
Elucidation of Agonist and Antagonist Dynamic Binding Patterns in ER-α by Integration of Molecular Docking, Molecular Dynamics Simulations and Quantum Mechanical Calculations


 ,
,
Abstract
:1. Introduction
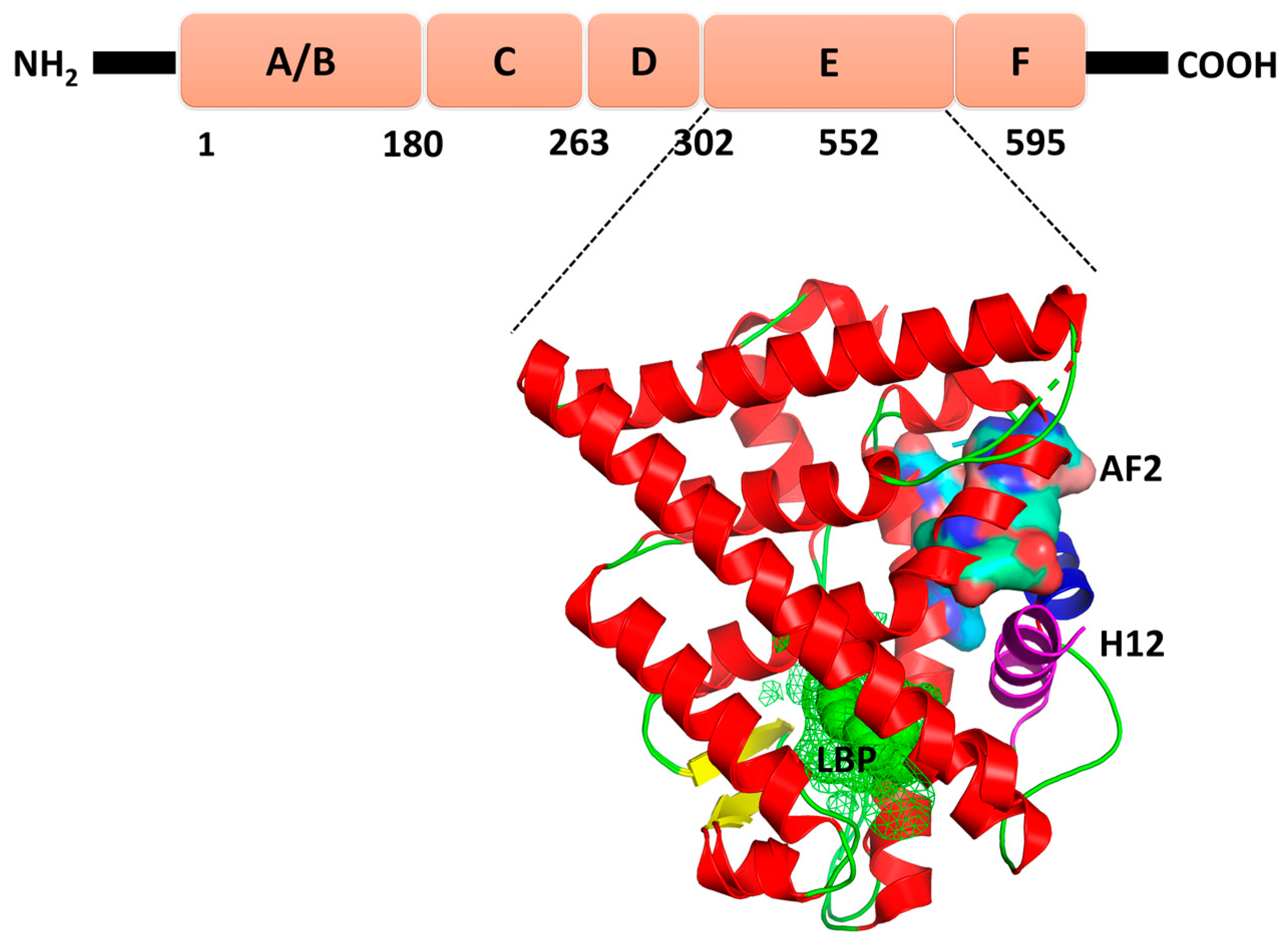
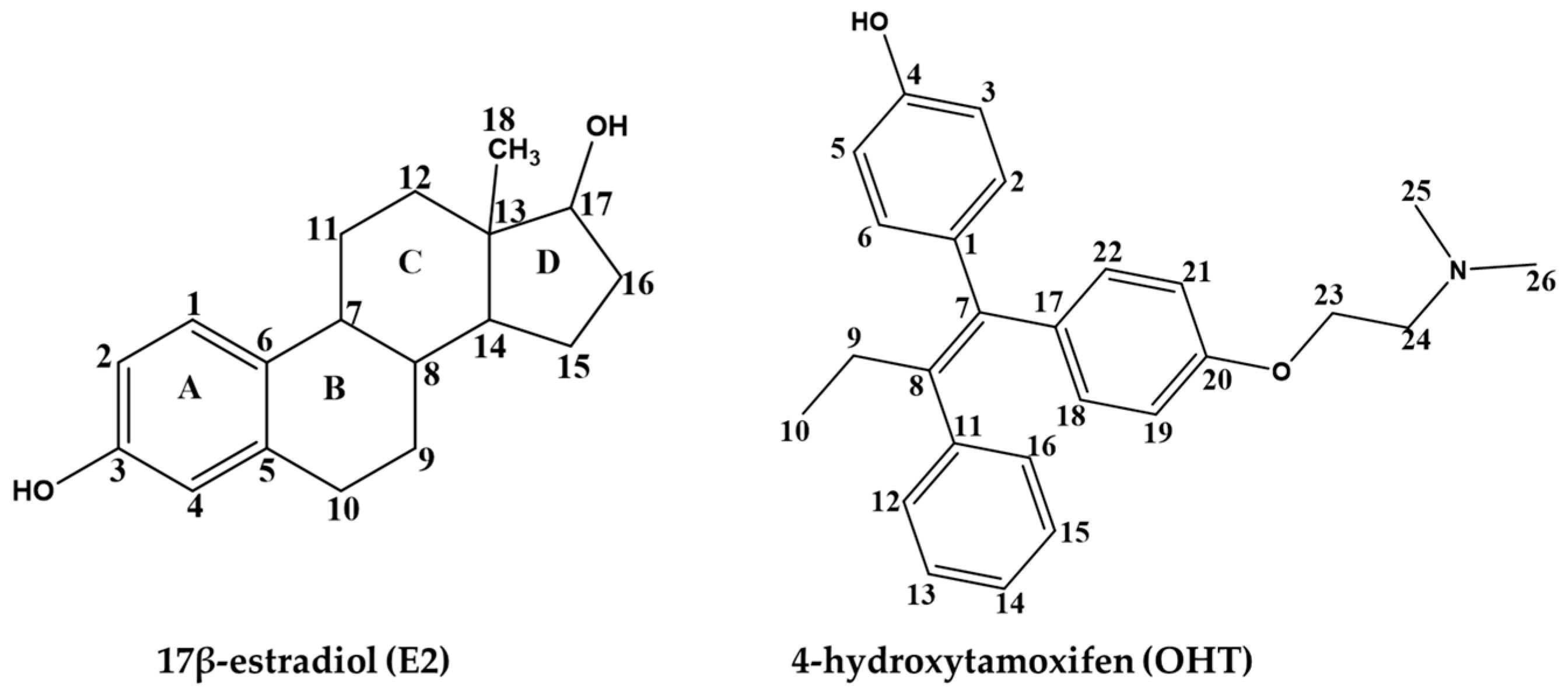
2. Results
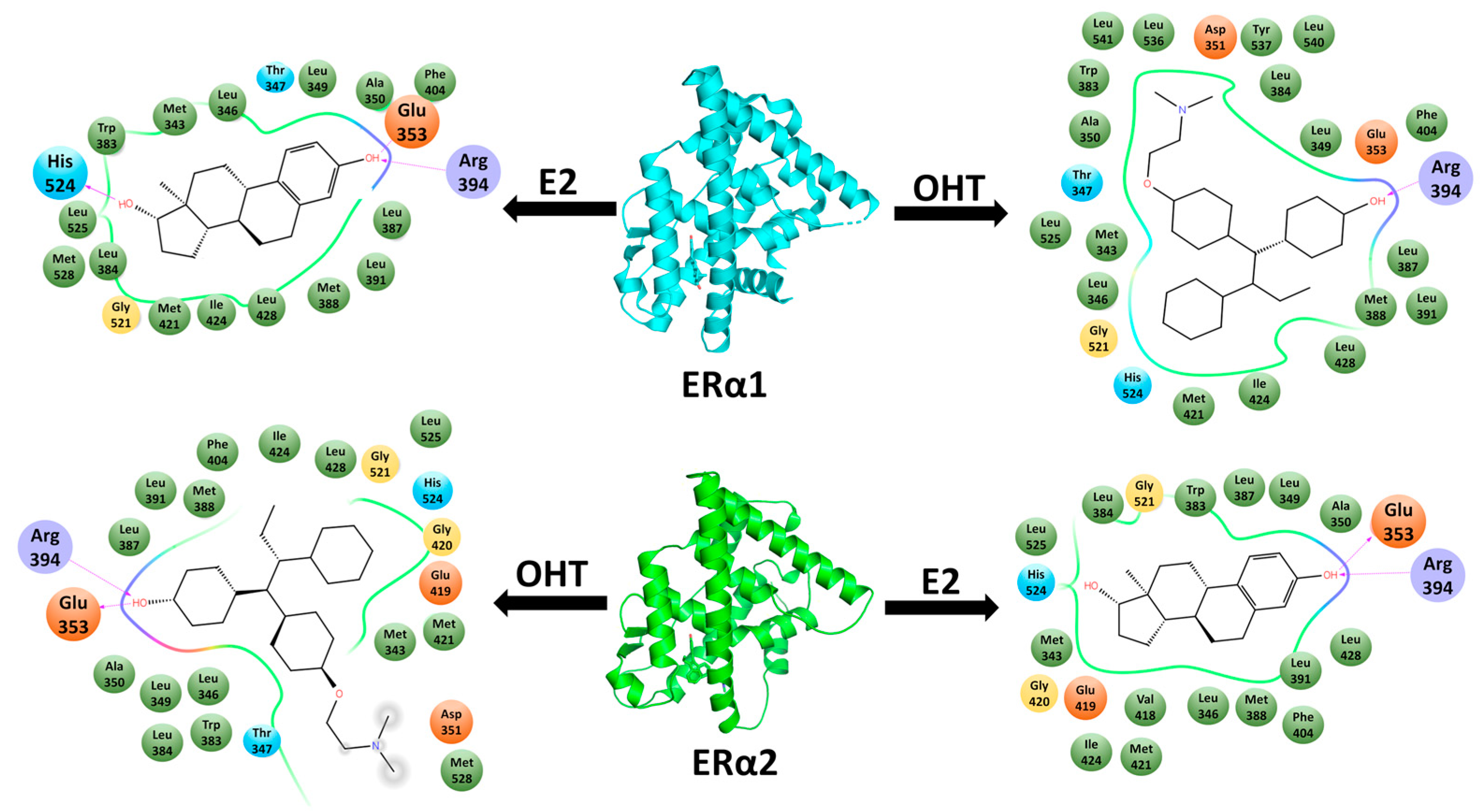
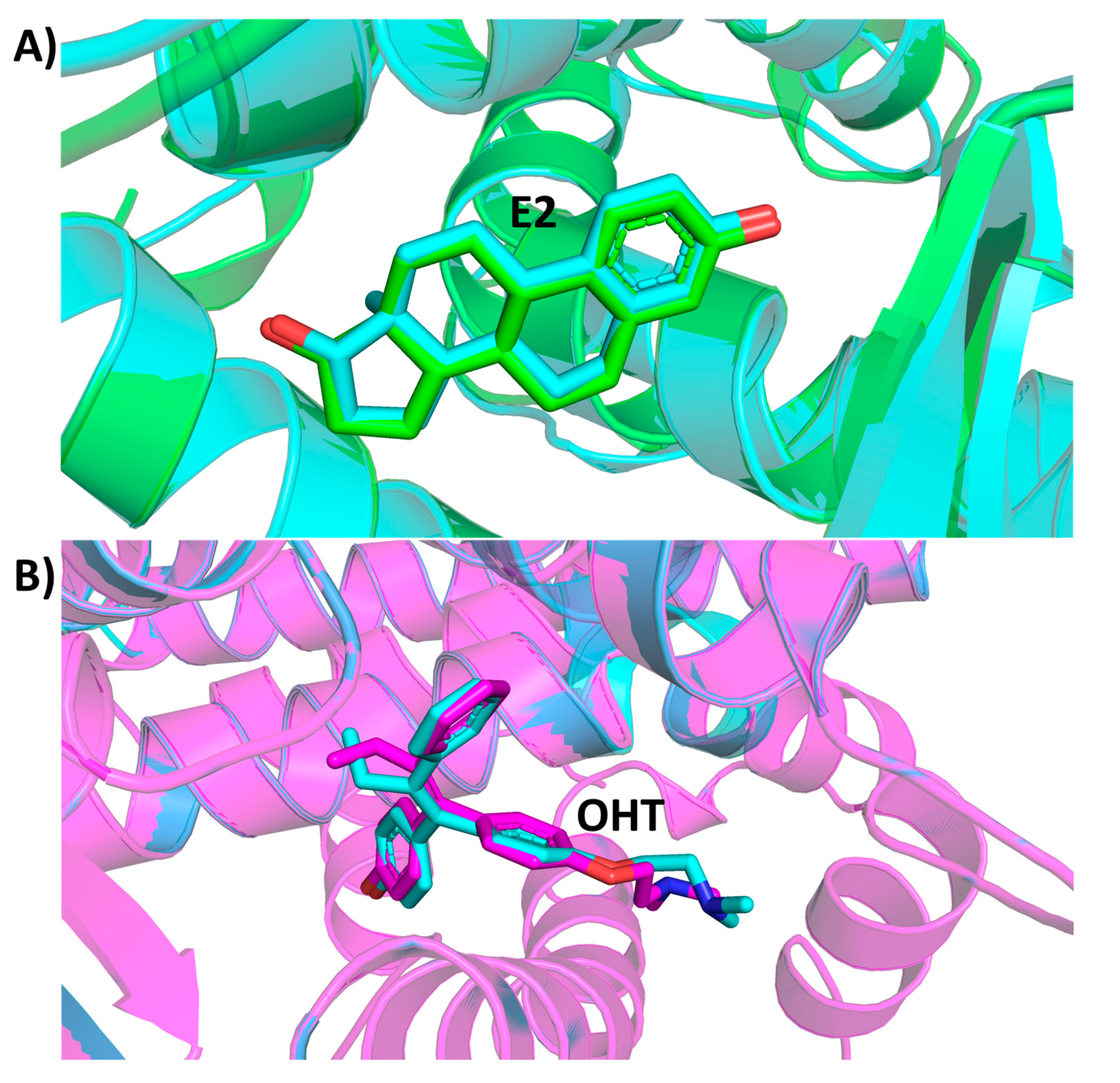
2.1. Molecular Docking
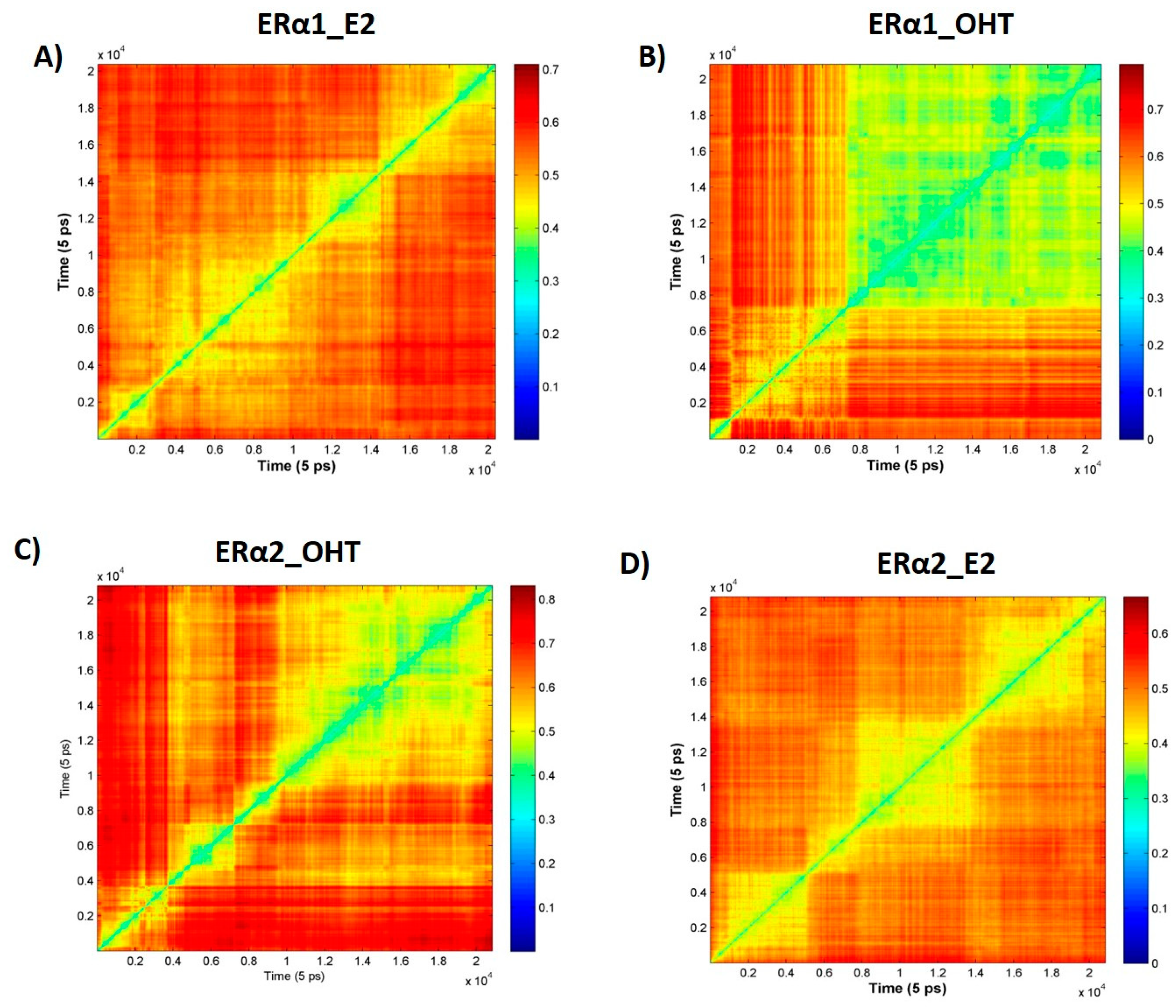
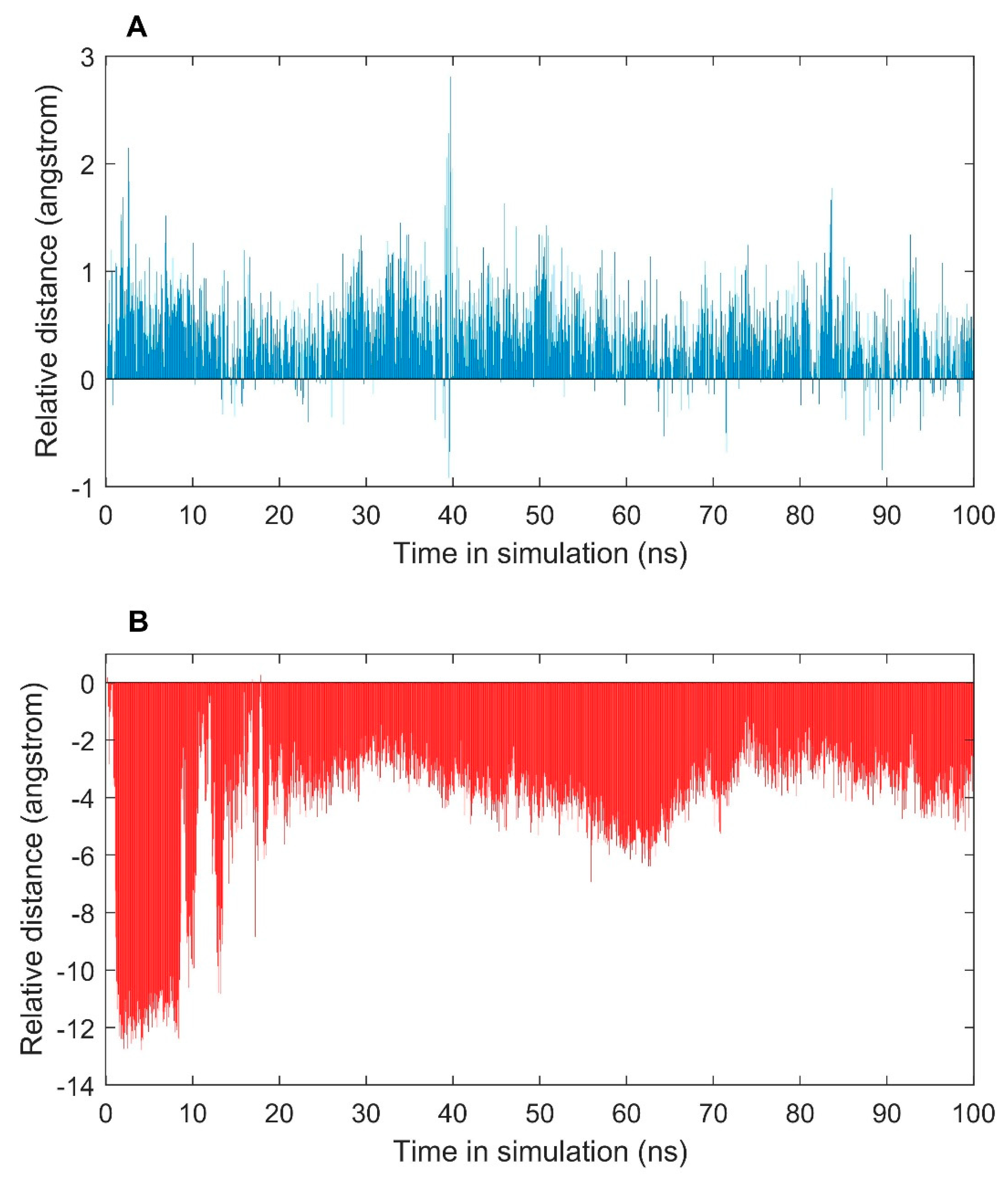
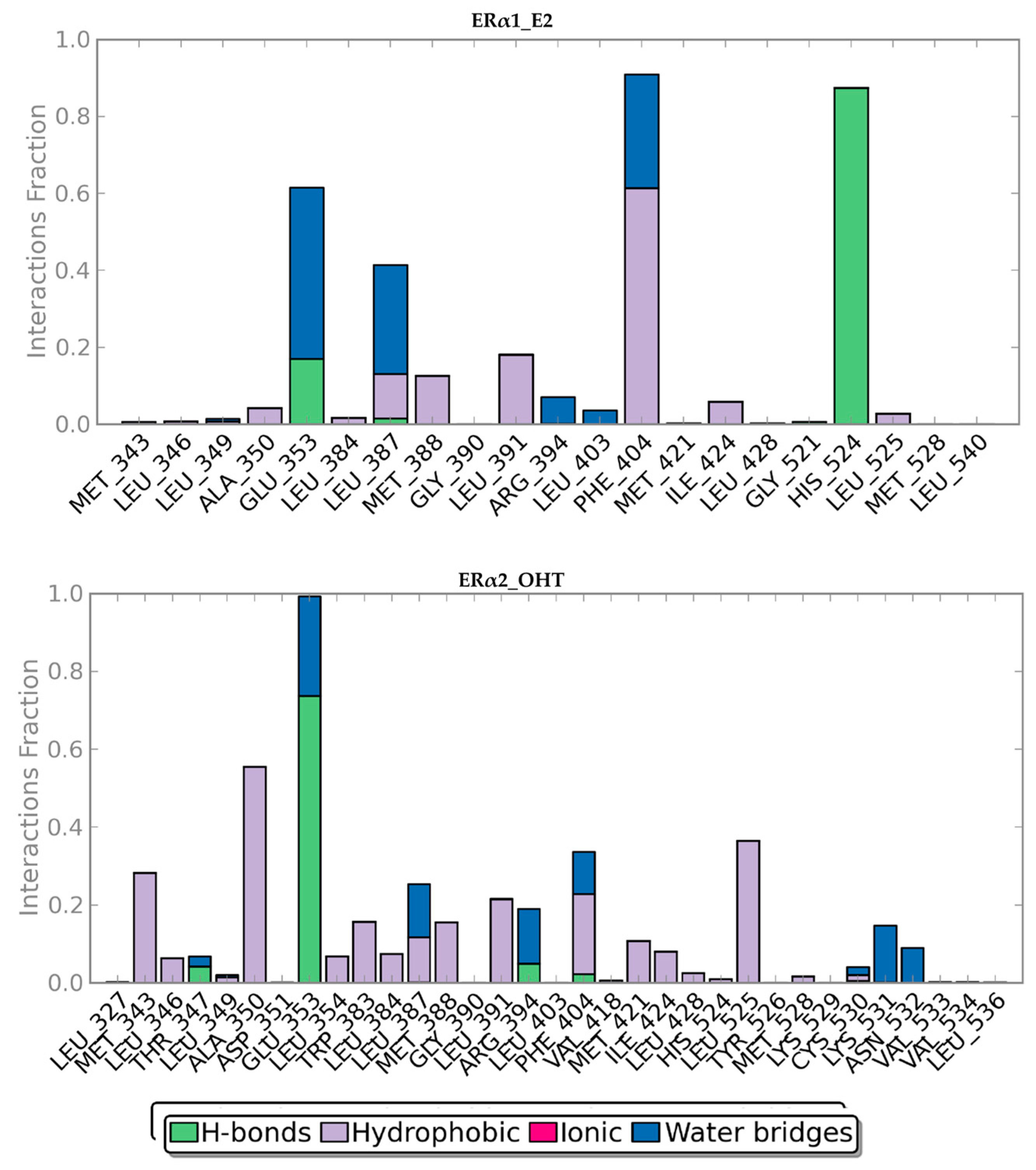
2.2. MD Simulations
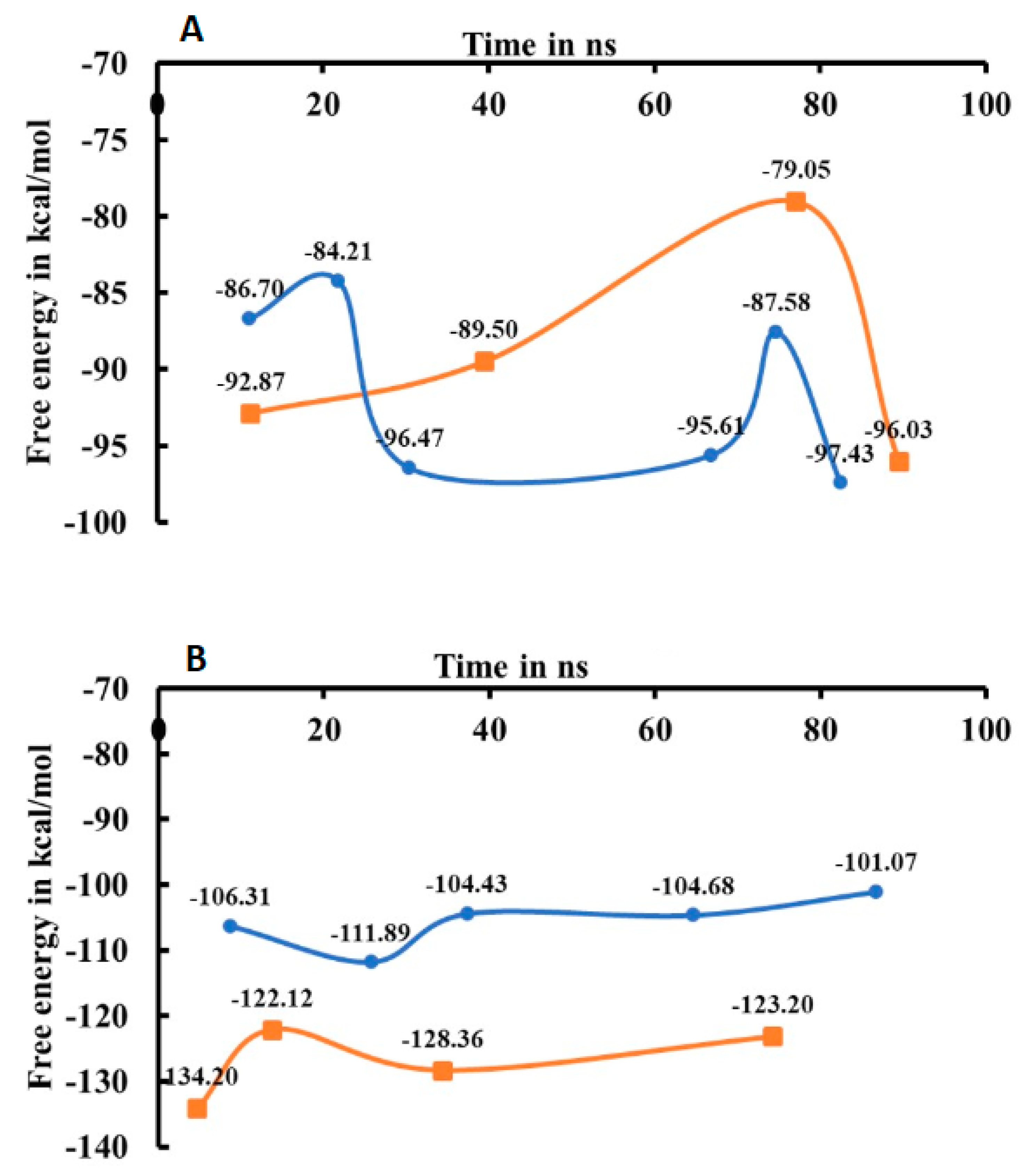
2.3. Energy Analysis
3. Discussion
4. Materials and Methods
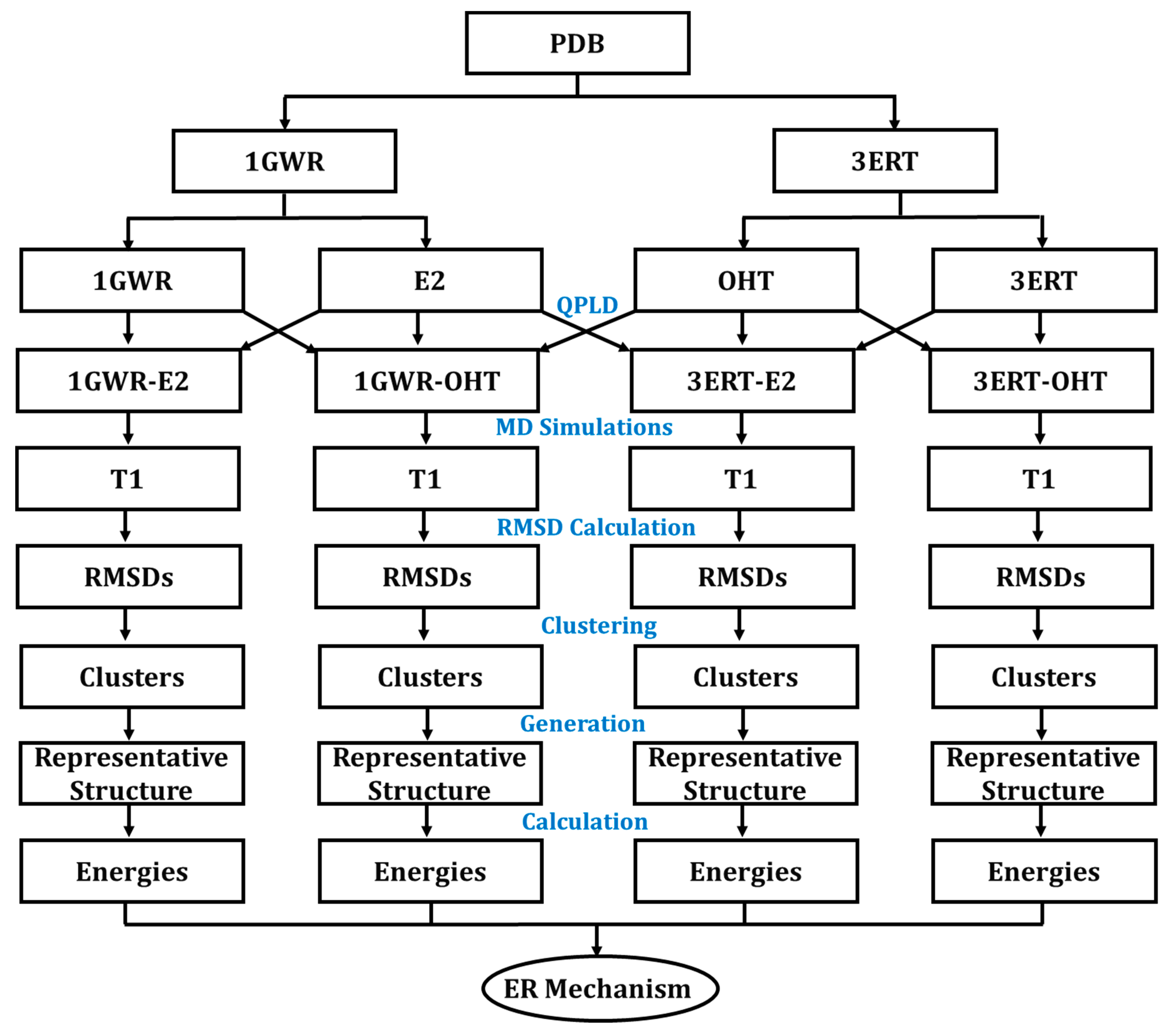
4.1. Study Design
4.2. Molecular Docking
4.2.1. Protein Preparation WIZARD
4.2.2. Ligand Preparation
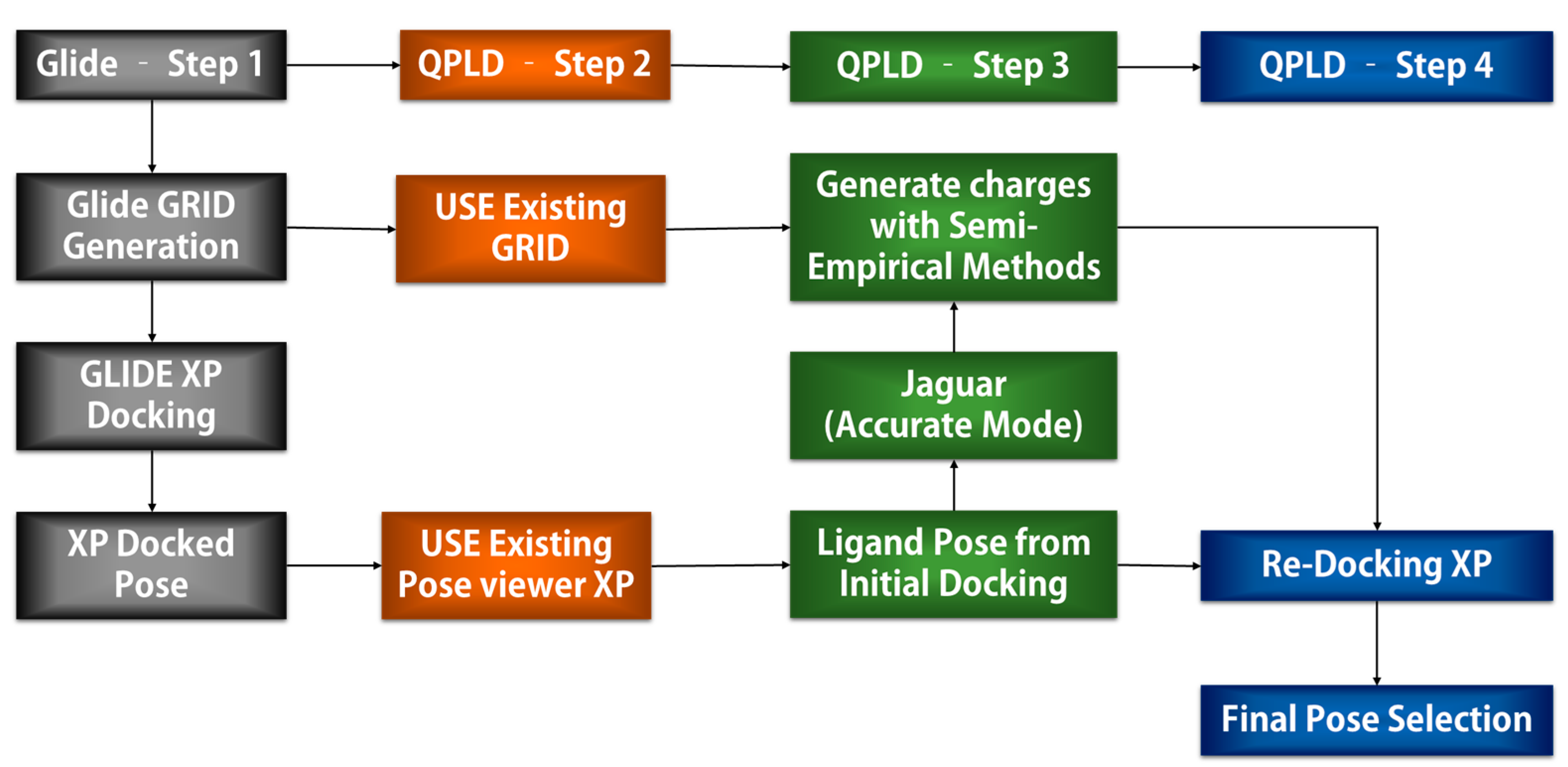
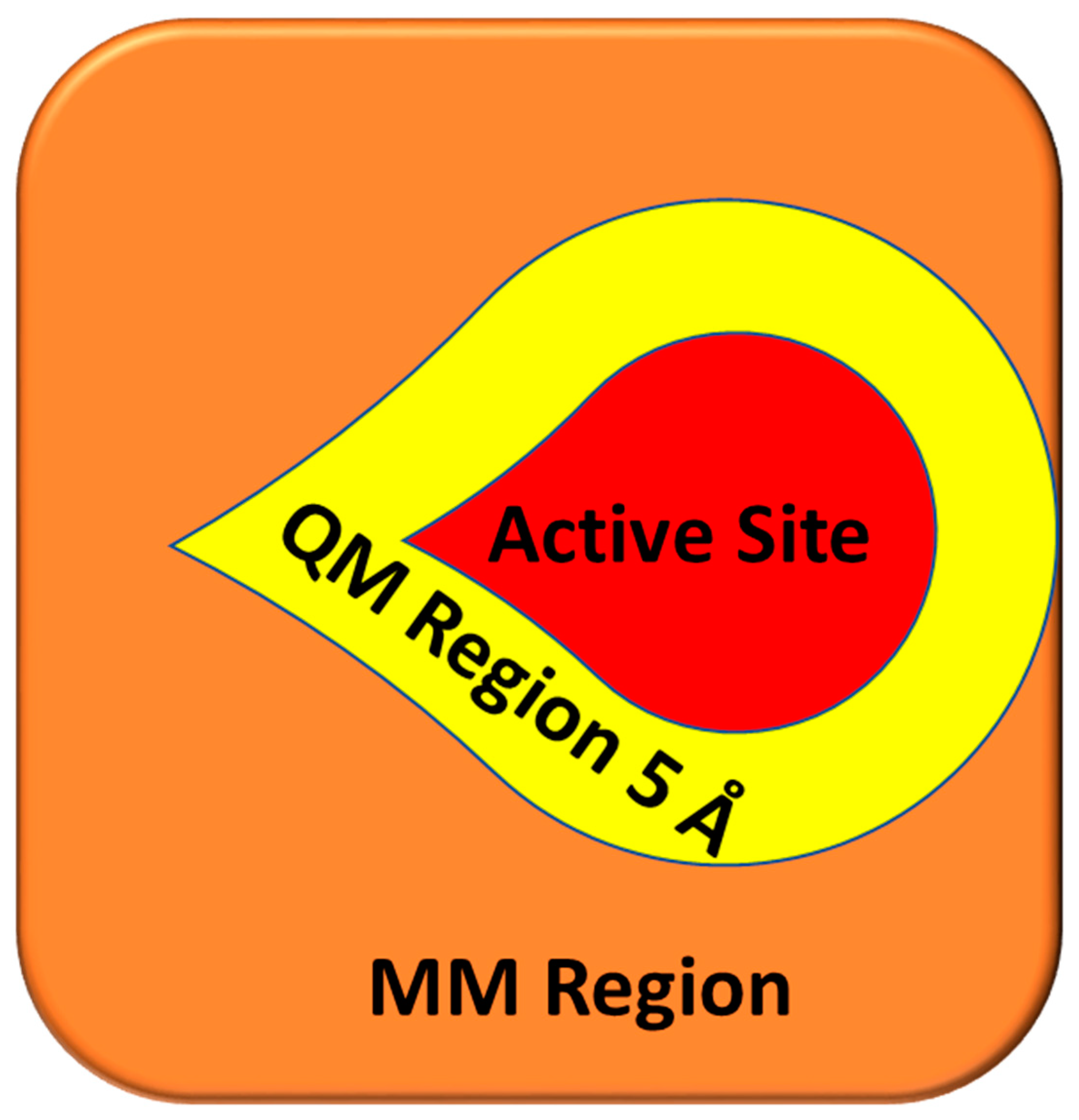
4.2.3. QPLD Molecular Docking
4.3. MD Simulations
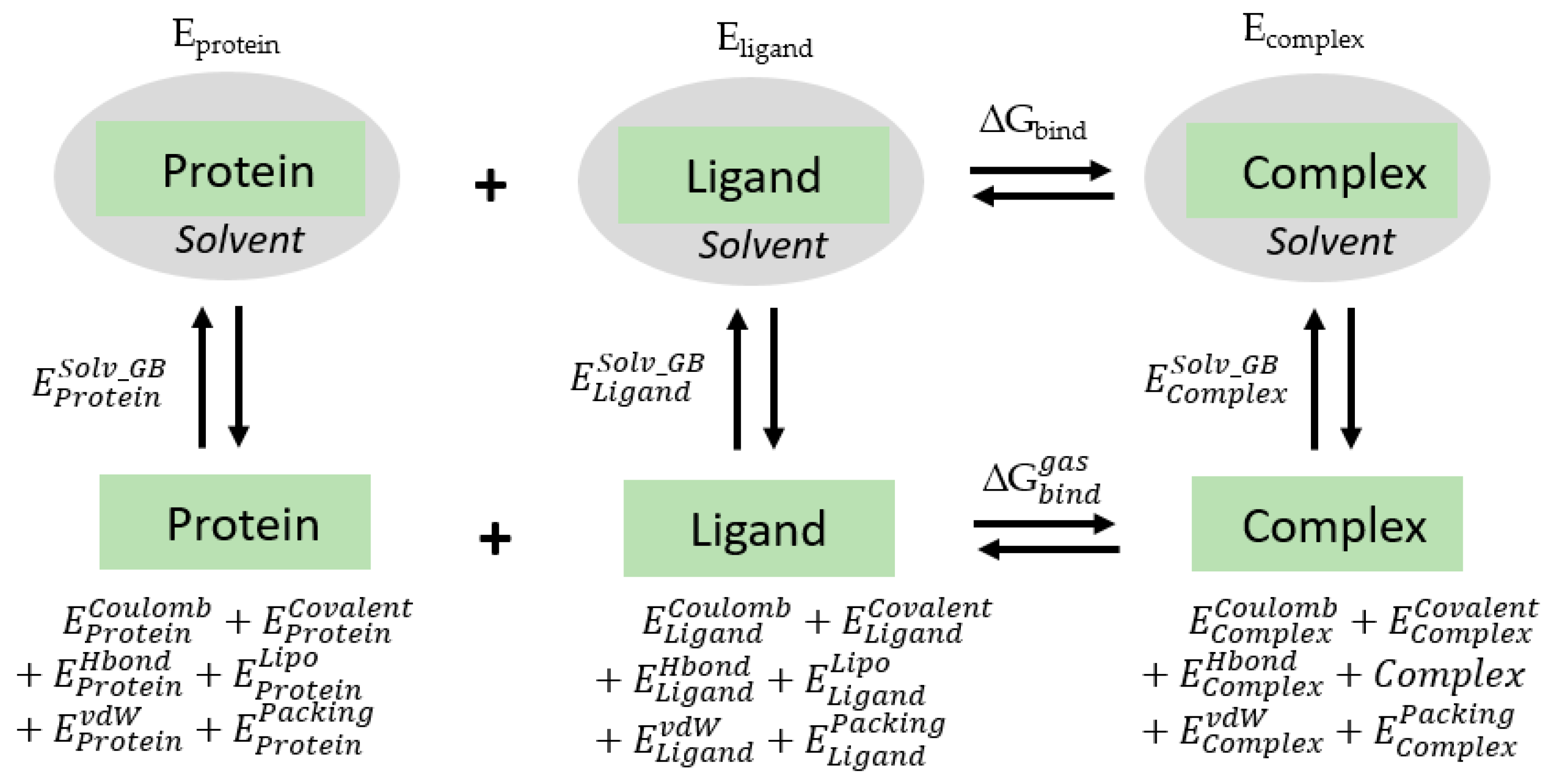
4.4. Energy Calculation
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Disclaimer
References
- Carroll, J.S.; Brown, M. Estrogen Receptor Target Gene: An Evolving Concept. Mol. Endocrinol. 2006, 20, 1707–1714. [Google Scholar] [CrossRef]
- Germain, P.; Staels, B.; Dacquet, C.; Spedding, M.; Laudet, V. Overview of nomenclature of nuclear receptors. Pharmacol. Rev. 2006, 58, 685–704. [Google Scholar] [CrossRef]
- Schug, T.T.; Janesick, A.; Blumberg, B.; Heindel, J.J. Endocrine disrupting chemicals and disease susceptibility. J. Steroid Biochem. Mol. Biol. 2011, 127, 204–215. [Google Scholar] [CrossRef] [Green Version]
- Kampa, M.; Pelekanou, V.; Notas, G.; Stathopoulos, E.N.; Castanas, E. The estrogen receptor: Two or more molecules, multiple variants, diverse localizations, signaling and functions. Are we undergoing a paradigm-shift as regards their significance in breast cancer? Hormones 2013, 12, 69–85. [Google Scholar] [CrossRef] [PubMed]
- Ascenzi, P.; Bocedi, A.; Marino, M. Structure-function relationship of estrogen receptor alpha and beta: Impact on human health. Mol. Asp. Med. 2006, 27, 299–402. [Google Scholar] [CrossRef] [PubMed]
- Shiau, A.K.; Barstad, D.; Loria, P.M.; Cheng, L.; Kushner, P.J.; Agard, D.A.; Greene, G.L. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell 1998, 95, 927–937. [Google Scholar] [CrossRef] [Green Version]
- Brzozowski, A.M.; Pike, A.C.W.; Dauter, Z.; Hubbard, R.E.; Bonn, T.; Engström, O.; Öhman, L.; Greene, G.L.; Gustafsson, J.-Å.; Carlquist, M. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature 1997, 389, 753–758. [Google Scholar] [CrossRef] [PubMed]
- Rastinejad, F.; Huang, P.; Chandra, V.; Khorasanizadeh, S. Understanding nuclear receptor form and function using structural biology. J. Mol. Endocrinol. 2013, 51, T1–T21. [Google Scholar] [CrossRef] [Green Version]
- Huang, P.; Chandra, V.; Rastinejad, F. Structural overview of the nuclear receptor superfamily: Insights into physiology and therapeutics. Annu. Rev. Physiol. 2010, 72, 247–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yaşar, P.; Ayaz, G.; User, S.D.; Güpür, G.; Muyan, M. Molecular mechanism of estrogen-estrogen receptor signaling. Reprod. Med. Biol. 2016, 16, 4–20. [Google Scholar] [CrossRef] [PubMed]
- Ng, H.W.; Perkins, R.; Tong, W.; Hong, H. Versatility or promiscuity: The estrogen receptors, control of ligand selectivity and an update on subtype selective ligands. Int. J. Environ. Res. Public Health 2014, 11, 8709–8742. [Google Scholar] [CrossRef] [Green Version]
- Pike, A.C. Lessons learnt from structural studies of the oestrogen receptor. Best Pract. Res. Clin. Endocrinol. Metab. 2006, 20, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Nettles, K.W.; Bruning, J.B.; Gil, G.; O’Neill, E.E.; Nowak, J.; Guo, Y.; Kim, Y.; DeSombre, E.R.; Dilis, R.; Hanson, R.N.; et al. Structural plasticity in the oestrogen receptor ligand-binding domain. EMBO Rep. 2007, 8, 563–568. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Zakharov, M.N.; Khan, S.H.; Miki, R.; Jang, H.; Toraldo, G.; Singh, R.; Bhasin, S.; Jasuja, R. The dynamic structure of the estrogen receptor. J. Amino Acids 2011, 2011, 812540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruning, J.B.; Parent, A.A.; Gil, G.; Zhao, M.; Nowak, J.; Pace, M.C.; Smith, C.L.; Afonine, P.V.; Adams, P.D.; Katzenellenbogen, J.A.; et al. Coupling of receptor conformation and ligand orientation determine graded activity. Nat. Chem. Biol. 2010, 6, 837–843. [Google Scholar] [CrossRef] [Green Version]
- Bjornstrom, L.; Sjoberg, M. Mechanisms of Estrogen Receptor Signaling: Convergence of Genomic and Nongenomic Actions on Target Genes. Mol. Endocrinol. 2005, 19, 833–842. [Google Scholar] [CrossRef] [Green Version]
- Pakdel, F. Molecular Pathways of Estrogen Receptor Action. Int. J. Mol. Sci. 2018, 19, 2591. [Google Scholar] [CrossRef] [Green Version]
- Ng, H.W.; Zhang, W.; Shu, M.; Luo, H.; Ge, W.; Perkins, R.; Tong, W.; Hong, H. Competitive molecular docking approach for predicting estrogen receptor subtype alpha agonists and antagonists. BMC Bioinform. 2014, 15 (Suppl. S11), S4. [Google Scholar] [CrossRef] [Green Version]
- Pang, X.; Fu, W.; Wang, J.; Kang, D.; Xu, L.; Zhao, Y.; Liu, A.L.; Du, G.H. Identification of Estrogen Receptor alpha Antagonists from Natural Products via In Vitro and In Silico Approaches. Oxidative Med. Cell. Longev. 2018, 2018, 6040149. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Sedykh, A.; Tripathi, A.; Zhu, H.; Afantitis, A.; Mouchlis, V.D.; Melagraki, G.; Rusyn, I.; Tropsha, A. Identification of putative estrogen receptor-mediated endocrine disrupting chemicals using QSAR- and structure-based virtual screening approaches. Toxicol. Appl. Pharmacol. 2013, 272, 67–76. [Google Scholar] [CrossRef] [Green Version]
- Muchtaridi, M.; Syahidah, H.N.; Subarnas, A.; Yusuf, M.; Bryant, S.D.; Langer, T. Molecular Docking and 3D-Pharmacophore Modeling to Study the Interactions of Chalcone Derivatives with Estrogen Receptor Alpha. Pharmaceuticals 2017, 10, 81. [Google Scholar] [CrossRef] [Green Version]
- Dutta, S.; Kharkar, P.S.; Sahu, N.U.; Khanna, A. Molecular docking prediction and in vitro studies elucidate anti-cancer activity of phytoestrogens. Life Sci. 2017, 185, 73–84. [Google Scholar] [CrossRef]
- Ksiazek, P.; Bryl, K. Molecular Docking Reveals Binding Features of Estrogen Receptor Beta Selective Ligands. Curr. Comput.-Aided Drug Des. 2015, 11, 137–151. [Google Scholar] [CrossRef]
- Powers, C.N.; Setzer, W.N. A molecular docking study of phytochemical estrogen mimics from dietary herbal supplements. Silico Pharmacol. 2015, 3, 4. [Google Scholar] [CrossRef] [Green Version]
- Fratev, F. Activation helix orientation of the estrogen receptor is mediated by receptor dimerization: Evidence from molecular dynamics simulations. Phys. Chem. Chem. Phys. 2015, 17, 13403–13420. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Wang, J. Ligand selectivity of estrogen receptors by a molecular dynamics study. Eur. J. Med. Chem. 2014, 74, 726–735. [Google Scholar] [CrossRef]
- Lu, Q.; Cai, Z.; Fu, J.; Luo, S.; Liu, C.; Li, X.; Zhao, D. Molecular docking and molecular dynamics studies on the interactions of hydroxylated polybrominated diphenyl ethers to estrogen receptor alpha. Ecotoxicol. Environ. Saf. 2014, 101, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Ng, H.L. Simulations reveal increased fluctuations in estrogen receptor-alpha conformation upon antagonist binding. J. Mol. Graph. Model. 2016, 69, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Sakkiah, S.; Kusko, R.; Tong, W.; Hong, H. Applications of Molecular Dynamics Simulations in Computational Toxicology. In Advances in Computational Toxicology: Methodologies and Applications in Regulatory Science; Hong, H., Ed.; Springer International Publishing: Cham, Switzerland, 2019; pp. 181–212. [Google Scholar] [CrossRef]
- Selvaraj, C.; Sakkiah, S.; Tong, W.; Hong, H. Molecular dynamics simulations and applications in computational toxicology and nanotoxicology. Food Chem. Toxicol. 2018, 112, 495–506. [Google Scholar] [CrossRef] [PubMed]
- Mansouri, K.; Abdelaziz, A.; Rybacka, A.; Roncaglioni, A.; Tropsha, A.; Varnek, A.; Zakharov, A.; Worth, A.; Richard, A.M.; Grulke, C.M.; et al. CERAPP: Collaborative Estrogen Receptor Activity Prediction Project. Environ. Health Perspect. 2016, 124, 1023–1033. [Google Scholar] [CrossRef]
- Sakkiah, S.; Guo, W.; Pan, B.; Kusko, R.; Tong, W.; Hong, H. Computational prediction models for assessing endocrine disrupting potential of chemicals. J. Environ. Sci. Health C Environ. Carcinog Ecotoxicol. Rev. 2018, 36, 192–218. [Google Scholar] [CrossRef]
- Norinder, U.; Boyer, S. Conformal Prediction Classification of a Large Data Set of Environmental Chemicals from ToxCast and Tox21 Estrogen Receptor Assays. Chem. Res. Toxicol. 2016, 29, 1003–1010. [Google Scholar] [CrossRef] [PubMed]
- Ng, H.W.; Doughty, S.W.; Luo, H.; Ye, H.; Ge, W.; Tong, W.; Hong, H. Development and Validation of Decision Forest Model for Estrogen Receptor Binding Prediction of Chemicals Using Large Data Sets. Chem. Res. Toxicol. 2015, 28, 2343–2351. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Peng, T.; Yang, X.; Liu, H. Development of QSAR models for predicting the binding affinity of endocrine disrupting chemicals to eight fish estrogen receptor. Ecotoxicol. Environ. Saf. 2018, 148, 211–219. [Google Scholar] [CrossRef]
- Ng, H.W.; Shu, M.; Luo, H.; Ye, H.; Ge, W.; Perkins, R.; Tong, W.; Hong, H. Estrogenic activity data extraction and in silico prediction show the endocrine disruption potential of bisphenol A replacement compounds. Chem. Res. Toxicol. 2015, 28, 1784–1795. [Google Scholar] [CrossRef] [PubMed]
- Bhhatarai, B.; Wilson, D.M.; Price, P.S.; Marty, S.; Parks, A.K.; Carney, E. Evaluation of OASIS QSAR Models Using ToxCast in Vitro Estrogen and Androgen Receptor Binding Data and Application in an Integrated Endocrine Screening Approach. Environ. Health Perspect. 2016, 124, 1453–1461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niu, A.Q.; Xie, L.J.; Wang, H.; Zhu, B.; Wang, S.Q. Prediction of selective estrogen receptor beta agonist using open data and machine learning approach. Drug Des. Dev. Ther. 2016, 10, 2323–2331. [Google Scholar] [CrossRef] [Green Version]
- Ribay, K.; Kim, M.T.; Wang, W.; Pinolini, D.; Zhu, H. Predictive Modeling of Estrogen Receptor Binding Agents Using Advanced Cheminformatics Tools and Massive Public Data. Front. Environ. Sci. 2016, 4, 12. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Q.; Lu, Y.; Zhao, Y.; Li, R.; Luan, F.; Cordeiro, M.N. Rational Design of Multi-Target Estrogen Receptors ERalpha and ERbeta by QSAR Approaches. Curr. Drug Targets 2017, 18, 576–591. [Google Scholar] [CrossRef] [PubMed]
- Browne, P.; Judson, R.S.; Casey, W.M.; Kleinstreuer, N.C.; Thomas, R.S. Screening Chemicals for Estrogen Receptor Bioactivity Using a Computational Model. Environ. Sci. Technol. 2015, 49, 8804–8814. [Google Scholar] [CrossRef]
- Sosnovcova, J.; Rucki, M.; Bendova, H. Estrogen Receptor Binding Affinity of Food Contact Material Components Estimated by QSAR. Cent. Eur. J. Public Health 2016, 24, 241–244. [Google Scholar] [CrossRef] [Green Version]
- Blair, R.M.; Fang, H.; Branham, W.S.; Hass, B.S.; Dial, S.L.; Moland, C.L.; Tong, W.; Shi, L.; Perkins, R.; Sheehan, D.M. The Estrogen Receptor Relative Binding Affinities of 188 Natural and Xenochemicals: Structural Diversity of Ligands. Toxicol. Sci. 2000, 54, 138–153. [Google Scholar] [CrossRef] [Green Version]
- Yang, R.; Li, N.; Rao, K.; Ma, M.; Wang, Z. Combined action of estrogen receptor agonists and antagonists in two-hybrid recombinant yeast in vitro. Ecotoxicol. Environ. Saf. 2015, 111, 228–235. [Google Scholar] [CrossRef]
- Puranik, N.V.; Srivastava, P.; Bhatt, G.; John Mary, D.J.S.; Limaye, A.M.; Sivaraman, J. Determination and analysis of agonist and antagonist potential of naturally occurring flavonoids for estrogen receptor (ERα) by various parameters and molecular modelling approach. Sci. Rep. 2019, 9, 7450. [Google Scholar] [CrossRef] [PubMed]
- Sakkiah, S.; Selvaraj, C.; Gong, P.; Zhang, C.; Tong, W.; Hong, H. Development of estrogen receptor beta binding prediction model using large sets of chemicals. Oncotarget 2017, 8, 92989–93000. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Xu, L.; Fang, H.; Richard, A.M.; Bray, J.D.; Judson, R.S.; Zhou, G.; Colatsky, T.J.; Aungst, J.L.; Teng, C.; et al. EADB: An estrogenic activity database for assessing potential endocrine activity. Toxicol. Sci. 2013, 135, 277–291. [Google Scholar] [CrossRef] [Green Version]
- Shi, L.; Tong, W.; Fang, H.; Xie, Q.; Hong, H.; Perkins, R.; Wu, J.; Tu, M.; Blair, R.M.; Branham, W.S.; et al. An integrated “4-phase” approach for setting endocrine disruption screening priorities--phase I and II predictions of estrogen receptor binding affinity. SAR QSAR Environ. Res. 2002, 13, 69–88. [Google Scholar] [CrossRef] [PubMed]
- Hong, H.; Tong, W.; Fang, H.; Shi, L.; Xie, Q.; Wu, J.; Perkins, R.; Walker, J.D.; Branham, W.; Sheehan, D.M. Prediction of estrogen receptor binding for 58,000 chemicals using an integrated system of a tree-based model with structural alerts. Environ. Health Perspect. 2002, 110, 29–36. [Google Scholar] [CrossRef] [Green Version]
- Jordan, V.C.; Collins, M.M.; Rowsby, L.; Prestwich, G. A monohydroxylated metabolite of tamoxifen with potent antioestrogenic activity. J. Endocrinol. 1977, 75, 305–316. [Google Scholar] [CrossRef]
- Wang, Y.; Chirgadze, N.Y.; Briggs, S.L.; Khan, S.; Jensen, E.V.; Burris, T.P. A second binding site for hydroxytamoxifen within the coactivator-binding groove of estrogen receptor β. Proc. Natl. Acad. Sci. USA 2006, 103, 9908–9911. [Google Scholar] [CrossRef] [Green Version]
- Cho, A.E.; Guallar, V.; Berne, B.J.; Friesner, R. Importance of accurate charges in molecular docking: Quantum mechanical/molecular mechanical (QM/MM) approach. J. Comput. Chem. 2005, 26, 915–931. [Google Scholar] [CrossRef] [Green Version]
- Zhong, H.; Kirschner, K.N.; Lee, M.; Bowen, J.P. Binding free energy calculation for duocarmycin/DNA complex based on the QPLD-derived partial charge model. Bioorg. Med. Chem. Lett. 2008, 18, 542–545. [Google Scholar] [CrossRef] [PubMed]
- Reema Abu, K.; Hamada Abd, E.-A.; Dima, S.; Ghadeer, A.; Ghassan Abu, S. CETP Inhibitory Activity of Chlorobenzyl Benzamides: QPLD Docking, Pharmacophore Mapping and Synthesis. Lett. Drug Des. Discov. 2017, 14, 1391–1400. [Google Scholar] [CrossRef]
- Udommaneethanakit, T.; Rungrotmongkol, T.; Frecer, V.; Seneci, P.; Miertus, S.; Bren, U. Drugs against avian influenza a virus: Design of novel sulfonate inhibitors of neuraminidase N1. Curr. Pharm. Des. 2014, 20, 3478–3487. [Google Scholar] [CrossRef] [PubMed]
- Furlan, V.; Bren, U. Insight into Inhibitory Mechanism of PDE4D by Dietary Polyphenols Using Molecular Dynamics Simulations and Free Energy Calculations. Biomolecules 2021, 11, 479. [Google Scholar] [CrossRef] [PubMed]
- Robertson, M.J.; Qian, Y.; Robinson, M.C.; Tirado-Rives, J.; Jorgensen, W.L. Development and Testing of the OPLS-AA/M Force Field for RNA. J. Chem. Theory Comput. 2019, 15, 2734–2742. [Google Scholar] [CrossRef] [PubMed]
- Lešnik, S.; Hodošček, M.; Bren, U.; Stein, C.; Bondar, A.-N. Potential Energy Function for Fentanyl-Based Opioid Pain Killers. J. Chem. Inf. Modeling 2020, 60, 3566–3576. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Storer, J.W.; Giesen, D.J.; Cramer, C.J.; Truhlar, D.G. Class IV charge models: A new semiempirical approach in quantum chemistry. J. Comput. Aided Mol. Des. 1995, 9, 87–110. [Google Scholar] [CrossRef]
- Shivakumar, D.; Harder, E.; Damm, W.; Friesner, R.A.; Sherman, W. Improving the Prediction of Absolute Solvation Free Energies Using the Next Generation OPLS Force Field. J. Chem. Theory Comput. 2012, 8, 2553–2558. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ER Complex | Glide | QPLD | ||
|---|---|---|---|---|
| XP Score Kcal/mol | Docking Energy | QPLD Score Kcal/mol | Docking Energy | |
| ERα1_E2 | −11.00 | −39.74 | −11.45 | −38.78 |
| ERα2_E2 | −9.65 | −32.54 | −9.82 | −33.41 |
| ERα1_OHT | −9.07 | −29.43 | −8.17 | −28.96 |
| ERα2_OHT | −8.59 | −35.81 | −10.94 | −38.23 |
| Complex | Atoms in Complex | Waters | Ions |
|---|---|---|---|
| ERα1_E2 | 3980 | 7890 | 30 Na+; 23 Cl− |
| ERα1_OHT | 3994 | 7890 | 29 Na+; 22 Cl− |
| ERα2_E2 | 3946 | 9138 | 36 Na+; 26 Cl− |
| ERα2_OHT | 3960 | 9138 | 35 Na+; 25 Cl− |
| Acronyms | ΔG_Bind | ΔG_Bind_Coulomb | ΔG_Bind_vdW | Ligand Energy | Complex Energy | Receptor Energy |
|---|---|---|---|---|---|---|
| ERα1_E2 | −44.10 | −7.73 | −22.17 | 1.73 | −9402.71 | −9360.34 |
| ERα1_OHT | −44.68 | −22.03 | −17.47 | 32.43 | −9352.97 | −9340.71 |
| ERα2_E2 | −24.69 | −9.19 | −7.13 | 1.72 | −9325.79 | −9302.81 |
| ERα2_OHT | −42.24 | −39.65 | −17.04 | 32.12 | −9342.13 | −9332.01 |
| ER Complex | Cluster Number | Number of Structures | Start Frame | End Frame | Representative Structure |
|---|---|---|---|---|---|
| ERα1_E2 | 1 | 4314 | 2 | 4328 | 2312 |
| 2 | 1012 | 4329 | 5346 | 4545 | |
| 3 | 1366 | 5364 | 6787 | 6339 | |
| 4 | 7027 | 7423 | 14,514 | 13,922 | |
| 5 | 1938 | 14,523 | 16,491 | 15,558 | |
| 6 | 874 | 16,730 | 17,629 | 17,171 | |
| ERα1_OHT | 1 | 1696 | 2 | 1698 | 979 |
| 2 | 860 | 2456 | 3422 | 2866 | |
| 3 | 1328 | 5894 | 7298 | 7154 | |
| 4 | 13,528 | 7306 | 20,835 | 15,478 | |
| ERα2_E2 | 1 | 5247 | 2 | 5451 | 2353 |
| 2 | 6915 | 5453 | 12,945 | 8216 | |
| 3 | 1294 | 15,187 | 16,740 | 16,064 | |
| 4 | 3786 | 16,735 | 20,835 | 18,646 | |
| ERα2_OHT | 1 | 3732 | 5 | 3745 | 1813 |
| 2 | 1531 | 4552 | 6086 | 5356 | |
| 3 | 3497 | 6087 | 9643 | 7771 | |
| 4 | 5916 | 9641 | 15,769 | 13,467 | |
| 5 | 2988 | 15,770 | 19,021 | 18,075 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sakkiah, S.; Selvaraj, C.; Guo, W.; Liu, J.; Ge, W.; Patterson, T.A.; Hong, H. Elucidation of Agonist and Antagonist Dynamic Binding Patterns in ER-α by Integration of Molecular Docking, Molecular Dynamics Simulations and Quantum Mechanical Calculations. Int. J. Mol. Sci. 2021, 22, 9371. https://doi.org/10.3390/ijms22179371
Sakkiah S, Selvaraj C, Guo W, Liu J, Ge W, Patterson TA, Hong H. Elucidation of Agonist and Antagonist Dynamic Binding Patterns in ER-α by Integration of Molecular Docking, Molecular Dynamics Simulations and Quantum Mechanical Calculations. International Journal of Molecular Sciences. 2021; 22(17):9371. https://doi.org/10.3390/ijms22179371
Chicago/Turabian StyleSakkiah, Sugunadevi, Chandrabose Selvaraj, Wenjing Guo, Jie Liu, Weigong Ge, Tucker A. Patterson, and Huixiao Hong. 2021. "Elucidation of Agonist and Antagonist Dynamic Binding Patterns in ER-α by Integration of Molecular Docking, Molecular Dynamics Simulations and Quantum Mechanical Calculations" International Journal of Molecular Sciences 22, no. 17: 9371. https://doi.org/10.3390/ijms22179371