
Local Therapies and Modulation of Tumor Surrounding Stroma in Malignant Pleural Mesothelioma: A Translational Approach

 , , , , , , , and
, , , , , , , and 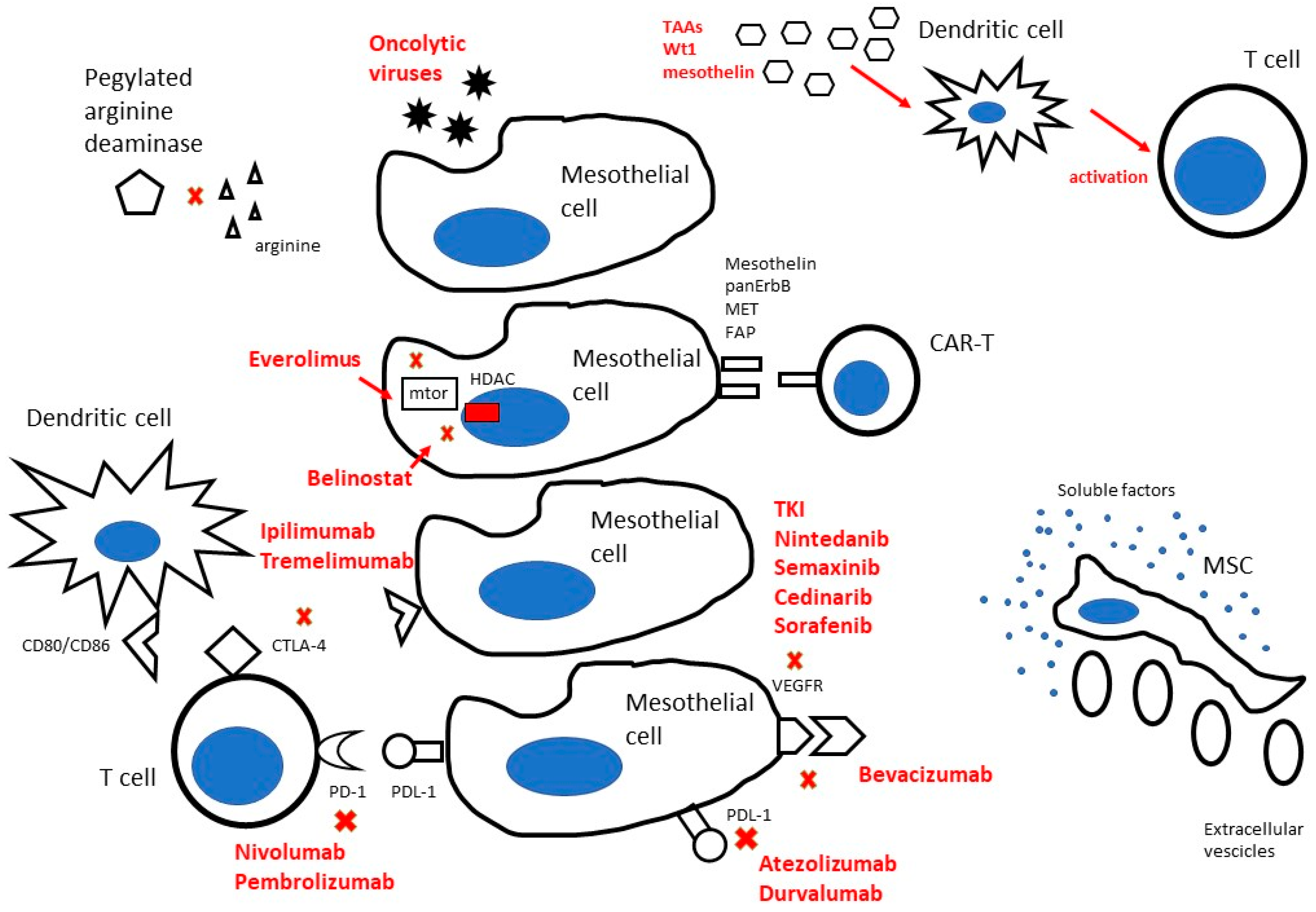{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Targeting MPM Stroma by Local Approach
2.1. Biologic Frame and Rationale
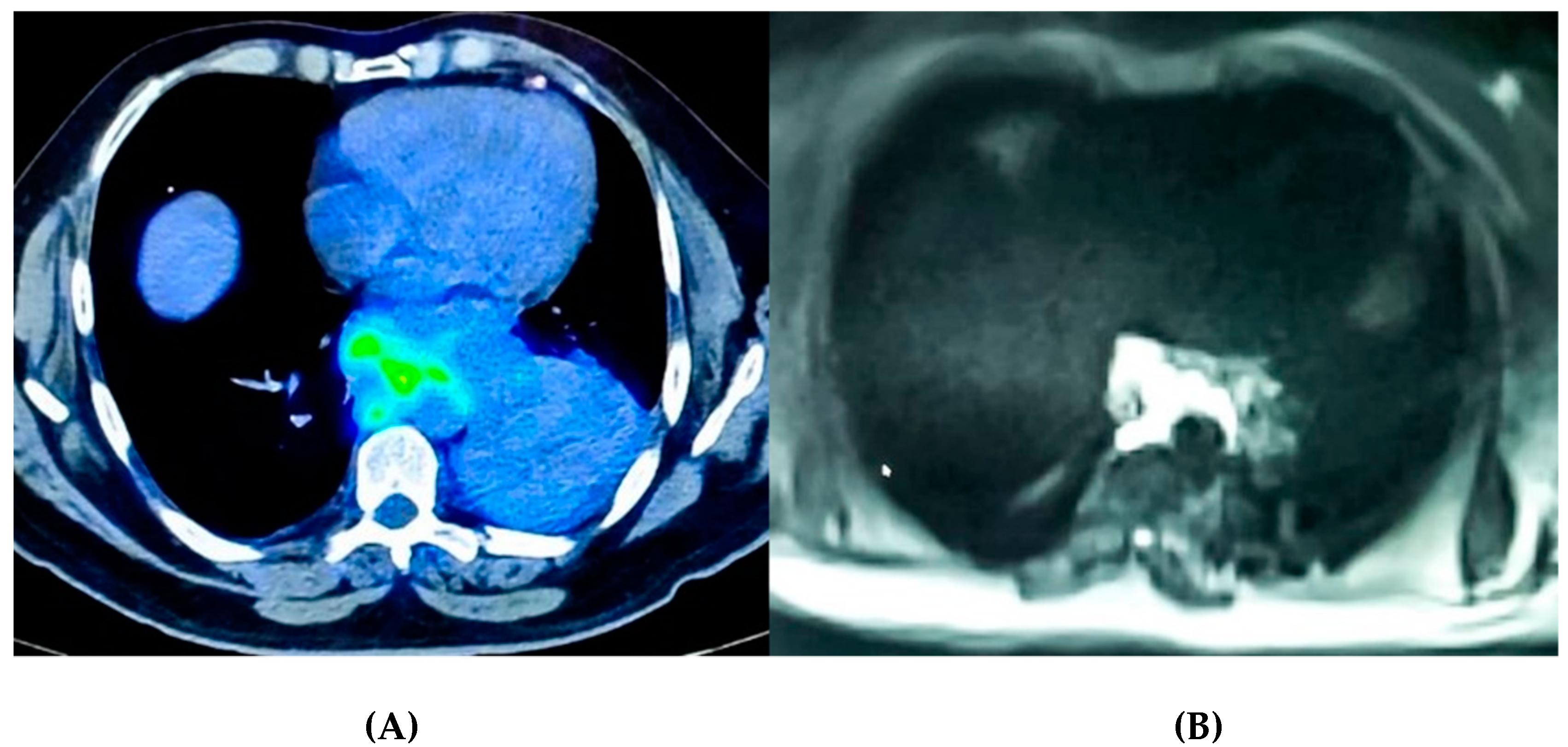
2.2. Imaging as a Tool to Assess Tumor Micro-Environment
3. How to Local Target MPM: Where We Are Going
3.1. Nanoparticles (NPs) as Novel Promise for MPM
3.2. Advanced Cell Therapy
3.2.1. Adoptive Cell Therapy for MPM
3.2.2. Drug Loading and Drug Delivery by Mesenchymal Stromal Cells (MSCs) and Their Extracellular Vesicles
3.3. Implanted Biocompatible Scaffolds and Biomaterials
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Britton, M. The epidemiology of mesothelioma. Semin. Oncol. 2002, 29, 18–25. [Google Scholar] [CrossRef]
- Abbott, D.M.; Bortolotto, C.; Benvenuti, S.; Lancia, A.; Filippi, A.R.; Stella, G.M. Malignant Pleural Mesothelioma: Genetic and Microenviromental Heterogeneity as an Unexpected Reading Frame and Therapeutic Challenge. Cancers 2020, 12, 1186. [Google Scholar] [CrossRef]
- Scherpereel, A.; Opitz, I.; Berghmans, T.; Psallidas, I.; Glatzer, M.; Rigau, D.; Astoul, P.; Bölükbas, S.; Boyd, J.; Coolen, J.; et al. ERS/ESTS/EACTS/ESTRO guidelines for the management of malignant pleural mesothelioma. Eur. Respir. J. 2020, 55, 1900953. [Google Scholar] [CrossRef]
- Garland, L.L.; Rankin, C.; Gandara, D.R.; Rivkin, S.E.; Scott, K.M.; Nagle, R.B.; Klein-Szanto, A.J.; Testa, J.R.; Altomare, D.A.; Borden, E.C. Phase II study of erlotinib in patients with malignant pleural mesothelioma: A Southwest Oncology Group Study. J. Clin. Oncol. 2007, 25, 2406–2413. [Google Scholar] [CrossRef]
- Govindan, R.; Kratzke, R.A.; Herndon, J.E.; Niehans, G.A.; Vollmer, R.; Watson, D.; Green, M.R.; Kindler, H.L. Cancer and Leukemia Group B (CALGB 30101). Gefitinib in patients with malignant mesothelioma: A phase II study by the Cancer and Leukemia Group B. Clin. Cancer Res. 2005, 11, 2300–2304. [Google Scholar] [CrossRef] [Green Version]
- Ou, W.B.; Hubert, C.; Corson, J.M.; Bueno, R.; Flynn, D.L.; Sugarbaker, D.J.; Fletcher, J.A. Targeted inhibition of multiple receptor tyrosine kinases in mesothelioma. Neoplasia 2011, 13, 12–22. [Google Scholar] [CrossRef] [Green Version]
- Gray, S.G.; Mutti, L. Immunotherapy for mesothelioma: A critical review of current clinical trials and future perspectives. Transl. Lung Cancer Res. 2020, 9 (Suppl. 1), S100–S119. [Google Scholar] [CrossRef]
- Terenziani, R.; Zoppi, S.; Fumarola, C.; Alfieri, R.; Bonelli, M. Immunotherapeutic Approaches in Malignant Pleural Mesothelioma. Cancers 2021, 13, 2793. [Google Scholar] [CrossRef]
- Scherpereel, A.; Mazieres, J.; Greillier, L.; Lantuejoul, S.; Dô, P.; Bylicki, O.; Monnet, I.; Corre, R.; Audigier-Valette, C.; Locatelli-Sanchez, M.; et al. Nivolumab or nivolumab plus ipilimumab in patients with relapsed malignant pleural mesothelioma (IFCT-1501 MAPS2): A multicentre, open-label, randomised, non-comparative, phase 2 trial. Lancet Oncol. 2019, 20, 239–253. [Google Scholar] [CrossRef]
- Maio, M.; Scherpereel, A.; Calabrò, L.; Aerts, J.; Perez, S.C.; Bearz, A.; Nackaerts, K.; Fennell, D.A.; Kowalski, D.; Tsao, A.S.; et al. Tremelimumab as second-line or third-line treatment in relapsed malignant mesothelioma (DETERMINE): A multicentre, international, randomised, double-blind, placebo-controlled phase 2b trial. Lancet Oncol. 2017, 18, 1261–1273. [Google Scholar] [CrossRef]
- Alcala, N.; Mangiante, L.; Le-Stang, N.; Gustafson, C.E.; Boyault, S.; Damiola, F.; Alcala, K.; Brevet, M.; Thivolet-Bejui, F.; Blanc-Fournier, C.; et al. Redefining malignant pleural mesothelioma types as a continuum uncovers immune-vascular interactions. EBioMedicine 2019, 48, 191–202. [Google Scholar] [CrossRef] [Green Version]
- Désage, A.L.; Karpathiou, G.; Peoc’h, M.; Froudarakis, M.E. The Immune Microenvironment of Malignant Pleural Mesothelioma: A Literature Review. Cancers 2021, 13, 3205. [Google Scholar] [CrossRef]
- Lorenzini, E.; Ciarrocchi, A.; Torricelli, F. Molecular Fingerprints of Malignant Pleural Mesothelioma: Not Just a Matter of Genetic Alterations. J. Clin. Med. 2021, 10, 2470. [Google Scholar] [CrossRef]
- Wadowski, B.; Bueno, R.; de Rienzo, A. Immune Microenvironment and Genetics in Malignant Pleural Mesothelioma. Front. Oncol. 2021, 11, 684025. [Google Scholar] [CrossRef]
- Napoli, F.; Listì, A.; Zambelli, V.; Witel, G.; Bironzo, P.; Papotti, M.; Volante, M.; Scagliotti, G.; Righi, L. Pathological Characterization of Tumor Immune Microenvironment (TIME) in Malignant Pleural Mesothelioma. Cancers 2021, 13, 2564. [Google Scholar] [CrossRef]
- Minnema-Luiting, J.; Vroman, H.; Aerts, J.; Cornelissen, R. Heterogeneity in Immune Cell Content in Malignant Pleural Mesothelioma. Int. J. Mol. Sci. 2018, 19, 1041. [Google Scholar] [CrossRef] [Green Version]
- Stella, G.M. Carbon nanotubes and pleural damage: Perspectives of nanosafety in the light of asbestos experience. Biointerphases 2011, 6, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Urso, L.; Cavallari, I.; Sharova, E.; Ciccarese, F.; Pasello, G.; Ciminale, V. Metabolic rewiring and redox alterations in malignant pleural mesothelioma. Br. J. Cancer 2020, 122, 52–61. [Google Scholar] [CrossRef]
- Menis, J.; Pasello, G.; Remon, J. Immunotherapy in malignant pleural mesothelioma: A review of literature data. Transl. Lung Cancer Res. 2021, 6, 2988–3000. [Google Scholar] [CrossRef]
- Patil, N.S.; Righi, L.; Koeppen, H.; Zou, W.; Izzo, S.; Grosso, F.; Libener, R.; Loiacono, M.; Monica, V.; Buttigliero, C.; et al. Molecular and Histopathological Characterization of the Tumor Immune Microenvironment in Advanced Stage of Malignant Pleural Mesothelioma. J. Thorac. Oncol. 2018, 13, 124–133. [Google Scholar] [CrossRef] [Green Version]
- Tolani, B.; Acevedo, L.A.; Hoang, N.T.; He, B. Heterogeneous Contributing Factors in MPM Disease Development and Progression: Biological Advances and Clinical Implications. Int. J. Mol. Sci. 2018, 19, 238. [Google Scholar] [CrossRef] [Green Version]
- Pasello, G.; Zago, G.; Lunardi, F.; Urso, L.; Kern, I.; Vlacic, G.; Grosso, F.; Mencoboni, M.; Ceresoli, G.L.; Schiavon, M.; et al. Malignant pleural mesothelioma immune microenvironment and checkpoint expression: Correlation with clinical-pathological features and intratumor heterogeneity over time. Ann. Oncol. 2018, 29, 1258–1265. [Google Scholar] [CrossRef]
- Pezzuto, F.; Lunardi, F.; Vedovelli, L.; Fortarezza, F.; Urso, L.; Grosso, F.; Ceresoli, G.L.; Kern, I.; Vlacic, G.; Faccioli, E.; et al. P14/ARF-Positive Malignant Pleural Mesothelioma: A Phenotype With Distinct Immune Microenvironment. Front. Oncol. 2021, 11, 653497. [Google Scholar] [CrossRef]
- Zhang, M.; Luo, J.L.; Sun, Q.; Harber, J.; Dawson, A.G.; Nakas, A.; Busacca, S.; Sharkey, A.J.; Waller, D.; Sheaff, M.T.; et al. Clonal architecture in mesothelioma is prognostic and shapes the tumour microenvironment. Nat. Commun. 2021, 12, 1751. [Google Scholar] [CrossRef]
- de Perrot, M. Prognostic role of PD-L1 in malignant pleural mesothelioma: Unraveling the complexity of the tumor microenvironment in mesothelioma. Ann. Thorac. Surg. 2021, 21. [Google Scholar] [CrossRef]
- Losi, L.; Bertolini, F.; Guaitoli, G.; Fabbiani, L.; Banchelli, F.; Ambrosini-Spaltro, A.; Botticelli, L.; Scurani, L.; Baldessari, C.; Barbieri, F.; et al. Role of evaluating tumor infiltrating lymphocytes, programmed death 1 ligand 1 and mismatch repair proteins expression in malignant mesothelioma. Int. J. Oncol. 2019, 55, 1157–1164. [Google Scholar] [CrossRef] [PubMed]
- Rrapaj, E.; Giacometti, L.; Spina, P.; Salvo, M.; Baselli, G.A.; Veggiani, C.; Rena, O.; Trisolini, E.; Boldorini, R.L. Programmed cell death 1 ligand 1 (PD-L1) expression is associated with poor prognosis of malignant pleural mesothelioma patients with good performance status. Pathology 2021, 53, 462–469. [Google Scholar] [CrossRef]
- Marcq, E.; Siozopoulou, V.; de Waele, J.; van Audenaerde, J.; Zwaenepoel, K.; Santermans, E.; Hens, N.; Pauwels, P.; van Meerbeeck, J.P.; Smits, E.L. Prognostic and predictive aspects of the tumor immune microenvironment and immune checkpoints in malignant pleural mesothelioma. Oncoimmunology 2016, 6, e1261241. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.C.; Hwang, S.H.; Kim, N.Y.; Lee, H.S.; Ji, S.; Yang, Y.; Kim, Y. Hypoxia promotes acquisition of aggressive phenotypes in human malignant mesothelioma. BMC Cancer 2018, 18, 819. [Google Scholar] [CrossRef]
- Noman, M.Z.; Desantis, G.; Janji, B.; Hasmim, M.; Karray, S.; Dessen, P.; Bronte, V.; Chouaib, S. PD-L1 is a novel direct target of HIF-1alpha, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J. Exp. Med. 2014, 211, 781–790. [Google Scholar] [CrossRef]
- Corzo, C.A.; Condamine, T.; Lu, L.; Cotter, M.J.; Youn, J.-I.; Cheng, P.; Cho, H.-I.; Celis, E.; Quiceno, D.G.; Padhya, T.; et al. HIF-1alpha regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J. Exp. Med. 2010, 207, 2439–2453. [Google Scholar] [CrossRef]
- Digifico, E.; Belgiovine, C.; Mantovani, A.; Allavena, P. Microenvironment and Immunology of the Human Pleural Malignant Mesothelioma. In Mesothelioma; Ceresoli, G., Bombardieri, E., D’Incalci, M., Eds.; Springer: Cham, Switzerland, 2019. [Google Scholar] [CrossRef]
- Carter, P.; Smith, L.; Ryan, M. Identification and validation of cell surface antigens for antibody targeting in oncology. Endocr Relat. Cancer. 2004, 11, 659–687. [Google Scholar] [CrossRef] [Green Version]
- Richter, M.; Zhang, H. Receptor-targeted cancer therapy. DNA Cell Biol. 2005, 24, 271–282. [Google Scholar] [CrossRef]
- Loo, D.T.; Mather, J.P. Antibody-based identification of cell surface antigens: Targets for cancer therapy. Curr. Opin. Pharmacol. 2008, 8, 627–631. [Google Scholar] [CrossRef]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef]
- Moses, M.A.; Brem, H.; Langer, R. Advancing the field of drug delivery: Taking aim at cancer. Cancer Cell. 2003, 4, 337–341. [Google Scholar] [CrossRef] [Green Version]
- Wahid, B.; Ali, A.; Rafique, S.; Waqar, M.; Wasim, M.; Wahid, K.; Idrees, M. An overview of cancer immunotherapeutic strategies. Immunotherapy 2018, 10, 999–1010. [Google Scholar] [CrossRef] [PubMed]
- Lérias, J.R.; de Sousa, E.; Paraschoudi, G.; Martins, J.; Condeço, C.; Figueiredo, N.; Carvalho, C.; Dodoo, E.; Maia, A.; Castillo-Martin, M.; et al. Trained Immunity for Personalized Cancer Immunotherapy: Current Knowledge and Future Opportunities. Front. Microbiol. 2020, 10, 2924. [Google Scholar] [CrossRef] [PubMed]
- Robert, C. A decade of immune-checkpoint inhibitors in cancer therapy. Nat. Commun. 2020, 11, 3801. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, H.; Nakagawa, K. Combination therapy with PD-1 or PD-L1 inhibitors for cancer. Int. J. Clin. Oncol. 2020, 25, 818–830. [Google Scholar] [CrossRef]
- Ramalingam, S.S.; Belani, C.P.; Ruel, C.; Frankel, P.; Gitlitz, B.; Koczywas, M.; Espinoza-Delgado, I.; Gandara, D. Phase II study of belinostat (PXD101), a histone deacetylase inhibitor, for second line therapy of advanced malignant pleural mesothelioma. J. Thorac. Oncol. 2009, 4, 97–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Londono, L.M.; Cowell, J.; Saatci, O.; Aras, M.; Ersan, P.G.; Serra, S.; Pei, H.; Clift, R.; Zhao, Q.; et al. Targeting Adenosine with Adenosine Deaminase 2 to Inhibit Growth of Solid Tumors. Cancer Res. 2021, 81, 3319–3332. [Google Scholar] [CrossRef] [PubMed]
- Lacerenza, S.; Ciregia, F.; Giusti, L.; Bonotti, A.; Greco, V.; Giannaccini, G.; D’Antongiovanni, V.; Fallahi, P.; Pieroni, L.; Cristaudo, A.; et al. Putative Biomarkers for Malignant Pleural Mesothelioma Suggested by Proteomic Analysis of Cell Secretome. Cancer Genom. Proteomics. 2020, 17, 225–236. [Google Scholar] [CrossRef]
- Stella, G.M.; Benvenuti, S.; Gentile, A.; Comoglio, P.M. MET Activation and Physical Dynamics of the Metastatic Process: The Paradigm of Cancers of Unknown Primary Origin. EBioMedicine 2017, 24, 34–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lolo, F.N.; Jiménez-Jiménez, V.; Sánchez-Álvarez, M.; del Pozo, M.Á. Tumor-stroma biomechanical crosstalk: A perspective on the role of caveolin-1 in tumor progression. Cancer Metastasis Rev. 2020, 39, 485–503. [Google Scholar] [CrossRef] [PubMed]
- Fiering, S.; Ang, L.H.; Lacoste, J.; Smith, T.D.; Griner, E. Reproducibility Project: Cancer Biology. Registered report: Biomechanical remodeling of the microenvironment by stromal caveolin-1 favors tumor invasion and metastasis. elife 2015, 4, e04796. [Google Scholar] [CrossRef] [Green Version]
- Gillies, R.J.; Kinahan, P.E.; Hricak, H. Radiomics: Images Are More Than Pictures, They Are Data. Radiology 2016, 278, 563–777. [Google Scholar] [CrossRef] [Green Version]
- Rizzo, S.; Botta, F.; Raimondi, S.; Origgi, D.; Fanciullo, C.; Morganti, A.G.; Bellomi, M. Radiomics: The Facts and the Challenges of Image Analysis. Eur. Radiol. Exp. 2018, 2, 36. [Google Scholar] [CrossRef]
- Sun, R.; Limkin, E.J.; Vakalopoulou, M.; Dercle, L.; Champiat, S.; Han, S.R.; Verlingue, L.; Brandao, D.; Lancia, A.; Ammari, S.; et al. A radiomics approach to assess tumour-infiltrating CD8 cells and response to anti-PD-1 or anti-PD-L1 immunotherapy: An imaging biomarker, retrospective multicohort study. Lancet Oncol. 2018, 19, 1180–1191. [Google Scholar] [CrossRef]
- Martini, K.; Frauenfelder, T. Old Borders and New Horizons in Multimodality Imaging of Malignant Pleural Mesothelioma. Thorac. Cardiovasc. Surg. 2021. [Google Scholar] [CrossRef]
- Armato, S.G.; Blyth, K.G.; Keating, J.J.; Katz, S.; Tsim, S.; Coolen, J.; Gudmundsson, E.; Opitz, I.; Nowak, A.K. Imaging in pleural mesothelioma: A review of the 13th International Conference of the International Mesothelioma Interest Group. Lung Cancer 2016, 101, 48–58. [Google Scholar] [CrossRef] [Green Version]
- Ciliberto, M.; Kishida, Y.; Seki, S.; Yoshikawa, T.; Ohno, Y. Update of MR Imaging for Evaluation of Lung Cancer. Radiol. Clin. N. Am. 2018, 56, 437–469. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Gong, X.Q.; Tao, Y.Y.; Wang, R.; Yang, G.; Li, J.D.; Ren, T.; Li, Z.M.; Yang, C.; Wang, W.C.; et al. Correlative Study Between IVIM-DWI Parameters and the Expression Levels of Ang-2 and TKT in Hepatocellular Carcinoma. Front. Oncol. 2021. [Google Scholar] [CrossRef]
- Meyer, H.J.; Wienke, A.; Surov, A. Association Between VEGF Expression and Diffusion Weighted Imaging in Several Tumors-A Systematic Review and Meta-Analysis. Diagnostics 2019, 9, 126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zarogoulidis, P.; Mavroudi, M.; Porpodis, K.; Domvri, K.; Sakkas, A.; Machairiotis, N.; Stylianaki, A.; Tsiotsios, A.; Courcoutsakis, N.; Zarogoulidis, K. Pegylated liposomal doxorubicin in malignant pleural mesothelioma: A possible guardian for long-term survival. Oncol. Targets Ther. 2012, 5, 231–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hillerdal, G.; Sorensen, J.B.; Sundström, S.; Riska, H.; Vikström, A.; Hjerpe, A. Treatment of malignant pleural mesothelioma with carboplatin, liposomized doxorubicin, and gemcitabine: A phase II study. J. Thorac. Oncol. 2008, 3, 1325–1331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ando, H.; Kobayashi, S.; Abu Lila, A.S.; Eldin, N.E.; Kato, C.; Shimizu, T.; Ukawa, M.; Kawazoe, K.; Ishida, T. Advanced therapeutic approach for the treatment of malignant pleural mesothelioma via the intrapleural administration of liposomal pemetrexed. J. Control. Release 2015, 220 Pt A, 29–36. [Google Scholar] [CrossRef]
- Eldin, N.E.; Abu Lila, A.S.; Kawazoe, K.; Elnahas, H.M.; Mahdy, M.A.; Ishida, T. Encapsulation in a rapid-release liposomal formulation enhances the anti-tumor efficacy of pemetrexed in a murine solid mesothelioma-xenograft model. Eur. J. Pharm. Sci. 2016, 81, 60–66. [Google Scholar] [CrossRef]
- Medina, L.A.; Calixto, S.M.; Klipper, R.; Phillips, W.T.; Goins, B. Avidin/biotin-liposome system injected in the pleural space for drug delivery to mediastinal lymph nodes. J. Pharm. Sci. 2004, 93, 2595–2608. [Google Scholar] [CrossRef]
- Marazioti, A.; Papadia, K.; Giannou, A.; Stathopoulos, G.T.; Antimisiaris, S.G. Prolonged retention of liposomes in the pleural cavity of normal mice and high tumor distribution in mice with malignant pleural effusion, after intrapleural injection. Int. J. Nanomed. 2019, 14, 3773–3784. [Google Scholar] [CrossRef] [Green Version]
- Cova, E.; Pandolfi, L.; Colombo, M.; Frangipane, V.; Inghilleri, S.; Morosini, M.; Mrakic-Sposta, S.; Moretti, S.; Monti, M.; Pignochino, Y.; et al. Pemetrexed-loaded nanoparticles targeted to malignant pleural mesothelioma cells: An in vitro study. Int. J. Nanomed. 2019, 14, 773–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulz, M.D.; Zubris, K.A.; Wade, J.E.; Padera, R.F.; Xu, X.; Grinstaff, M.W.; Colson, Y.L. Paclitaxel-loaded expansile nanoparticles in a multimodal treatment model of malignant mesothelioma. Ann. Thorac. Surg. 2011, 92, 2007–2014. [Google Scholar] [CrossRef]
- Kanai, O.; Fujita, K.; Nakatani, K.; Mio, T. Repetitive responses to nanoparticle albumin-bound paclitaxel and carboplatin in malignant pleural mesothelioma. Respirol. Case Rep. 2016, 4, 28–31. [Google Scholar] [CrossRef] [Green Version]
- Sakurai, Y.; Kato, A.; Hida, Y.; Hamada, J.; Maishi, N.; Hida, K.; Harashima, H. Synergistic Enhancement of Cellular Uptake With CD44-Expressing Malignant Pleural Mesothelioma by Combining Cationic Liposome and Hyaluronic Acid-Lipid Conjugate. J. Pharm. Sci. 2019, 108, 3218–3224. [Google Scholar] [CrossRef] [PubMed]
- Oble, D.A.; Loewe, R.; Yu, P.; Mihm, M.C., Jr. Focus on TILs: Prognostic significance of tumor infiltrating lymphocytes in human melanoma. Cancer Immun. 2009, 9, 3. [Google Scholar] [PubMed]
- Dafni, U.; Michielin, O.; Lluesma, S.M.; Tsourti, Z.; Polydoropoulou, V.; Karlis, D.; Besser, M.J.; Haanen, J.; Svane, I.M.; Ohashi, P.S.; et al. Efficacy of adoptive therapy with tumor-infiltrating lymphocytes and recombinant interleukin-2 in advanced cutaneous melanoma: A systematic review and meta-analysis. Ann. Oncol. 2019, 30, 1902–1913. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Deng, H.; Zhou, Y.; Ye, Y.; Zhao, S.; Liang, S.; Cai, S.; Lin, J.; Tang, Y.; Wu, Y. Expression and clinical significance of CXC chemokines in the glioblastoma microenvironment. Life Sci. 2020, 261, 118486. [Google Scholar] [CrossRef]
- Hargadon, K.M. Strategies to Improve the Efficacy of Dendritic Cell-Based Immunotherapy for Melanoma. Front Immunol. 2017, 8, 1594. [Google Scholar] [CrossRef] [Green Version]
- Brossart, P. Dendritic cells in vaccination therapies of malignant diseases. Transfus Apher Sci. 2002, 27, 183–186. [Google Scholar] [CrossRef]
- Mastelic-Gavillet, B.; Navarro Rodrigo, B.; Décombaz, L.; Wang, H.; Ercolano, G.; Ahmed, R.; Lozano, L.E.; Ianaro, A.; Derré, L.; Valerio, M.; et al. Adenosine mediates functional and metabolic suppression of peripheral and tumor-infiltrating CD8+ T cells. J. Immunother Cancer 2019, 7, 257. [Google Scholar] [CrossRef]
- Belderbos, R.A.; Vroman, H.; Aerts, J.G.J.V. Cellular Immunotherapy and Locoregional Administration of CAR T-Cells in Malignant Pleural Mesothelioma. Front. Oncol. 2020, 10, 777. [Google Scholar] [CrossRef]
- van Gulijk, M.; Dammeijer, F.; Aerts, J.G.J.V.; Vroman, H. Combination Strategies to Optimize Efficacy of Dendritic Cell-Based Immunotherapy. Front. Immunol. 2018, 9, 2759. [Google Scholar] [CrossRef] [Green Version]
- Garg, A.D.; Vara Perez, M.; Schaaf, M.; Agostinis, P.; Zitvogel, L.; Kroemer, G.; Galluzzi, L. Trial watch: Dendritic cell-based anticancer immunotherapy. Oncoimmunology 2017, 6, e1328341. [Google Scholar] [CrossRef]
- Dumoulin, D.W.; Cornelissen, R.; Bezemer, K.; Baart, S.J.; Aerts, J.G.J.V. Long-Term Follow-Up of Mesothelioma Patients Treated with Dendritic Cell Therapy in Three Phase I/II Trials. Vaccines 2021, 9, 525. [Google Scholar] [CrossRef] [PubMed]
- Chintala, N.K.; Restle, D.; Quach, H.; Saini, J.; Bellis, R.; Offin, M.; Beattie, J.; Adusumilli, P.S. CAR T-cell therapy for pleural mesothelioma: Rationale, preclinical development, and clinical trials. Lung Cancer 2021, 157, 48–59. [Google Scholar] [CrossRef] [PubMed]
- Klampatsa, A.; Dimou, V.; Albelda, S.M. Mesothelin-targeted CAR-T cell therapy for solid tumors. Expert Opin. Biol. Ther. 2021, 21, 473–486. [Google Scholar] [CrossRef] [PubMed]
- Gray, S.G. Emerging avenues in immunotherapy for the management of malignant pleural mesothelioma. BMC Pulm. Med. 2021, 21, 148. [Google Scholar] [CrossRef]
- Czapla, J.; Matuszczak, S.; Kulik, K.; Wiśniewska, E.; Pilny, E.; Jarosz-Biej, M.; Smolarczyk, R.; Sirek, T.; Zembala, M.O.; Zembala, M.; et al. The effect of culture media on large-scale expansion and characteristic of adipose tissue-derived mesenchymal stromal cells. Stem Cell Res. Ther. 2019, 10, 235. [Google Scholar] [CrossRef] [Green Version]
- Facchetti, G.; Petrella, F.; Spaggiari, L.; Rimoldi, I. Malignant Pleural Mesothelioma: State of the art and advanced cell therapy. Eur. J. Med. Chem. 2017, 142, 266–270. [Google Scholar] [CrossRef]
- Petrella, F.; Rimoldi, I.; Rizzo, S.; Spaggiari, L. Mesenchymal Stromal Cells for Antineoplastic Drug Loading and Delivery. Medicines 2017, 4, 87. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Q.; Ding, H.; Wang, L.; Yan, Y.; Wan, Y.; Yi, Y.; Tao, L.; Zhu, C. Circulating Exosomal miR-96 as a Novel Biomarker for Radioresistant Non-Small-Cell Lung Cancer. J. Oncol. 2021, 2021, 5893981. [Google Scholar] [CrossRef]
- Markov, A.; Thangavelu, L.; Aravindhan, S.; Zekiy, A.O.; Jarahian, M.; Chartrand, M.S.; Pathak, Y.; Marofi, F.; Shamlou, S.; Hassanzadeh, A. Mesenchymal stem/stromal cells as a valuable source for the treatment of immune-mediated disorders. Stem Cell Res. Ther. 2021, 12, 192. [Google Scholar] [CrossRef] [PubMed]
- Burgio, S.; Noori, L.; Marino Gammazza, A.; Campanella, C.; Logozzi, M.; Fais, S.; Bucchieri, F.; Cappello, F.; Caruso Bavisotto, C. Extracellular Vesicles-Based Drug Delivery Systems: A New Challenge and the Exemplum of Malignant Pleural Mesothelioma. Int. J. Mol. Sci. 2020, 21, 5432. [Google Scholar] [CrossRef] [PubMed]
- Coccè, V.; Franzè, S.; Brini, A.T.; Giannì, A.B.; Pascucci, L.; Ciusani, E.; Alessandri, G.; Farronato, G.; Cavicchini, L.; Sordi, V.; et al. In Vitro Anticancer Activity of Extracellular Vesicles (EVs) Secreted by Gingival Mesenchymal Stromal Cells Primed with Paclitaxel. Pharmaceutics 2019, 11, 61. [Google Scholar] [CrossRef] [Green Version]
- Crivelli, B.; Chlapanidas, T.; Perteghella, S.; Lucarelli, E.; Pascucci, L.; Brini, A.T.; Ferrero, I.; Marazzi, M.; Pessina, A.; Torre, M.L.; et al. Mesenchymal stem/stromal cell extracellular vesicles: From active principle to next generation drug delivery system. J. Control. Release 2017, 262, 104–117. [Google Scholar] [CrossRef] [PubMed]
- El-Sherbiny, I.M.; Yacoub, M.H. Hydrogel scaffolds for tissue engineering: Progress and challenges. Glob. Cardiol. Sci. Pract. 2013, 2013, 316–342. [Google Scholar] [CrossRef] [Green Version]
- Pharaon, M.R.; Scholz, T.; Evans, G.R.D. Tissue Engineering. In Plastic and Reconstructive Surgery; Siemionow, M.Z., Eisenmann-Klein, M., Eds.; Springer Specialist Surgery Series; Springer: London, UK, 2010. [Google Scholar] [CrossRef]
- Ricci, C.; Moroni, L.; Danti, S. Cancer tissue engineering—New perspectives in understanding the biology of solid tumours—A critical review. OA Tissue Eng. 2013, 1, 4. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Blum, N.T.; Lin, J.; Qu, J.; Huang, P. Biomaterial scaffold-based local drug delivery systems for cancer immunotherapy. Sci. Bull. 2020, 65, 1489–1504. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lisini, D.; Lettieri, S.; Nava, S.; Accordino, G.; Frigerio, S.; Bortolotto, C.; Lancia, A.; Filippi, A.R.; Agustoni, F.; Pandolfi, L.; et al. Local Therapies and Modulation of Tumor Surrounding Stroma in Malignant Pleural Mesothelioma: A Translational Approach. Int. J. Mol. Sci. 2021, 22, 9014. https://doi.org/10.3390/ijms22169014
Lisini D, Lettieri S, Nava S, Accordino G, Frigerio S, Bortolotto C, Lancia A, Filippi AR, Agustoni F, Pandolfi L, et al. Local Therapies and Modulation of Tumor Surrounding Stroma in Malignant Pleural Mesothelioma: A Translational Approach. International Journal of Molecular Sciences. 2021; 22(16):9014. https://doi.org/10.3390/ijms22169014
Chicago/Turabian StyleLisini, Daniela, Sara Lettieri, Sara Nava, Giulia Accordino, Simona Frigerio, Chandra Bortolotto, Andrea Lancia, Andrea Riccardo Filippi, Francesco Agustoni, Laura Pandolfi, and et al. 2021. "Local Therapies and Modulation of Tumor Surrounding Stroma in Malignant Pleural Mesothelioma: A Translational Approach" International Journal of Molecular Sciences 22, no. 16: 9014. https://doi.org/10.3390/ijms22169014