
Structural Water Stabilizes Protein Motifs in Liquid Protein Phase: The Folding Mechanism of Short β-Sheets Coupled to Phase Transition

 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
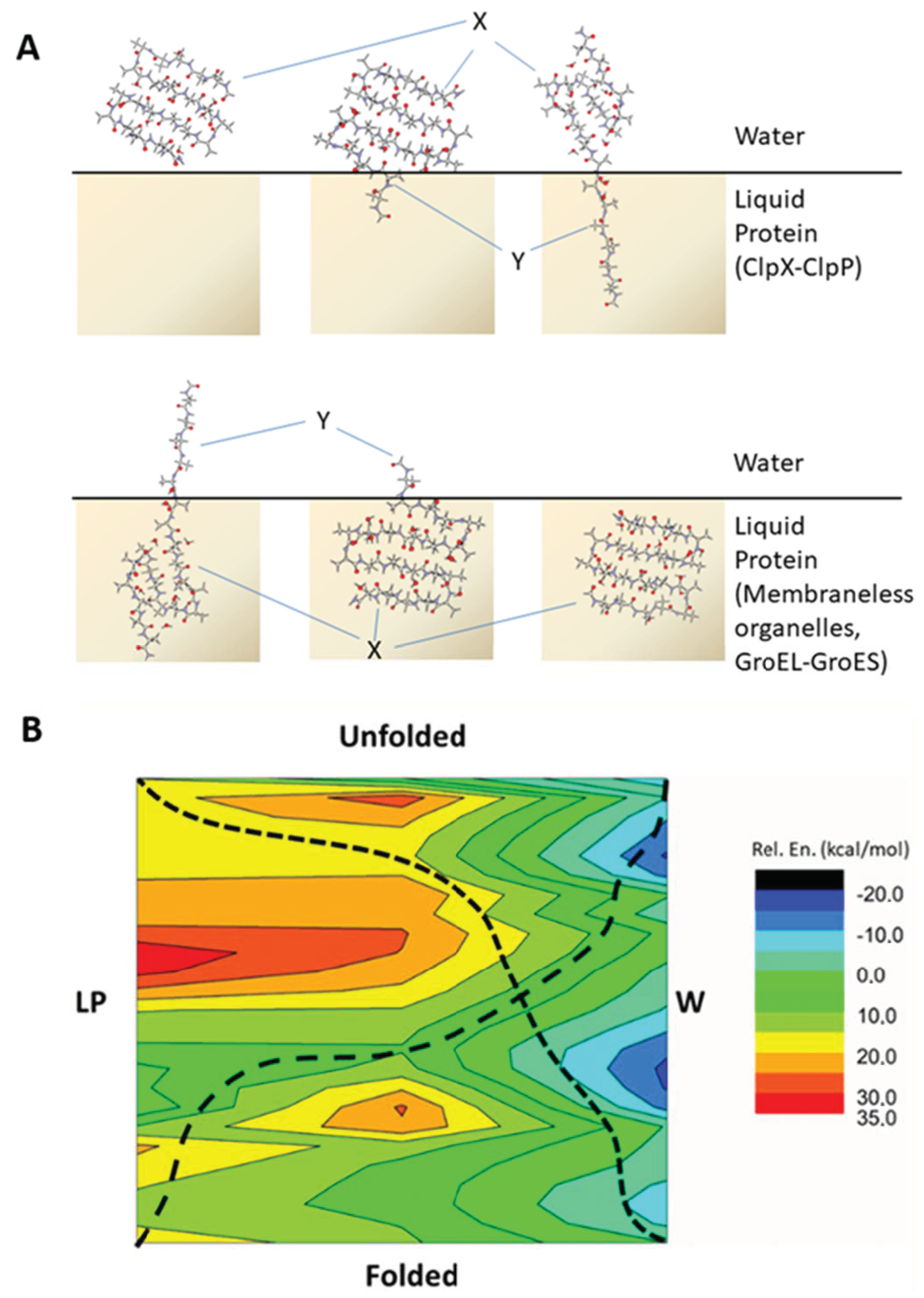{kind=link}
1. Introduction
2. Results and Discussions
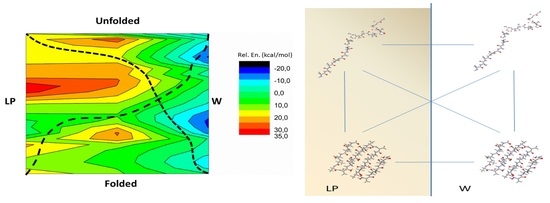
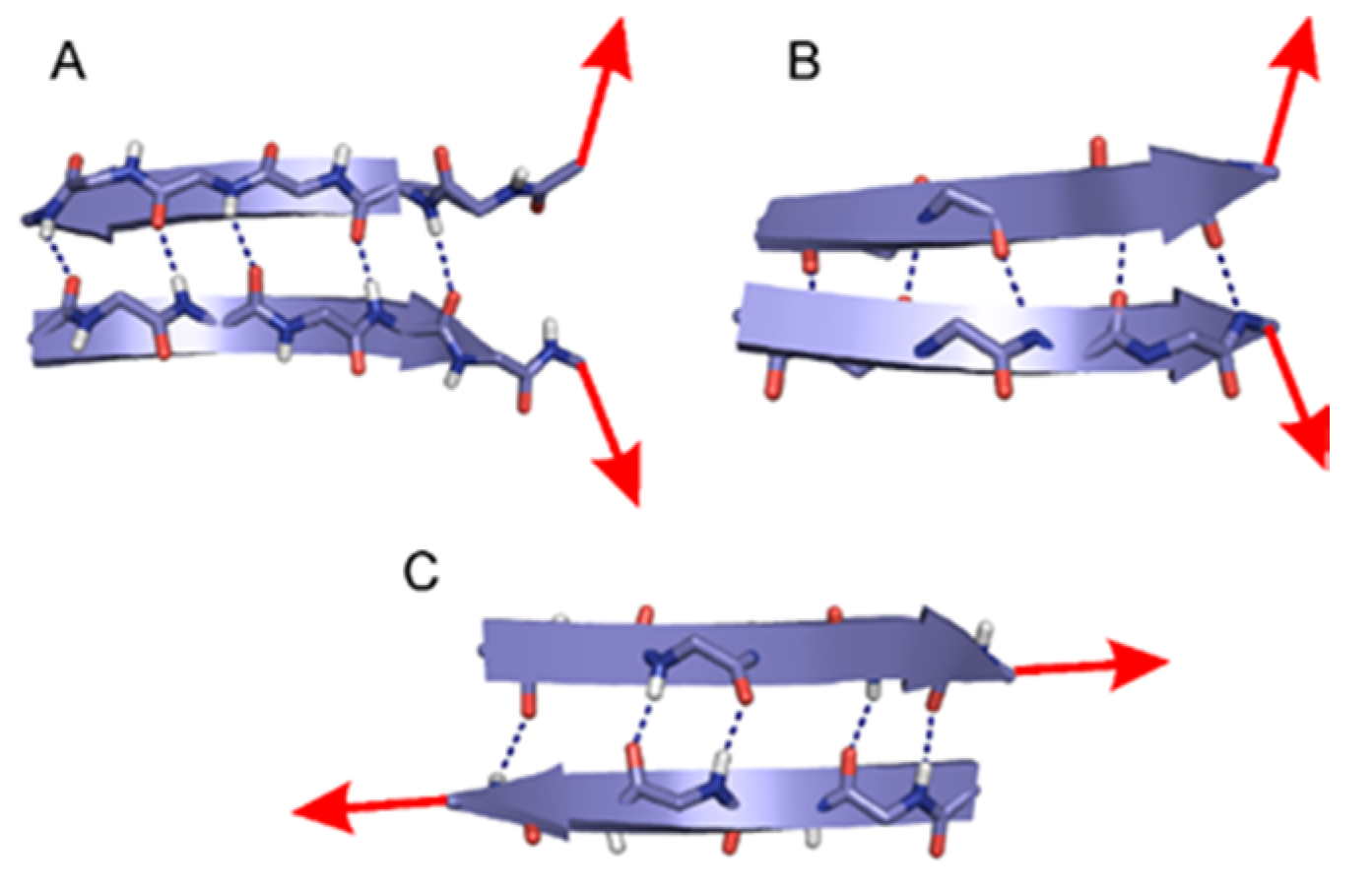
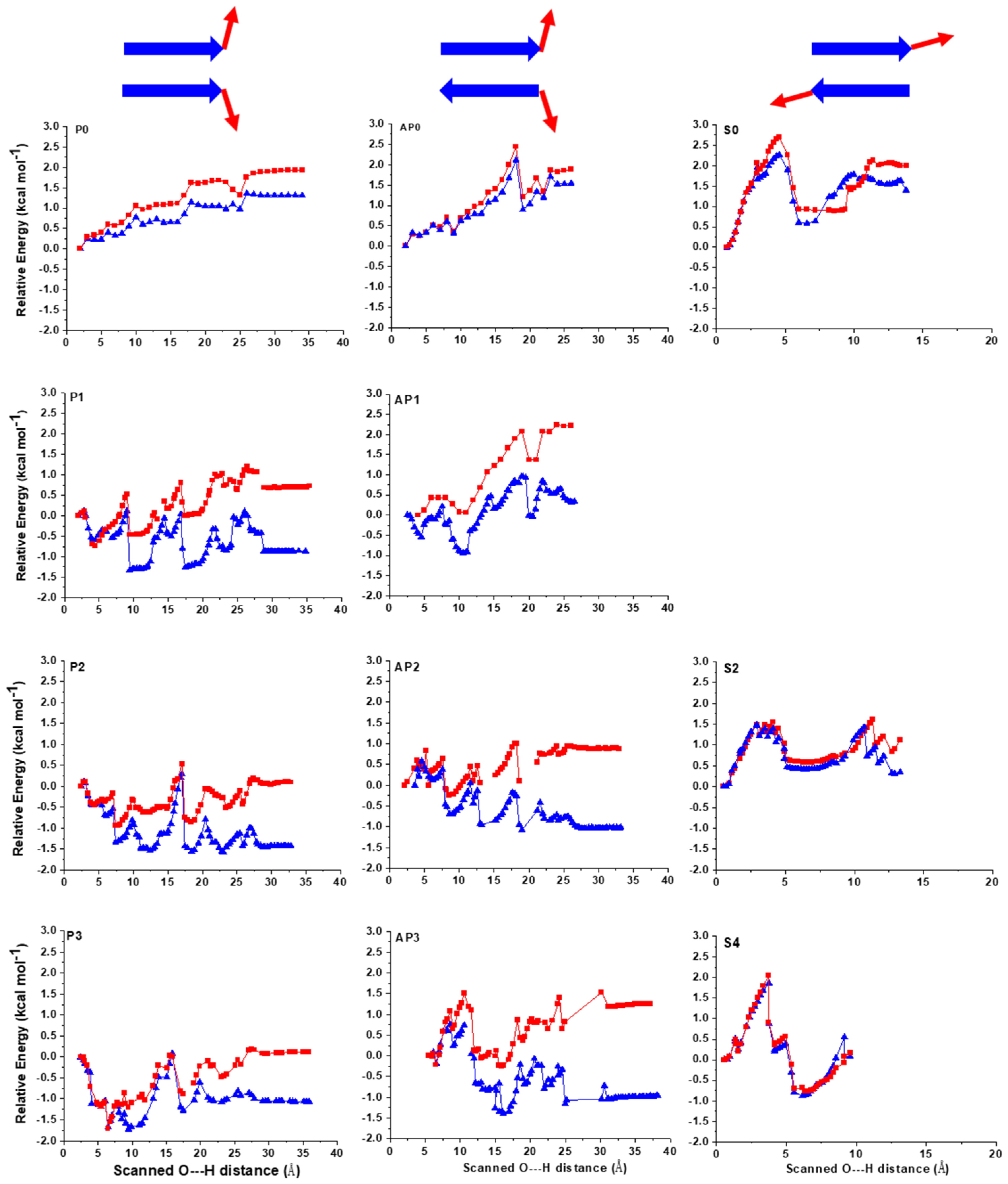
2.1. The Energy Profile of Lateral Unfolding in W and LP Environments
2.2. Sheared Unfolding in Different Surrounding Media
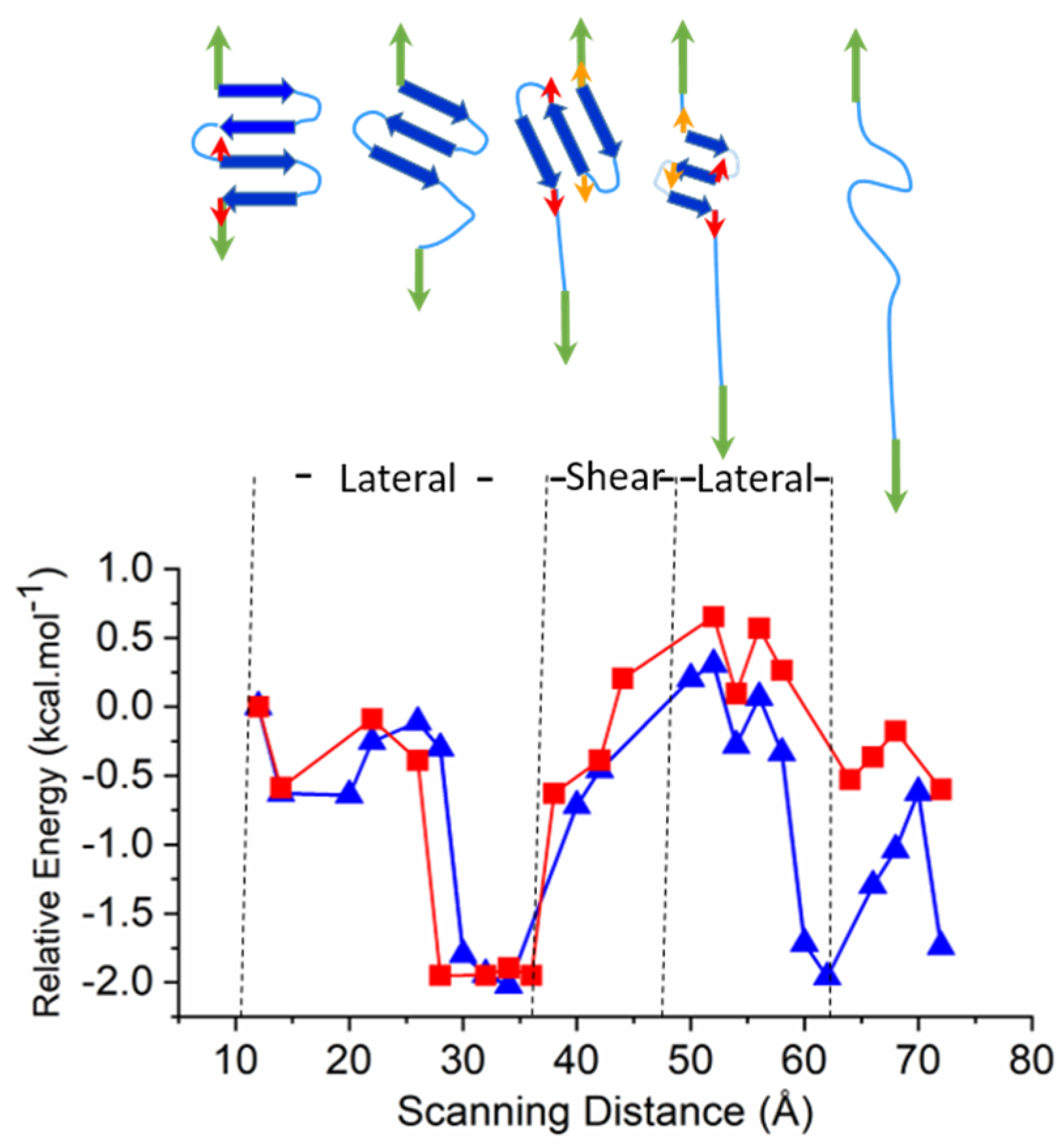
2.3. Larger Models—Consecutive Lateral and Sheared Unfolding Results in Strand Shortening
2.4. Energetic Aspects of Coupling Phase Changes to Folding/Unfolding Events
2.5. Comparison to Experiments
3. Materials and Methods
3.1. Quantum Mechanical Calculations
3.2. Employed Models
3.2.1. Lateral Unfolding
3.2.2. Sheared Unfolding
3.2.3. Larger Models
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Boeynaems, S.; Alberti, S.; Fawzi, N.L.; Mittag, T.; Polymenidou, M.; Rousseau, F.; Schymkowitz, J.; Shorter, J.; Wolozin, B.; Bosch, L.V.D.; et al. Protein Phase Separation: A New Phase in Cell Biology. Trends Cell Biol. 2018, 28, 420–435. [Google Scholar] [CrossRef] [Green Version]
- Gomes, E.; Shorter, J. The molecular language of membraneless organelles. J. Biol. Chem. 2019, 294, 7115–7127. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Horwich, A.L.; Sigler, P.B. The crystal structure of the asymmetric GroEL–GroES–(ADP) 7 chaperonin complex. Nat. Cell Biol. 1997, 388, 741–750. [Google Scholar] [CrossRef]
- Hanson, P.I.; Whiteheart, S. AAA+ proteins: Have engine, will work. Nat. Rev. Mol. Cell Biol. 2005, 6, 519–529. [Google Scholar] [CrossRef]
- Zozulia, O.; Korendovych, I.V. Semi-Rationally Designed Short Peptides Self-Assemble and Bind Hemin to Promote Cyclopropanation. Angew. Chem. 2020, 132, 8185–8189. [Google Scholar] [CrossRef]
- Brangwynne, C.P.; Eckmann, C.R.; Courson, D.S.; Rybarska, A.; Hoege, C.; Gharakhani, J.; Jülicher, F.; Hyman, A.A. Germline P Granules Are Liquid Droplets That Localize by Controlled Dissolution/Condensation. Science 2009, 324, 1729–1732. [Google Scholar] [CrossRef] [PubMed]
- Hughes, M.P.; Sawaya, M.R.; Boyer, D.R.; Goldschmidt, L.; Rodriguez, J.A.; Cascio, D.; Chong, L.; Gonen, T.; Eisenberg, D.S. Atomic structures of low-complexity protein segments reveal kinked β sheets that assemble networks. Science 2018, 359, 698–701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Booth, D.R.; Sunde, M.; Bellotti, V.; Robinson, C.V.; Hutchinson, W.L.; Fraser, P.E.; Hawkins, P.N.; Dobson, C.M.; Radford, S.; Blake, C.C.F.; et al. Instability, unfolding and aggregation of human lysozyme variants underlying amyloid fibrillogenesis. Nat. Cell Biol. 1997, 385, 787–793. [Google Scholar] [CrossRef]
- Laganowsky, A.; Liu, C.; Sawaya, M.R.; Whitelegge, J.P.; Park, J.; Zhao, M.; Pensalfini, A.; Soriaga, A.B.; Landau, M.; Teng, P.K.; et al. Atomic View of a Toxic Amyloid Small Oligomer. Science 2012, 335, 1228–1231. [Google Scholar] [CrossRef] [Green Version]
- Chuang, E.; Hori, A.; Hesketh, C.D.; Shorter, J. Amyloid assembly and disassembly. J. Cell Sci. 2018, 131, jcs189928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, B.; Sosa, R.P.; Mårtensson, A.K.F.; Jiang, K.; Tong, A.; Dorfman, K.D.; Takahashi, M.; Lincoln, P.; Bustamante, C.J.; Westerlund, F.; et al. Hydrophobic catalysis and a potential biological role of DNA unstacking induced by environment effects. Proc. Natl. Acad. Sci. USA 2019, 116, 17169–17174. [Google Scholar] [CrossRef] [Green Version]
- Starikov, E.B.; Nordén, B. Enthalpy−Entropy Compensation: A Phantom or Something Useful? J. Phys. Chem. B 2007, 111, 14431–14435. [Google Scholar] [CrossRef]
- Mason, T.O.; Michaels, T.C.T.; Levin, A.; Dobson, C.M.; Gazit, E.; Knowles, T.P.J.; Buell, A.K. Thermodynamics of Polypeptide Supramolecular Assembly in the Short-Chain Limit. J. Am. Chem. Soc. 2017, 139, 16134–16142. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, A.J.; Knowles, T.; Tartaglia, G.G.; Fitzpatrick, A.W.; Devlin, G.L.; Shammas, S.L.; Waudby, C.; Mossuto, M.F.; Meehan, S.; Gras, S.; et al. Metastability of Native Proteins and the Phenomenon of Amyloid Formation. J. Am. Chem. Soc. 2011, 133, 14160–14163. [Google Scholar] [CrossRef]
- Dobson, C.M. Protein folding and misfolding. Nature 2003, 426, 884–890. [Google Scholar] [CrossRef] [PubMed]
- Legname, G.; Baskakov, I.V.; Nguyen, H.-O.B.; Riesner, D.; Cohen, F.E.; DeArmond, S.J.; Prusiner, S.B. Synthetic Mammalian Prions. Science 2004, 305, 673–676. [Google Scholar] [CrossRef] [PubMed]
- Wimley, W.C. The versatile β-barrel membrane protein. Curr. Opin. Struct. Biol. 2003, 13, 404–411. [Google Scholar] [CrossRef]
- Perczel, A.; Hudáky, A.P.; Pálfi, V.K. Dead-End Street of Protein Folding: Thermodynamic Rationale of Amyloid Fibril Formation. J. Am. Chem. Soc. 2007, 129, 14959–14965. [Google Scholar] [CrossRef] [PubMed]
- Navizet, L.; Cailliez, F.; Lavery, R. Probing Protein Mechanics: Residue-Level Properties and Their Use in Defining Domains. Biophys. J. 2004, 87, 1426–1435. [Google Scholar] [CrossRef]
- Dougan, L.; Feng, G.; Lu, H.; Fernandez, J.M. Solvent molecules bridge the mechanical unfolding transition state of a protein. Proc. Natl. Acad. Sci. USA 2008, 105, 3185–3190. [Google Scholar] [CrossRef] [Green Version]
- Morozov, A.V.; Kortemme, T. Potential Functions for Hydrogen Bonds in Protein Structure Prediction and Design. In Advances in Protein Chemistry; Peptide Solvation and HBonds; Academic Press: Cambridge, MA, USA, 2005; Volume 72, pp. 1–38. [Google Scholar] [CrossRef]
- Paci, E.; Karplus, M. Forced unfolding of fibronectin type 3 modules: An analysis by biased molecular dynamics simulations. J. Mol. Biol. 1999, 288, 441–459. [Google Scholar] [CrossRef] [Green Version]
- Beke-Somfai, T.; Perczel, A. Zipper-Like Unfolding of β-Sheets Accessed by Pioneer Water Molecules: Atomic Resolution of Forced Unfold Reveals Different Mechanisms for Parallel and Antiparallel Motifs. J. Phys. Chem. Lett. 2010, 1, 1341–1345. [Google Scholar] [CrossRef]
- Morozov, A.V.; Kortemme, T.; Tsemekhman, K.; Baker, D. Close agreement between the orientation dependence of hydrogen bonds observed in protein structures and quantum mechanical calculations. Proc. Natl. Acad. Sci. USA 2004, 101, 6946–6951. [Google Scholar] [CrossRef] [Green Version]
- Pohl, G.; Beke, T.; Borbély, J.; Perczel, A. Prediction of Folding Preference of 10 kDa Silk-like Proteins Using a Lego Approach and ab Initio Calculations. J. Am. Chem. Soc. 2006, 128, 14548–14559. [Google Scholar] [CrossRef]
- Kortelainen, M.; Suhonen, A.; Hamza, A.; Papai, I.; Nauha, E.; Yliniemelä-Sipari, S.; Nissinen, M.; Pihko, P.M. Folding Patterns in a Family of Oligoamide Foldamers. Chem. A Eur. J. 2015, 21, 9493–9504. [Google Scholar] [CrossRef] [PubMed]
- Johansson, J.R.; Hermansson, E.; Nordén, B.; Kann, N.; Beke-Somfai, T. δ-Peptides from RuAAC-Derived 1,5-Disubstituted Triazole Units. Eur. J. Org. Chem. 2014, 2014, 2703–2713. [Google Scholar] [CrossRef]
- Himo, F. Recent Trends in Quantum Chemical Modeling of Enzymatic Reactions. J. Am. Chem. Soc. 2017, 139, 6780–6786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, D.; Perišić, O.; Peng, Q.; Cao, Y.; Lam, C.; Lu, H.; Li, H. Single-molecule force spectroscopy reveals a mechanically stable protein fold and the rational tuning of its mechanical stability. Proc. Natl. Acad. Sci. USA 2007, 104, 9278–9283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banerjee, P.R.; Deniz, A.A. Shedding Light on Protein Folding Landscapes by Single-molecule Fluorescence. Chem. Soc. Rev. 2013, 43, 1172–1188. [Google Scholar] [CrossRef] [Green Version]
- Narimoto, T.; Sakurai, K.; Okamoto, A.; Chatani, E.; Hoshino, M.; Hasegawa, K.; Naiki, H.; Goto, Y. Conformational stability of amyloid fibrils of β2 -microglobulin probed by guanidine-hydrochloride-induced unfolding. FEBS Lett. 2004, 576, 313–319. [Google Scholar] [CrossRef] [Green Version]
- Mardirossian, N.; Head-Gordon, M. Thirty years of density functional theory in computational chemistry: An overview and extensive assessment of 200 density functionals. Mol. Phys. 2017, 115, 2315–2372. [Google Scholar] [CrossRef]
- Papp, D.; Rovó, P.; Jákli, I.; Császár, A.G.; Perczel, A. Four faces of the interaction between ions and aromatic rings. J. Comput. Chem. 2017, 38, 1762–1773. [Google Scholar] [CrossRef]
- Shu, C.; Jiang, Z.; Biczysko, M. Toward accurate prediction of amino acid derivatives structure and energetics from DFT: Glycine conformers and their interconversions. J. Mol. Model. 2020, 26, 1–13. [Google Scholar] [CrossRef]
- Alabugin, I.V.; Manoharan, M.; Peabody, S.; Weinhold, F. Electronic Basis of Improper Hydrogen Bonding: A Subtle Balance of Hyperconjugation and Rehybridization. J. Am. Chem. Soc. 2003, 125, 5973–5987. [Google Scholar] [CrossRef]
- Rabuck, A.D.; Scuseria, G.E. Performance of recently developed kinetic energy density functionals for the calculation of hydrogen binding strengths and hydrogen-bonded structures. Theor. Chem. Acc. 2000, 104, 439–444. [Google Scholar] [CrossRef]
- Boese, A.D. Density Functional Theory and Hydrogen Bonds: Are We There Yet? ChemPhysChem 2015, 16, 978–985. [Google Scholar] [CrossRef] [PubMed]
- Luccarelli, J.; Paton, R.S. Hydrogen-Bond-Dependent Conformational Switching: A Computational Challenge from Experimental Thermochemistry. J. Org. Chem. 2018, 84, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Pepin, R.; Laszlo, K.J.; Marek, A.; Peng, B.; Bush, M.F.; Lavanant, H.; Afonso, C.; Tureček, F. Toward a Rational Design of Highly Folded Peptide Cation Conformations. 3D Gas-Phase Ion Structures and Ion Mobility Characterization. J. Am. Soc. Mass Spectrom. 2016, 27, 1647–1660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perczel, A.; Gáspári, Z.; Csizmadia, I.G. Structure and stability of β-pleated sheets. J. Comput. Chem. 2005, 26, 1155–1168. [Google Scholar] [CrossRef]
- Perczel, A.; Angyan, J.G.; Kajtar, M.; Viviani, W.; Rivail, J.L.; Marcoccia, J.F.; Csizmadia, I.G. Peptide models. 1. Topology of selected peptide conformational potential energy surfaces (glycine and alanine derivatives). J. Am. Chem. Soc. 1991, 113, 6256–6265. [Google Scholar] [CrossRef]
- Beke, T.; Czajlik, A.; Csizmadia, I.G.; Perczel, A. Determining suitablelego-structures to estimate stability of larger peptide nanostructures using computational methods. Phys. Biol. 2006, 3, S26–S39. [Google Scholar] [CrossRef] [PubMed]
- Sastre, S.; Casasnovas, R.; Muñoz, F.; Frau, J. Isodesmic reaction for accurate theoretical pK a calculations of amino acids and peptides. Phys. Chem. Chem. Phys. 2016, 18, 11202–11212. [Google Scholar] [CrossRef]
- Wimley, W.C.; White, S. Experimentally determined hydrophobicity scale for proteins at membrane interfaces. Nat. Struct. Mol. Biol. 1996, 3, 842–848. [Google Scholar] [CrossRef] [PubMed]
- Wimley, W.C.; Creamer, T.P.; White, S. Solvation Energies of Amino Acid Side Chains and Backbone in a Family of Host–Guest Pentapeptides. Biochemistry 1996, 35, 5109–5124. [Google Scholar] [CrossRef]
- Baker, T.A.; Sauer, R.T. ClpXP, an ATP-powered unfolding and protein-degradation machine. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2012, 1823, 15–28. [Google Scholar] [CrossRef] [Green Version]
- Hill, K.; Model, K.; Ryan, M.T.; Dietmeier, K.; Martin, F.; Wagner, R.; Pfanner, N. Tom40 forms the hydrophilic channel of the mitochondrial import pore for preproteins. Nature 1998, 395, 516–521. [Google Scholar] [CrossRef]
- Fei, X.; Bell, T.A.; Jenni, S.; Stinson, B.M.; Baker, T.A.; Harrison, S.C.; Sauer, R.T. Structures of the ATP-fueled ClpXP proteolytic machine bound to protein substrate. eLife 2020, 9, e52774. [Google Scholar] [CrossRef]
- Buell, A.K.; Dhulesia, A.; Mossuto, M.F.; Cremades, N.; Kumita, J.R.; Dumoulin, M.; Welland, M.E.; Knowles, T.; Salvatella, X.; Dobson, C.M. Population of Nonnative States of Lysozyme Variants Drives Amyloid Fibril Formation. J. Am. Chem. Soc. 2011, 133, 7737–7743. [Google Scholar] [CrossRef] [Green Version]
- Beke, T.; Csizmadia, I.G.; Perczel, A. Theoretical Study on Tertiary Structural Elements of β-peptides: Nanotubes Formed from Parallel-Sheet-Derived Assemblies of β-Peptides. J. Am. Chem. Soc. 2006, 128, 5158–5167. [Google Scholar] [CrossRef] [PubMed]
- And, R.F.W.B.; Bayles, D. Properties of Atoms in Molecules: Group Additivity. J. Phys. Chem. A 2000, 104, 5579–5589. [Google Scholar] [CrossRef]
- Cances, E.; Mennucci, B.; Tomasi, J. A new integral equation formalism for the polarizable continuum model: Theoretical background and applications to isotropic and anisotropic dielectrics. J. Chem. Phys. 1997, 107, 3032–3041. [Google Scholar] [CrossRef]
- Stewart, J.J.P. Optimization of parameters for semiempirical methods VI: More modifications to the NDDO approximations and re-optimization of parameters. J. Mol. Model. 2012, 19, 1–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Řezáč, J.; Fanfrlik, J.; Salahub, D.; Hobza, P. Semiempirical Quantum Chemical PM6 Method Augmented by Dispersion and H-Bonding Correction Terms Reliably Describes Various Types of Noncovalent Complexes. J. Chem. Theory Comput. 2009, 5, 1749–1760. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Papp, D.; Szigyártó, I.C.; Nordén, B.; Perczel, A.; Beke-Somfai, T. Structural Water Stabilizes Protein Motifs in Liquid Protein Phase: The Folding Mechanism of Short β-Sheets Coupled to Phase Transition. Int. J. Mol. Sci. 2021, 22, 8595. https://doi.org/10.3390/ijms22168595
Papp D, Szigyártó IC, Nordén B, Perczel A, Beke-Somfai T. Structural Water Stabilizes Protein Motifs in Liquid Protein Phase: The Folding Mechanism of Short β-Sheets Coupled to Phase Transition. International Journal of Molecular Sciences. 2021; 22(16):8595. https://doi.org/10.3390/ijms22168595
Chicago/Turabian StylePapp, Dóra, Imola Csilla Szigyártó, Bengt Nordén, András Perczel, and Tamás Beke-Somfai. 2021. "Structural Water Stabilizes Protein Motifs in Liquid Protein Phase: The Folding Mechanism of Short β-Sheets Coupled to Phase Transition" International Journal of Molecular Sciences 22, no. 16: 8595. https://doi.org/10.3390/ijms22168595