrigues, J.A.; Dotson, B.; Brooks, M.D.; Zilberman, D. Local DNA hypomethylation activates genes in rice endosperm. Proc. Natl. Acad. Sci. USA 2010, 107, 18729–18734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Xie, S.; Dong, X.; Zhao, X.; Zeng, B.; Chen, J.; Yang, W.; Zhao, H.; Wang, G.; Chen, Z. Genome-wide high resolution parental-specific DNA and histone methylation maps uncover patterns of imprinting regulation in maize. Genome Res. 2014, 24, 167–176. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.Y.; Le, B.H.; Chen, M.; Henry, K.F.; Hur, J.; Hsieh, T.F.; Chen, P.Y.; Pelletier, J.M.; Pellegrini, M.; Fischer, R.L.; et al. Similarity between soybean and Arabidopsis seed methylomes and loss of non-CG methylation does not affect seed development. Proc. Natl. Acad. Sci. USA 2017, 114, E9730–E9739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gehring, M.; Bubb, K.L.; Henikoff, S. Extensive demethylation of repetitive elements during seed development underlies gene imprinting. Science 2009, 324, 1447–1451. [Google Scholar] [CrossRef] [Green Version]
- Gehring, M.; Satyaki, P.R. Endosperm and imprinting, inextricably linked. Plant Physiol. 2017, 173, 143–154. [Google Scholar] [CrossRef] [Green Version]
- Zuo, J.; Wang, Y.; Zhu, B.; Luo, Y.; Wang, Q.; Gao, L. Comparative analysis of DNA methylation reveals specific regulations on ethylene pathway in tomato fruit. Genes 2018, 9, 266. [Google Scholar] [CrossRef] [Green Version]
- Lang, Z.; Wang, Y.; Tang, K.; Tang, D.; Datsenka, T.; Cheng, J.; Zhang, Y.; Handa, A.K.; Zhu, J.K. Critical roles of DNA demethylation in the activation of ripening-induced genes and inhibition of ripening-repressed genes in tomato fruit. Proc. Natl. Acad. Sci. USA 2017, 114, E4511–E4519. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.; How-Kit, A.; Stammitti, L.; Teyssier, E.; Rolin, D.; Mortain-Bertrand, A. A DEMETER-like DNA demethylase governs tomato fruit ripening. Proc. Natl. Acad. Sci. USA 2015, 112, 10804–10809. [Google Scholar] [CrossRef] [Green Version]
- Xing, M.Q.; Zhang, Y.J.; Zhou, S.R.; Hu, W.Y.; Wu, X.T.; Ye, Y.J.; Wu, X.X.; Xiao, Y.P.; Li, X.; Xue, H.W. Global analysis reveals the crucial roles of DNA methylation during rice seed development. Plant Physiol. 2015, 168, 1417–1432. [Google Scholar] [CrossRef] [Green Version]
- Kawakatsu, T.; Nery, J.R.; Castanon, R.; Ecker, J.R. Dynamic DNA methylation reconfiguration during seed development and germination. Genome Biol. 2017, 18, 171. [Google Scholar] [CrossRef] [Green Version]
- Dempsey, D.M.A.; Klessig, D.F. How does the multifaceted plant hormone salicylic acid combat disease in plants and are similar mechanisms utilized in humans? BMC Biol. 2017, 15, 23. [Google Scholar] [CrossRef] [Green Version]
- Pasternak, T.; Groot, E.P.; Kazantsev, F.V.; Teale, W.; Omelyanchuk, N.; Kovrizhnykh, V.; Palme, K.; Mironova, V.V. Salicylic acid affects root meristem patterning via auxin distribution in a concentration-dependent manner. Plant Physiol. 2019, 180, 1725–1739. [Google Scholar] [CrossRef]
- Arıkan, B.; Özden, S.; Turgut-Kara, N. DNA methylation related gene expression and morphophysiological response to abiotic stresses in Arabidopsis thaliana. Environ. Exp. Bot. 2018, 149, 17–26. [Google Scholar] [CrossRef]
- Li, J.F.; Park, E.; von Arnim, A.G.; Nebenführ, A. The FAST technique: A simplified Agrobacterium-based transformation method for transient gene expression analysis in seedlings of Arabidopsis and other plant species. Plant Methods 2009, 5, 6. [Google Scholar] [CrossRef] [Green Version]
- Clough, S.J.; Bent, A.F. Floral dip: A simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant J. 1998, 16, 735–743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jefferson, R.A.; Kavanagh, T.A.; Bevan, M.W. GUS fusions: Beta-glucuronidase as a sensitive and versatile gene fusion marker in higher plants. EMBO J. 1987, 6, 3901–3907. [Google Scholar] [CrossRef]
- Stangeland, B.; Salehian, Z. An improved clearing method for GUS assay in Arabidopsis endosperm and seeds. Plant Mol. Biol. Rep. 2002, 20, 107–114. [Google Scholar] [CrossRef]
- Smalle, J.; Haegman, M.; Kurepa, J.; Van Montagu, M.; Van Der Straeten, D. Ethylene can stimulate Arabidopsis hypocotyl elongation in the light. Proc. Natl. Acad. Sci. USA 1997, 94, 2756–2761. [Google Scholar] [CrossRef] [Green Version]
- Fujii, H.; Zhu, J.K. Arabidopsis mutant deficient in 3 abscisic acid-activated protein kinases reveals critical roles in growth, reproduction, and stress. Proc. Natl. Acad. Sci. USA 2009, 106, 8380–8385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bidadi, H.; Yamaguchi, S.; Asahina, M.; Satoh, S. Effects of shoot-applied gibberellin/gibberellin-biosynthesis inhibitors on root growth and expression of gibberellin biosynthesis genes in Arabidopsis thaliana. Plant Root. 2010, 4, 4–11. [Google Scholar] [CrossRef] [Green Version]
- Khokon, M.A.R.; Okuma, E.I.J.I.; Hossain, M.A.; Munemasa, S.; Uraji, M.; Nakamura, Y.; Mori, I.C.; Murata, Y. Involvement of extracellular oxidative burst in salicylic acid-induced stomatal closure in Arabidopsis. Plant Cell Environ. 2011, 34, 434–443. [Google Scholar] [CrossRef]
- Piya, S.; Binder, B.M.; Hewezi, T. Canonical and noncanonical ethylene signaling pathways that regulate Arabidopsis susceptibility to the cyst nematode Heterodera schachtii. New Phytol. 2019, 221, 946–959. [Google Scholar] [CrossRef] [Green Version]
- Piya, S.; Liu, J.; Burch-Smith, T.; Baum, T.J.; Hewezi, T. A role for Arabidopsis growth-regulating factors 1 and 3 in growth–stress antagonism. J. Exp. Bot. 2020, 71, 1402–1417. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bennett, M.; Cleaves, K.; Hewezi, T. Expression Patterns of DNA Methylation and Demethylation Genes during Plant Development and in Response to Phytohormones. Int. J. Mol. Sci. 2021, 22, 9681. https://doi.org/10.3390/ijms22189681
Bennett M, Cleaves K, Hewezi T. Expression Patterns of DNA Methylation and Demethylation Genes during Plant Development and in Response to Phytohormones. International Journal of Molecular Sciences. 2021; 22(18):9681. https://doi.org/10.3390/ijms22189681
Chicago/Turabian StyleBennett, Morgan, Kailyn Cleaves, and Tarek Hewezi. 2021. "Expression Patterns of DNA Methylation and Demethylation Genes during Plant Development and in Response to Phytohormones" International Journal of Molecular Sciences 22, no. 18: 9681. https://doi.org/10.3390/ijms22189681