
The Influence of Microbiome Dysbiosis and Bacterial Biofilms on Epidermal Barrier Function in Atopic Dermatitis—An Update


 ,
, {kind=link}
{kind=link}
Abstract
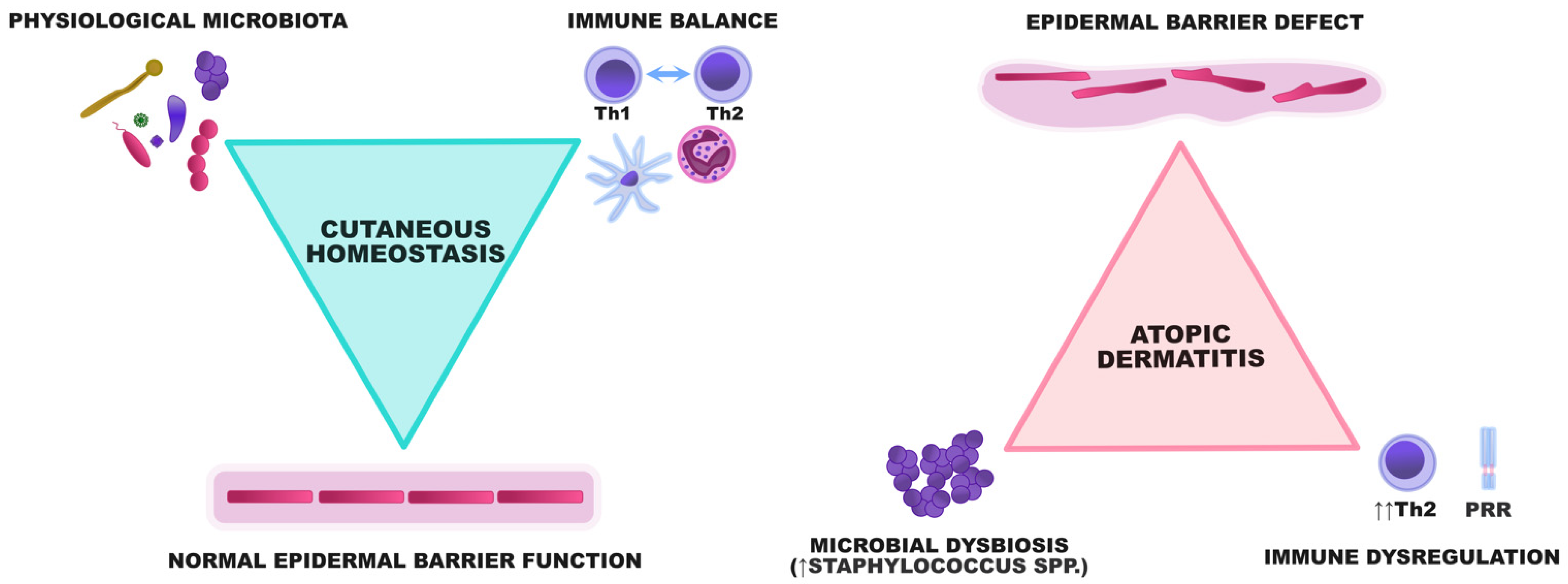
:1. Introduction
- An epidermal barrier defect;
- Immune dysregulation;
- Skin microbial dysbiosis;
- The itch/scratch cycle.
2. Materials and Methods
3. The Function and Structure of the Physiological Epidermal Barrier
3.1. Tight Junctions
3.2. Filaggrin
3.3. Endogenous Proteases
3.4. Epidermal Lipids
4. Immune Dysregulation in Atopic Dermatitis
5. The Skin Microbiome in Atopic Dermatitis
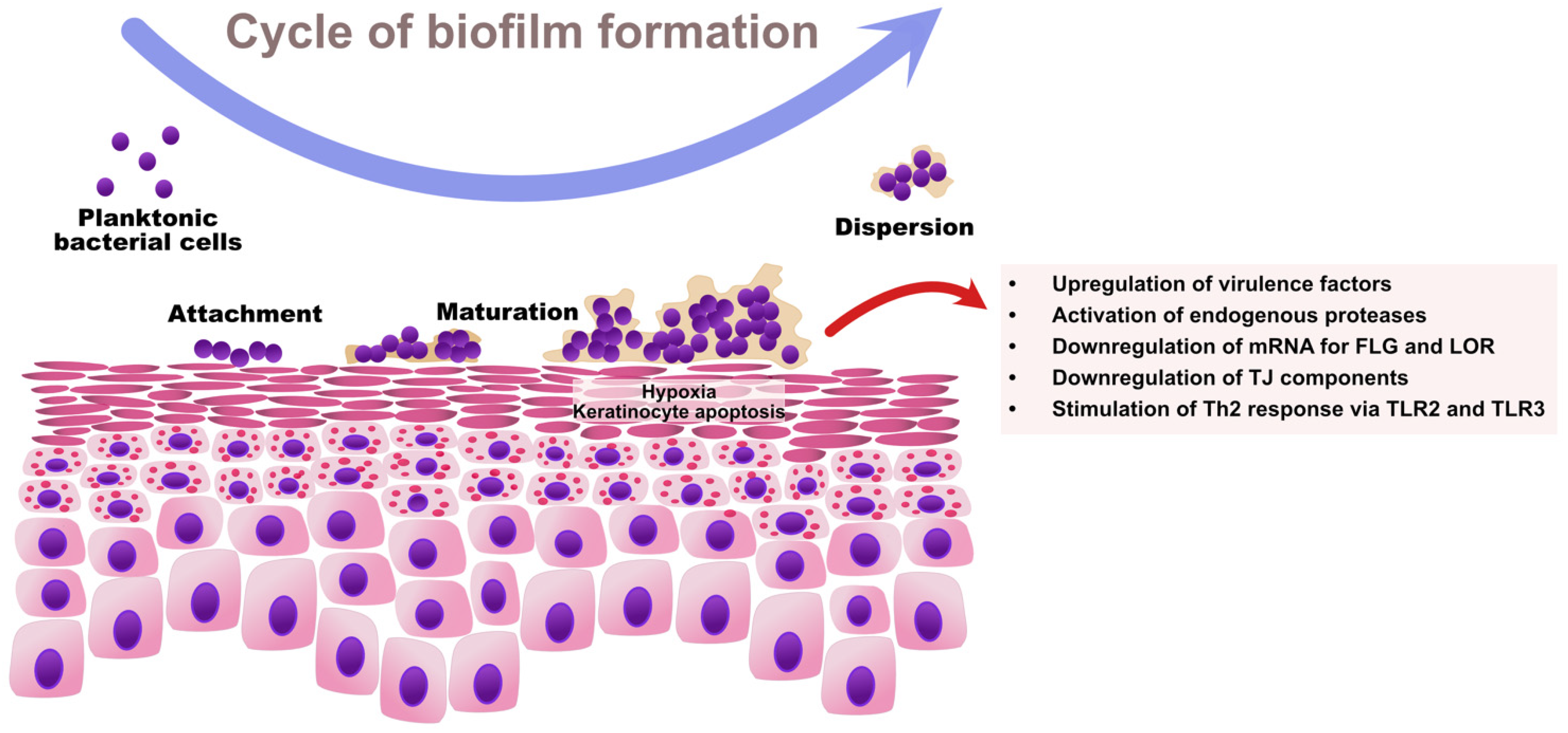
6. Biofilms
7. The Influence of Barrier Disruption on Skin Microbiome Dysbiosis
8. The Influence of Skin Microbiome Dysbiosis on the Skin Barrier
8.1. Effect on Tight Junctions
8.2. The Influence on Filaggrin and Epidermal Differentiation
8.3. Skin Barrier Disruption Mediated by Proteases
9. The Influence of Skin Microbiome Dysbiosis on Itch
10. The Effect of Bacterial Biofilms on the Skin Barrier in Atopic Dermatitis
11. Therapeutic Methods with the Potential of Restoring the Normal Skin Microbiota
12. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Wollenberg, A.; Barbarot, S.; Bieber, T.; Christen-Zaech, S.; Deleuran, M.; Fink-Wagner, A.; Gieler, U.; Girolomoni, G.; Lau, S.; Muraro, A.; et al. Consensus-Based European Guidelines for Treatment of Atopic Eczema (Atopic Dermatitis) in Adults and Children: Part I. J. Eur. Acad. Dermatol. Venereol. 2018, 32, 657–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langan, S.M.; Irvine, A.D.; Weidinger, S. Atopic Dermatitis. Lancet 2020, 396, 345–360. [Google Scholar] [CrossRef]
- Williams, H.C.; Burney, P.G.; Pembroke, A.C.; Hay, R.J. Validation of the U.K. Diagnostic Criteria for Atopic Dermatitis in a Population Setting. U.K. Diagnostic Criteria for Atopic Dermatitis Working Party. Br. J. Dermatol. 1996, 135, 12–17. [Google Scholar] [CrossRef]
- Rajka Georg, H.; Jon, M. Diagnostic Features of Atopic Dermatitis. Acta Derm. Venereol. 1980, 60, 44–47. [Google Scholar]
- Weidinger, S.; Beck, L.A.; Bieber, T.; Kabashima, K.; Irvine, A.D. Atopic Dermatitis. Nat. Rev. Dis. Primers 2018, 4, 1. [Google Scholar] [CrossRef] [PubMed]
- Nettis, E.; Ortoncelli, M.; Pellacani, G.; Foti, C.; Di Leo, E.; Patruno, C.; Rongioletti, F.; Argenziano, G.; Ferrucci, S.; Macchia, L.; et al. A Multicenter Study on the Prevalence of Clinical Patterns and Clinical Phenotypes in Adult Atopic Dermatitis. J. Investig. Allergol. Clin. Immunol. 2020, 30, 448–450. [Google Scholar] [CrossRef]
- Sacotte, R.; Silverberg, J.I. Epidemiology of Adult Atopic Dermatitis. Clin. Dermatol. 2018, 36, 595–605. [Google Scholar] [CrossRef]
- Odhiambo, J.A.; Williams, H.C.; Clayton, T.O.; Robertson, C.F.; Asher, M.I.; ISAAC Phase Three Study Group. Global Variations in Prevalence of Eczema Symptoms in Children from ISAAC Phase Three. J. Allergy Clin. Immunol. 2009, 124, 1251–1258. [Google Scholar] [CrossRef] [PubMed]
- David Boothe, W.; Tarbox, J.A.; Tarbox, M.B. Atopic Dermatitis: Pathophysiology. Adv. Exp. Med. Biol. 2017, 1027, 21–37. [Google Scholar] [CrossRef] [PubMed]
- Pavlis, J.; Yosipovitch, G. Management of Itch in Atopic Dermatitis. Am. J. Clin. Dermatol. 2018, 19, 319–332. [Google Scholar] [CrossRef] [PubMed]
- Leung, D.Y.M.; Berdyshev, E.; Goleva, E. Cutaneous Barrier Dysfunction in Allergic Diseases. J. Allergy Clin. Immunol. 2020, 145, 1485–1497. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Seok, J.K.; Kang, H.C.; Cho, Y.-Y.; Lee, H.S.; Lee, J.Y. Skin Barrier Abnormalities and Immune Dysfunction in Atopic Dermatitis. Int. J. Mol. Sci. 2020, 21, 2867. [Google Scholar] [CrossRef] [Green Version]
- Liang, Y.; Chang, C.; Lu, Q. The Genetics and Epigenetics of Atopic Dermatitis-Filaggrin and Other Polymorphisms. Clin. Rev. Allergy Immunol. 2016, 51, 315–328. [Google Scholar] [CrossRef] [PubMed]
- Egawa, G.; Kabashima, K. Multifactorial Skin Barrier Deficiency and Atopic Dermatitis: Essential Topics to Prevent the Atopic March. J. Allergy Clin. Immunol. 2016, 138, 350–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kishibe, M. Physiological and Pathological Roles of Kallikrein-Related Peptidases in the Epidermis. J. Dermatol. Sci. 2019, 95, 50–55. [Google Scholar] [CrossRef] [Green Version]
- Tokumasu, R.; Yamaga, K.; Yamazaki, Y.; Murota, H.; Suzuki, K.; Tamura, A.; Bando, K.; Furuta, Y.; Katayama, I.; Tsukita, S. Dose-Dependent Role of Claudin-1 in Vivo in Orchestrating Features of Atopic Dermatitis. Proc. Natl. Acad. Sci. USA 2016, 113, E4061–E4068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Benedetto, A.; Rafaels, N.M.; McGirt, L.Y.; Ivanov, A.I.; Georas, S.N.; Cheadle, C.; Berger, A.E.; Zhang, K.; Vidyasagar, S.; Yoshida, T.; et al. Tight Junction Defects in Patients with Atopic Dermatitis. J. Allergy Clin. Immunol. 2011, 127, 773–786. [Google Scholar] [CrossRef] [Green Version]
- Egawa, G.; Kabashima, K. Barrier Dysfunction in the Skin Allergy. Allergol. Int. 2018, 67, 3–11. [Google Scholar] [CrossRef]
- Lee, J.; Jang, A.; Seo, S.J.; Myung, S.C. Epigenetic Regulation of Filaggrin Gene Expression in Human Epidermal Keratinocytes. Ann. Dermatol. 2020, 32, 122. [Google Scholar] [CrossRef] [PubMed]
- Dréno, B.; Araviiskaia, E.; Berardesca, E.; Gontijo, G.; Sanchez Viera, M.; Xiang, L.F.; Martin, R.; Bieber, T. Microbiome in Healthy Skin, Update for Dermatologists. J. Eur. Acad. Dermatol. Venereol. 2016, 30, 2038–2047. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Nakamura, Y.; Núñez, G. Role of the Microbiota in Skin Immunity and Atopic Dermatitis. Allergol. Int. 2017, 66, 539–544. [Google Scholar] [CrossRef]
- Byrd, A.L.; Belkaid, Y.; Segre, J.A. The Human Skin Microbiome. Nat. Rev. Microbiol. 2018, 16, 143–155. [Google Scholar] [CrossRef]
- Kumar, A.; Alam, A.; Rani, M.; Ehtesham, N.Z.; Hasnain, S.E. Biofilms: Survival and Defense Strategy for Pathogens. Int. J. Med. Microbiol. 2017, 307, 481–489. [Google Scholar] [CrossRef]
- Dréno, B.; Pécastaings, S.; Corvec, S.; Veraldi, S.; Khammari, A.; Roques, C. Cutibacterium Acnes (Propionibacterium Acnes) and Acne Vulgaris: A Brief Look at the Latest Updates. J. Eur. Acad. Dermatol. Venereol. 2018, 32, 5–14. [Google Scholar] [CrossRef] [Green Version]
- Roy, S.; Elgharably, H.; Sinha, M.; Ganesh, K.; Chaney, S.; Mann, E.; Miller, C.; Khanna, S.; Bergdall, V.K.; Powell, H.M.; et al. Mixed-Species Biofilm Compromises Wound Healing by Disrupting Epidermal Barrier Function. J. Pathol. 2014, 233, 331–343. [Google Scholar] [CrossRef]
- Brandner, J.M.; Zorn-Kruppa, M.; Yoshida, T.; Moll, I.; Beck, L.A.; De Benedetto, A. Epidermal Tight Junctions in Health and Disease. Tissue Barriers 2015, 3, e974451. [Google Scholar] [CrossRef] [Green Version]
- Brettmann, E.A.; de Guzman Strong, C. Recent Evolution of the Human Skin Barrier. Exp. Dermatol. 2018, 27, 859–866. [Google Scholar] [CrossRef]
- He, H.; Bissonnette, R.; Wu, J.; Diaz, A.; Saint-Cyr Proulx, E.; Maari, C.; Jack, C.; Louis, M.; Estrada, Y.; Krueger, J.G.; et al. Tape Strips Detect Distinct Immune and Barrier Profiles in Atopic Dermatitis and Psoriasis. J. Allergy Clin. Immunol. 2021, 147, 199–212. [Google Scholar] [CrossRef] [PubMed]
- Murphrey, M.B.; Miao, J.H.; Zito, P.M. Histology, Stratum Corneum. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- de Benedetto, A. Tight Junctions in the Skin: Still a Lot to Learn. Br. J. Dermatol. 2021, 184, 388–389. [Google Scholar] [CrossRef]
- Wertz, P. Epidermal Lamellar Granules. Skin Pharmacol. Physiol. 2018, 31, 262–268. [Google Scholar] [CrossRef]
- Otani, T.; Furuse, M. Tight Junction Structure and Function Revisited. Trends Cell Biol. 2020, 30, 805–817. [Google Scholar] [CrossRef]
- Zihni, C.; Mills, C.; Matter, K.; Balda, M.S. Tight Junctions: From Simple Barriers to Multifunctional Molecular Gates. Nat. Rev. Mol. Cell Biol. 2016, 17, 564–580. [Google Scholar] [CrossRef]
- Zheng, M.; Sun, S.; Zhou, J.; Liu, M. Virulence Factors Impair Epithelial Junctions during Bacterial Infection. J. Clin. Lab. Anal. 2021, 35, e23627. [Google Scholar] [CrossRef]
- Bin, L.; Leung, D.Y.M. Genetic and Epigenetic Studies of Atopic Dermatitis. Allergy Asthma Clin. Immunol. 2016, 12, 52. [Google Scholar] [CrossRef] [Green Version]
- Freeman, S.C.; Sonthalia, S. Histology, Keratohyalin Granules. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Drislane, C.; Irvine, A.D. The Role of Filaggrin in Atopic Dermatitis and Allergic Disease. Ann. Allergy Asthma Immunol. 2020, 124, 36–43. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.-Y. Molecular Mechanism of Epidermal Barrier Dysfunction as Primary Abnormalities. Int. J. Mol. Sci. 2020, 21, 1194. [Google Scholar] [CrossRef] [Green Version]
- Leyvraz, C.; Charles, R.-P.; Rubera, I.; Guitard, M.; Rotman, S.; Breiden, B.; Sandhoff, K.; Hummler, E. The Epidermal Barrier Function Is Dependent on the Serine Protease CAP1/Prss8. J. Cell Biol. 2005, 170, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Thyssen, J.P.; Jakasa, I.; Riethmüller, C.; Schön, M.P.; Braun, A.; Haftek, M.; Fallon, P.G.; Wróblewski, J.; Jakubowski, H.; Eckhart, L.; et al. Filaggrin Expression and Processing Deficiencies Impair Corneocyte Surface Texture and Stiffness in Mice. J. Investig. Dermatol. 2020, 140, 615–623. [Google Scholar] [CrossRef] [PubMed]
- Donovan, M.; Salamito, M.; Thomas-Collignon, A.; Simonetti, L.; Desbouis, S.; Rain, J.-C.; Formstecher, E.; Bernard, D. Filaggrin and Filaggrin 2 Processing Are Linked Together through Skin Aspartic Acid Protease Activation. PLoS ONE 2020, 15, e0232679. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Lim, K.-M. Skin Barrier Dysfunction and Filaggrin. Arch. Pharm. Res. 2021, 44, 36–48. [Google Scholar] [CrossRef] [PubMed]
- Moch, M.; Schwarz, N.; Windoffer, R.; Leube, R.E. The Keratin-Desmosome Scaffold: Pivotal Role of Desmosomes for Keratin Network Morphogenesis. Cell. Mol. Life Sci. 2020, 77, 543–558. [Google Scholar] [CrossRef] [Green Version]
- Brown, S.J.; McLean, W.H.I. One Remarkable Molecule: Filaggrin. J. Investig. Dermatol. 2012, 132, 751–762. [Google Scholar] [CrossRef] [Green Version]
- Zeeuwen, P.L.J.M. Epidermal Differentiation: The Role of Proteases and Their Inhibitors. Eur. J. Cell Biol. 2004, 83, 761–773. [Google Scholar] [CrossRef]
- Martin, M.J.; Estravís, M.; García-Sánchez, A.; Dávila, I.; Isidoro-García, M.; Sanz, C. Genetics and Epigenetics of Atopic Dermatitis: An Updated Systematic Review. Genes 2020, 11, 442. [Google Scholar] [CrossRef] [Green Version]
- Cui, L.; Jia, Y.; Cheng, Z.-W.; Gao, Y.; Zhang, G.-L.; Li, J.-Y.; He, C.-F. Advancements in the Maintenance of Skin Barrier/Skin Lipid Composition and the Involvement of Metabolic Enzymes. J. Cosmet. Dermatol. 2016, 15, 549–558. [Google Scholar] [CrossRef]
- de Veer, S.J.; Furio, L.; Swedberg, J.E.; Munro, C.A.; Brattsand, M.; Clements, J.A.; Hovnanian, A.; Harris, J.M. Selective Substrates and Inhibitors for Kallikrein-Related Peptidase 7 (KLK7) Shed Light on KLK Proteolytic Activity in the Stratum Corneum. J. Investig. Dermatol. 2017, 137, 430–439. [Google Scholar] [CrossRef] [Green Version]
- Sarri, C.A.; Roussaki-Schulze, A.; Vasilopoulos, Y.; Zafiriou, E.; Patsatsi, A.; Stamatis, C.; Gidarokosta, P.; Sotiriadis, D.; Sarafidou, T.; Mamuris, Z. Netherton Syndrome: A Genotype-Phenotype Review. Mol. Diagn. Ther. 2017, 21, 137–152. [Google Scholar] [CrossRef]
- Proksch, E. PH in Nature, Humans and Skin. J. Dermatol. 2018, 45, 1044–1052. [Google Scholar] [CrossRef] [PubMed]
- Chng, K.R.; Tay, A.S.L.; Li, C.; Ng, A.H.Q.; Wang, J.; Suri, B.K.; Matta, S.A.; McGovern, N.; Janela, B.; Wong, X.F.C.C.; et al. Whole Metagenome Profiling Reveals Skin Microbiome-Dependent Susceptibility to Atopic Dermatitis Flare. Nat. Microbiol. 2016, 1, 16106. [Google Scholar] [CrossRef]
- Jang, H.; Matsuda, A.; Jung, K.; Karasawa, K.; Matsuda, K.; Oida, K.; Ishizaka, S.; Ahn, G.; Amagai, Y.; Moon, C.; et al. Skin PH Is the Master Switch of Kallikrein 5-Mediated Skin Barrier Destruction in a Murine Atopic Dermatitis Model. J. Investig. Dermatol. 2016, 136, 127–135. [Google Scholar] [CrossRef]
- van Smeden, J.; Bouwstra, J.A. Stratum Corneum Lipids: Their Role for the Skin Barrier Function in Healthy Subjects and Atopic Dermatitis Patients. Curr. Probl. Dermatol. 2016, 49, 8–26. [Google Scholar] [CrossRef]
- Bhattacharya, N.; Sato, W.J.; Kelly, A.; Ganguli-Indra, G.; Indra, A.K. Epidermal Lipids: Key Mediators of Atopic Dermatitis Pathogenesis. Trends Mol. Med. 2019, 25, 551–562. [Google Scholar] [CrossRef]
- Li, S.; Teegarden, A.; Bauer, E.M.; Choi, J.; Messaddeq, N.; Hendrix, D.A.; Ganguli-Indra, G.; Leid, M.; Indra, A.K. Transcription Factor CTIP1/ BCL11A Regulates Epidermal Differentiation and Lipid Metabolism During Skin Development. Sci. Rep. 2017, 7, 13427. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Kirkwood, J.S.; Taylor, A.W.; Stevens, J.F.; Leid, M.; Ganguli-Indra, G.; Indra, A.K. Transcription Factor Ctip2 Controls Epidermal Lipid Metabolism and Regulates Expression of Genes Involved in Sphingolipid Biosynthesis during Skin Development. J. Investig. Dermatol. 2013, 133, 668–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berdyshev, E.; Goleva, E.; Bronova, I.; Dyjack, N.; Rios, C.; Jung, J.; Taylor, P.; Jeong, M.; Hall, C.F.; Richers, B.N.; et al. Lipid Abnormalities in Atopic Skin Are Driven by Type 2 Cytokines. JCI Insight 2018, 3, 98006. [Google Scholar] [CrossRef] [Green Version]
- Harris, I.R.; Farrell, A.M.; Grunfeld, C.; Holleran, W.M.; Elias, P.M.; Feingold, K.R. Permeability Barrier Disruption Coordinately Regulates MRNA Levels for Key Enzymes of Cholesterol, Fatty Acid, and Ceramide Synthesis in the Epidermis. J. Investig. Dermatol. 1997, 109, 783–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheung Lam, A.H.; Sandoval, N.; Wadhwa, R.; Gilkes, J.; Do, T.Q.; Ernst, W.; Chiang, S.-M.; Kosina, S.; Xu, H.H.; Fujii, G.; et al. Assessment of Free Fatty Acids and Cholesteryl Esters Delivered in Liposomes as Novel Class of Antibiotic. BMC Res. Notes 2016, 9, 337. [Google Scholar] [CrossRef] [Green Version]
- Macheleidt, O.; Kaiser, H.W.; Sandhoff, K. Deficiency of Epidermal Protein-Bound Omega-Hydroxyceramides in Atopic Dermatitis. J. Investig. Dermatol. 2002, 119, 166–173. [Google Scholar] [CrossRef] [Green Version]
- Hato, T.; Dagher, P.C. How the Innate Immune System Senses Trouble and Causes Trouble. Clin. J. Am. Soc. Nephrol. 2015, 10, 1459–1469. [Google Scholar] [CrossRef] [Green Version]
- Herwald, H.; Egesten, A. On PAMPs and DAMPs. J. Innate Immun. 2016, 8, 427–428. [Google Scholar] [CrossRef]
- Kaesler, S.; Volz, T.; Skabytska, Y.; Köberle, M.; Hein, U.; Chen, K.-M.; Guenova, E.; Wölbing, F.; Röcken, M.; Biedermann, T. Toll-like Receptor 2 Ligands Promote Chronic Atopic Dermatitis through IL-4–Mediated Suppression of IL-10. J. Allergy Clin. Immunol. 2014, 134, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Weidinger, S.; Klopp, N.; Rummler, L.; Wagenpfeil, S.; Novak, N.; Baurecht, H.-J.; Groer, W.; Darsow, U.; Heinrich, J.; Gauger, A.; et al. Association of NOD1 Polymorphisms with Atopic Eczema and Related Phenotypes. J. Allergy Clin. Immunol. 2005, 116, 177–184. [Google Scholar] [CrossRef] [PubMed]
- De Benedetto, A.; Agnihothri, R.; McGirt, L.Y.; Bankova, L.G.; Beck, L.A. Atopic Dermatitis: A Disease Caused by Innate Immune Defects? J. Investig. Dermatol. 2009, 129, 14–30. [Google Scholar] [CrossRef] [Green Version]
- Czarnowicki, T.; He, H.; Krueger, J.G.; Guttman-Yassky, E. Atopic Dermatitis Endotypes and Implications for Targeted Therapeutics. J. Allergy Clin. Immunol. 2019, 143, 1–11. [Google Scholar] [CrossRef]
- Renert-Yuval, Y.; Guttman-Yassky, E. New Treatments for Atopic Dermatitis Targeting beyond IL-4/IL-13 Cytokines. Ann. Allergy Asthma Immunol. 2020, 124, 28–35. [Google Scholar] [CrossRef] [Green Version]
- Brunner, P.M.; Guttman-Yassky, E.; Leung, D.Y.M. The Immunology of Atopic Dermatitis and Its Reversibility with Broad-Spectrum and Targeted Therapies. J. Allergy Clin. Immunol. 2017, 139, S65–S76. [Google Scholar] [CrossRef] [Green Version]
- Oliva, M.; Renert-Yuval, Y.; Guttman-Yassky, E. The ‘Omics’ Revolution: Redefining the Understanding and Treatment of Allergic Skin Diseases. Curr. Opin. Allergy Clin. Immunol. 2016, 16, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Grice, E.A.; Kong, H.H.; Conlan, S.; Deming, C.B.; Davis, J.; Young, A.C.; NISC Comparative Sequencing Program; Bouffard, G.G.; Blakesley, R.W.; Murray, P.R.; et al. Topographical and Temporal Diversity of the Human Skin Microbiome. Science 2009, 324, 1190–1192. [Google Scholar] [CrossRef] [Green Version]
- Grice, E.A.; Segre, J.A. The Skin Microbiome. Nat. Rev. Microbiol. 2011, 9, 244–253. [Google Scholar] [CrossRef] [PubMed]
- Paller, A.S.; Kong, H.H.; Seed, P.; Naik, S.; Scharschmidt, T.C.; Gallo, R.L.; Luger, T.; Irvine, A.D. The Microbiome in Patients with Atopic Dermatitis. J. Allergy Clin. Immunol. 2019, 143, 26–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, H.H.; Oh, J.; Deming, C.; Conlan, S.; Grice, E.A.; Beatson, M.A.; Nomicos, E.; Polley, E.C.; Komarow, H.D.; NISC Comparative Sequence Program; et al. Temporal Shifts in the Skin Microbiome Associated with Disease Flares and Treatment in Children with Atopic Dermatitis. Genome Res. 2012, 22, 850–859. [Google Scholar] [CrossRef] [Green Version]
- Brown, M.M.; Horswill, A.R. Staphylococcus Epidermidis—Skin Friend or Foe? PLoS Pathog. 2020, 16, e1009026. [Google Scholar] [CrossRef]
- Cogen, A.L.; Yamasaki, K.; Sanchez, K.M.; Dorschner, R.A.; Lai, Y.; MacLeod, D.T.; Torpey, J.W.; Otto, M.; Nizet, V.; Kim, J.E.; et al. Selective Antimicrobial Action Is Provided by Phenol-Soluble Modulins Derived from Staphylococcus Epidermidis, a Normal Resident of the Skin. J. Investig. Dermatol. 2010, 130, 192–200. [Google Scholar] [CrossRef] [Green Version]
- Otto, M. Staphylococcus Colonization of the Skin and Antimicrobial Peptides. Expert Rev. Dermatol. 2010, 5, 183–195. [Google Scholar] [CrossRef] [Green Version]
- Hon, K.L.; Tsang, Y.C.K.; Pong, N.H.; Leung, T.F.; Ip, M. Exploring Staphylococcus Epidermidis in Atopic Eczema: Friend or Foe? Clin. Exp. Dermatol. 2016, 41, 659–663. [Google Scholar] [CrossRef] [PubMed]
- Totté, J.E.E.; Pardo, L.M.; Fieten, K.B.; Vos, M.C.; Broek, T.J.; Schuren, F.H.J.; Pasmans, S.G.M.A. Nasal and Skin Microbiomes Are Associated with Disease Severity in Paediatric Atopic Dermatitis. Br. J. Dermatol. 2019, 181, 796–804. [Google Scholar] [CrossRef] [PubMed]
- Loomis, K.H.; Wu, S.K.; Ernlund, A.; Zudock, K.; Reno, A.; Blount, K.; Karig, D.K. A Mixed Community of Skin Microbiome Representatives Influences Cutaneous Processes More than Individual Members. Microbiome 2021, 9, 22. [Google Scholar] [CrossRef]
- Byrd, A.L.; Deming, C.; Cassidy, S.K.B.; Harrison, O.J.; Ng, W.-I.; Conlan, S.; NISC Comparative Sequencing Program; Belkaid, Y.; Segre, J.A.; Kong, H.H. Staphylococcus aureus and Staphylococcus epidermidis Strain Diversity Underlying Pediatric Atopic Dermatitis. Sci. Transl. Med. 2017, 9, eaal4651. [Google Scholar] [CrossRef] [Green Version]
- Leung, D.Y.M.; Calatroni, A.; Zaramela, L.S.; LeBeau, P.K.; Dyjack, N.; Brar, K.; David, G.; Johnson, K.; Leung, S.; Ramirez-Gama, M.; et al. The Nonlesional Skin Surface Distinguishes Atopic Dermatitis with Food Allergy as a Unique Endotype. Sci. Transl. Med. 2019, 11, eaav2685. [Google Scholar] [CrossRef]
- Tauber, M.; Balica, S.; Hsu, C.-Y.; Jean-Decoster, C.; Lauze, C.; Redoules, D.; Viodé, C.; Schmitt, A.-M.; Serre, G.; Simon, M.; et al. Staphylococcus aureus Density on Lesional and Nonlesional Skin Is Strongly Associated with Disease Severity in Atopic Dermatitis. J. Allergy Clin. Immunol. 2016, 137, 1272–1274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blicharz, L.; Usarek, P.; Młynarczyk, G.; Skowroński, K.; Rudnicka, L.; Samochocki, Z. Nasal Colonization by Staphylococci and Severity of Atopic Dermatitis. Dermatitis 2020, 31, 215–222. [Google Scholar] [CrossRef]
- Flemming, H.-C.; Wingender, J.; Szewzyk, U.; Steinberg, P.; Rice, S.A.; Kjelleberg, S. Biofilms: An Emergent Form of Bacterial Life. Nat. Rev. Microbiol. 2016, 14, 563–575. [Google Scholar] [CrossRef] [PubMed]
- Rabin, N.; Zheng, Y.; Opoku-Temeng, C.; Du, Y.; Bonsu, E.; Sintim, H.O. Biofilm Formation Mechanisms and Targets for Developing Antibiofilm Agents. Future Med. Chem. 2015, 7, 493–512. [Google Scholar] [CrossRef]
- Koo, H.; Allan, R.N.; Howlin, R.P.; Stoodley, P.; Hall-Stoodley, L. Targeting Microbial Biofilms: Current and Prospective Therapeutic Strategies. Nat. Rev. Microbiol. 2017, 15, 740–755. [Google Scholar] [CrossRef]
- Yin, W.; Wang, Y.; Liu, L.; He, J. Biofilms: The Microbial “Protective Clothing” in Extreme Environments. Int. J. Mol. Sci. 2019, 20, 3423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukherjee, S.; Bassler, B.L. Bacterial Quorum Sensing in Complex and Dynamically Changing Environments. Nat. Rev. Microbiol. 2019, 17, 371–382. [Google Scholar] [CrossRef]
- Schilcher, K.; Horswill, A.R. Staphylococcal Biofilm Development: Structure, Regulation, and Treatment Strategies. Microbiol. Mol. Biol. Rev. 2020, 84, e00026-19. [Google Scholar] [CrossRef]
- Di Domenico, E.G.; Cavallo, I.; Capitanio, B.; Ascenzioni, F.; Pimpinelli, F.; Morrone, A.; Ensoli, F. Staphylococcus aureus and the Cutaneous Microbiota Biofilms in the Pathogenesis of Atopic Dermatitis. Microorganisms 2019, 7, 301. [Google Scholar] [CrossRef] [Green Version]
- Le, K.Y.; Dastgheyb, S.; Ho, T.V.; Otto, M. Molecular Determinants of Staphylococcal Biofilm Dispersal and Structuring. Front. Cell. Infect. Microbiol. 2014, 4, 167. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.O.; Lee, S.; Lee, J.E.; Song, K.-H.; Kang, C.K.; Wi, Y.M.; San-Juan, R.; López-Cortés, L.E.; Lacoma, A.; Prat, C.; et al. Dysfunctional Accessory Gene Regulator (Agr) as a Prognostic Factor in Invasive Staphylococcus aureus Infection: A Systematic Review and Meta-Analysis. Sci. Rep. 2020, 10, 20697. [Google Scholar] [CrossRef]
- Le, K.Y.; Otto, M. Quorum-Sensing Regulation in Staphylococci—an Overview. Front. Microbiol. 2015, 6, 1174. [Google Scholar] [CrossRef] [Green Version]
- Blicharz, L.; Michalak, M.; Szymanek-Majchrzak, K.; Młynarczyk, G.; Skowroński, K.; Rudnicka, L.; Samochocki, Z. The Propensity to Form Biofilm In Vitro by Staphylococcus aureus Strains Isolated from the Anterior Nares of Patients with Atopic Dermatitis: Clinical Associations. Dermatology 2021, 237, 528–534. [Google Scholar] [CrossRef]
- Foster, T.J.; Geoghegan, J.A.; Ganesh, V.K.; Höök, M. Adhesion, Invasion and Evasion: The Many Functions of the Surface Proteins of Staphylococcus aureus. Nat. Rev. Microbiol. 2014, 12, 49–62. [Google Scholar] [CrossRef] [Green Version]
- Cho, S.-H.; Strickland, I.; Boguniewicz, M.; Leung, D.Y.M. Fibronectin and Fibrinogen Contribute to the Enhanced Binding of Staphylococcus aureus to Atopic Skin. J. Allergy Clin. Immunol. 2001, 108, 269–274. [Google Scholar] [CrossRef]
- Clausen, M.-L.; Edslev, S.M.; Andersen, P.S.; Clemmensen, K.; Krogfelt, K.A.; Agner, T. Staphylococcus aureus Colonization in Atopic Eczema and Its Association with Filaggrin Gene Mutations. Br. J. Dermatol. 2017, 177, 1394–1400. [Google Scholar] [CrossRef] [PubMed]
- Cai, S.C.S.; Chen, H.; Koh, W.-P.; Common, J.E.A.; van Bever, H.P.; McLean, W.H.I.; Lane, E.B.; Giam, Y.C.; Tang, M.B.Y. Filaggrin Mutations Are Associated with Recurrent Skin Infection in Singaporean Chinese Patients with Atopic Dermatitis. Br. J. Dermatol. 2012, 166, 200–203. [Google Scholar] [CrossRef]
- Nakatsuji, T.; Chen, T.H.; Two, A.M.; Chun, K.A.; Narala, S.; Geha, R.S.; Hata, T.R.; Gallo, R.L. Staphylococcus aureus Exploits Epidermal Barrier Defects in Atopic Dermatitis to Trigger Cytokine Expression. J. Investig. Dermatol. 2016, 136, 2192–2200. [Google Scholar] [CrossRef] [Green Version]
- Simpson, E.L.; Villarreal, M.; Jepson, B.; Rafaels, N.; David, G.; Hanifin, J.; Taylor, P.; Boguniewicz, M.; Yoshida, T.; De Benedetto, A.; et al. Patients with Atopic Dermatitis Colonized with Staphylococcus aureus Have a Distinct Phenotype and Endotype. J. Investig. Dermatol. 2018, 138, 2224–2233. [Google Scholar] [CrossRef] [Green Version]
- Feuillie, C.; Vitry, P.; McAleer, M.A.; Kezic, S.; Irvine, A.D.; Geoghegan, J.A.; Dufrêne, Y.F. Adhesion of Staphylococcus aureus to Corneocytes from Atopic Dermatitis Patients Is Controlled by Natural Moisturizing Factor Levels. mBio 2018, 9, e01184-18. [Google Scholar] [CrossRef] [Green Version]
- Towell, A.M.; Feuillie, C.; Vitry, P.; Da Costa, T.M.; Mathelié-Guinlet, M.; Kezic, S.; Fleury, O.M.; McAleer, M.A.; Dufrêne, Y.F.; Irvine, A.D.; et al. Staphylococcus aureus Binds to the N-Terminal Region of Corneodesmosin to Adhere to the Stratum Corneum in Atopic Dermatitis. Proc. Natl. Acad. Sci. USA. 2021, 118, e2014444118. [Google Scholar] [CrossRef]
- Igawa, S.; Ohzono, A.; Pham, P.; Wang, Z.; Nakatsuji, T.; Dokoshi, T.; Di Nardo, A. Sphingosine 1-Phosphate Receptor 2 Is Central to Maintaining Epidermal Barrier Homeostasis. J. Investig. Dermatol. 2021, 141, 1188–1197. [Google Scholar] [CrossRef]
- Lipsky, Z.W.; Marques, C.N.H.; German, G.K. Lipid Depletion Enables Permeation of Staphylococcus aureus Bacteria through Human Stratum Corneum. Tissue Barriers 2020, 8, 1754706. [Google Scholar] [CrossRef]
- Baurecht, H.; Rühlemann, M.C.; Rodríguez, E.; Thielking, F.; Harder, I.; Erkens, A.-S.; Stölzl, D.; Ellinghaus, E.; Hotze, M.; Lieb, W.; et al. Epidermal Lipid Composition, Barrier Integrity, and Eczematous Inflammation Are Associated with Skin Microbiome Configuration. J. Allergy Clin. Immunol. 2018, 141, 1668–1676. [Google Scholar] [CrossRef] [Green Version]
- Cleary, J.M.; Lipsky, Z.W.; Kim, M.; Marques, C.N.H.; German, G.K. Heterogeneous Ceramide Distributions Alter Spatially Resolved Growth of Staphylococcus aureus on Human Stratum Corneum. J. R. Soc. Interface 2018, 15, 20170848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Villarreal, M.; Stewart, S.; Choi, J.; Ganguli-Indra, G.; Babineau, D.C.; Philpot, C.; David, G.; Yoshida, T.; Boguniewicz, M.; et al. Altered Composition of Epidermal Lipids Correlates with Staphylococcus aureus Colonization Status in Atopic Dermatitis. Br. J. Dermatol. 2017, 177, e125–e127. [Google Scholar] [CrossRef] [PubMed]
- Meisel, J.S.; Sfyroera, G.; Bartow-McKenney, C.; Gimblet, C.; Bugayev, J.; Horwinski, J.; Kim, B.; Brestoff, J.R.; Tyldsley, A.S.; Zheng, Q.; et al. Commensal Microbiota Modulate Gene Expression in the Skin. Microbiome 2018, 6, 20. [Google Scholar] [CrossRef]
- Ohnemus, U.; Kohrmeyer, K.; Houdek, P.; Rohde, H.; Wladykowski, E.; Vidal, S.; Horstkotte, M.A.; Aepfelbacher, M.; Kirschner, N.; Behne, M.J.; et al. Regulation of Epidermal Tight-Junctions (TJ) during Infection with Exfoliative Toxin-Negative Staphylococcus Strains. J. Investig. Dermatol. 2008, 128, 906–916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bäsler, K.; Galliano, M.-F.; Bergmann, S.; Rohde, H.; Wladykowski, E.; Vidal-y-Sy, S.; Guiraud, B.; Houdek, P.; Schüring, G.; Volksdorf, T.; et al. Biphasic Influence of Staphylococcus aureus on Human Epidermal Tight Junctions. Ann. N. Y. Acad. Sci. 2017, 1405, 53–70. [Google Scholar] [CrossRef]
- Wang, B.; McHugh, B.J.; Qureshi, A.; Campopiano, D.J.; Clarke, D.J.; Fitzgerald, J.R.; Dorin, J.R.; Weller, R.; Davidson, D.J. IL-1β-Induced Protection of Keratinocytes against Staphylococcus aureus-Secreted Proteases Is Mediated by Human β-Defensin 2. J. Investig. Dermatol. 2017, 137, 95–105. [Google Scholar] [CrossRef] [Green Version]
- Kuo, I.-H.; Carpenter-Mendini, A.; Yoshida, T.; McGirt, L.Y.; Ivanov, A.I.; Barnes, K.C.; Gallo, R.L.; Borkowski, A.W.; Yamasaki, K.; Leung, D.Y.; et al. Activation of Epidermal Toll-like Receptor 2 Enhances Tight Junction Function: Implications for Atopic Dermatitis and Skin Barrier Repair. J. Investig. Dermatol. 2013, 133, 988–998. [Google Scholar] [CrossRef] [Green Version]
- Martens, K.; Seys, S.F.; Alpizar, Y.A.; Schrijvers, R.; Bullens, D.M.A.; Breynaert, C.; Lebeer, S.; Steelant, B. Staphylococcus aureus Enterotoxin B Disrupts Nasal Epithelial Barrier Integrity. Clin. Exp. Allergy 2021, 51, 87–98. [Google Scholar] [CrossRef]
- Mitamura, Y.; Nunomura, S.; Nanri, Y.; Ogawa, M.; Yoshihara, T.; Masuoka, M.; Tsuji, G.; Nakahara, T.; Hashimoto-Hachiya, A.; Conway, S.J.; et al. The IL-13/Periostin/IL-24 Pathway Causes Epidermal Barrier Dysfunction in Allergic Skin Inflammation. Allergy 2018, 73, 1881–1891. [Google Scholar] [CrossRef] [PubMed]
- Ryu, W.-I.; Lee, H.; Bae, H.C.; Ryu, H.J.; Son, S.W. IL-33 down-Regulates Filaggrin Expression by Inducing STAT3 and ERK Phosphorylation in Human Keratinocytes. J. Dermatol. Sci. 2016, 82, 131–134. [Google Scholar] [CrossRef]
- Imai, Y. Interleukin-33 in Atopic Dermatitis. J. Dermatol. Sci. 2019, 96, 2–7. [Google Scholar] [CrossRef] [Green Version]
- Park, K.-D.; Pak, S.; Park, K.-K. The Pathogenetic Effect of Natural and Bacterial Toxins on Atopic Dermatitis. Toxins 2016, 9, 3. [Google Scholar] [CrossRef] [Green Version]
- Nygaard, U.; van den Bogaard, E.H.; Niehues, H.; Hvid, M.; Deleuran, M.; Johansen, C.; Vestergaard, C. The “Alarmins” HMBG1 and IL-33 Downregulate Structural Skin Barrier Proteins and Impair Epidermal Growth. Acta Derm.-Venereol. 2017, 97, 305–312. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.S.; Choi, J.K.; Jung, H.J.; Park, K.H.; Jang, Y.H.; Lee, W.J.; Lee, S.-J.; Kim, S.-H.; Kang, H.Y.; Kim, J.M.; et al. Effects of Topical Application of a Recombinant Staphylococcal Enterotoxin A on DNCB and Dust Mite Extract-Induced Atopic Dermatitis-like Lesions in a Murine Model. Eur. J. Dermatol. EJD 2014, 24, 186–193. [Google Scholar] [CrossRef]
- Al Kindi, A.; Williams, H.; Matsuda, K.; Alkahtani, A.M.; Saville, C.; Bennett, H.; Alshammari, Y.; Tan, S.Y.; O’Neill, C.; Tanaka, A.; et al. Staphylococcus aureus Second Immunoglobulin-Binding Protein Drives Atopic Dermatitis via IL-33. J. Allergy Clin. Immunol. 2021, 147, 1354–1368. [Google Scholar] [CrossRef]
- Jeon, Y.J.; Gil, C.H.; Won, J.; Jo, A.; Kim, H.J. Symbiotic Microbiome Staphylococcus aureus from Human Nasal Mucus Modulates IL-33-Mediated Type 2 Immune Responses in Allergic Nasal Mucosa. BMC Microbiol. 2020, 20, 301. [Google Scholar] [CrossRef]
- Brauweiler, A.M.; Leung, D.Y.M.; Goleva, E. The Transcription Factor P63 Is a Direct Effector of IL-4- and IL-13-Mediated Repression of Keratinocyte Differentiation. J. Investig. Dermatol. 2021, 141, 770–778. [Google Scholar] [CrossRef]
- Brauweiler, A.M.; Goleva, E.; Leung, D.Y.M. Staphylococcus aureus Lipoteichoic Acid Damages the Skin Barrier through an IL-1-Mediated Pathway. J. Investig. Dermatol. 2019, 139, 1753–1761. [Google Scholar] [CrossRef]
- Simon, M. Effects of Environmental Skin Stressors on Filaggrin Degradation Products: Importance for Eczema. Br. J. Dermatol. 2018, 179, 560–561. [Google Scholar] [CrossRef]
- Son, E.D.; Kim, H.-J.; Park, T.; Shin, K.; Bae, I.-H.; Lim, K.-M.; Cho, E.-G.; Lee, T.R. Staphylococcus aureus Inhibits Terminal Differentiation of Normal Human Keratinocytes by Stimulating Interleukin-6 Secretion. J. Dermatol. Sci. 2014, 74, 64–71. [Google Scholar] [CrossRef]
- Williams, M.R.; Nakatsuji, T.; Sanford, J.A.; Vrbanac, A.F.; Gallo, R.L. Staphylococcus aureus Induces Increased Serine Protease Activity in Keratinocytes. J. Investig. Dermatol. 2017, 137, 377–384. [Google Scholar] [CrossRef] [Green Version]
- Williams, M.R.; Costa, S.K.; Zaramela, L.S.; Khalil, S.; Todd, D.A.; Winter, H.L.; Sanford, J.A.; O’Neill, A.M.; Liggins, M.C.; Nakatsuji, T.; et al. Quorum Sensing between Bacterial Species on the Skin Protects against Epidermal Injury in Atopic Dermatitis. Sci. Transl. Med. 2019, 11, eaat8329. [Google Scholar] [CrossRef] [PubMed]
- Elmwall, J.; Kwiecinski, J.; Na, M.; Ali, A.A.; Osla, V.; Shaw, L.N.; Wang, W.; Sävman, K.; Josefsson, E.; Bylund, J.; et al. Galectin-3 Is a Target for Proteases Involved in the Virulence of Staphylococcus aureus. Infect. Immun. 2017, 85, e00177-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cau, L.; Williams, M.R.; Butcher, A.M.; Nakatsuji, T.; Kavanaugh, J.S.; Cheng, J.Y.; Shafiq, F.; Higbee, K.; Hata, T.R.; Horswill, A.R.; et al. Staphylococcus Epidermidis Protease EcpA Can Be a Deleterious Component of the Skin Microbiome in Atopic Dermatitis. J. Allergy Clin. Immunol. 2021, 147, 955–966. [Google Scholar] [CrossRef] [PubMed]
- Ridder, M.J.; Daly, S.M.; Triplett, K.D.; Seawell, N.A.; Hall, P.R.; Bose, J.L. Staphylococcus aureus Fatty Acid Kinase FakA Modulates Pathogenesis during Skin Infection via Proteases. Infect. Immun. 2020, 88, e00163-20. [Google Scholar] [CrossRef]
- Pietrocola, G.; Nobile, G.; Rindi, S.; Speziale, P. Staphylococcus aureus Manipulates Innate Immunity through Own and Host-Expressed Proteases. Front. Cell. Infect. Microbiol. 2017, 7, 166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.S.; Yosipovitch, G. The Skin Microbiota and Itch: Is There a Link? J. Clin. Med. 2020, 9, 1190. [Google Scholar] [CrossRef]
- Cevikbas, F.; Wang, X.; Akiyama, T.; Kempkes, C.; Savinko, T.; Antal, A.; Kukova, G.; Buhl, T.; Ikoma, A.; Buddenkotte, J.; et al. A Sensory Neuron-Expressed IL-31 Receptor Mediates T Helper Cell-Dependent Itch: Involvement of TRPV1 and TRPA1. J. Allergy Clin. Immunol. 2014, 133, 448–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buhl, T.; Ikoma, A.; Kempkes, C.; Cevikbas, F.; Sulk, M.; Buddenkotte, J.; Akiyama, T.; Crumrine, D.; Camerer, E.; Carstens, E.; et al. Protease-Activated Receptor-2 Regulates Neuro-Epidermal Communication in Atopic Dermatitis. Front. Immunol. 2020, 11, 1740. [Google Scholar] [CrossRef]
- Kempkes, C.; Buddenkotte, J.; Cevikbas, F.; Buhl, T.; Steinhoff, M. Role of PAR-2 in Neuroimmune Communication and Itch. In Itch: Mechanisms and Treatment; Carstens, E., Akiyama, T., Eds.; Frontiers in Neuroscience; CRC Press/Taylor & Francis: Boca Raton, FL, USA, 2014; ISBN 978-1-4665-0543-8. [Google Scholar]
- Williams, M.R.; Nakatsuji, T.; Gallo, R.L. Staphylococcus aureus: Master Manipulator of the Skin. Cell Host Microbe 2017, 22, 579–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, Y.; Oscherwitz, J.; Cease, K.B.; Chan, S.M.; Muñoz-Planillo, R.; Hasegawa, M.; Villaruz, A.E.; Cheung, G.Y.C.; McGavin, M.J.; Travers, J.B.; et al. Staphylococcus δ-Toxin Induces Allergic Skin Disease by Activating Mast Cells. Nature 2013, 503, 397–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiu, I.M.; Heesters, B.A.; Ghasemlou, N.; Von Hehn, C.A.; Zhao, F.; Tran, J.; Wainger, B.; Strominger, A.; Muralidharan, S.; Horswill, A.R.; et al. Bacteria Activate Sensory Neurons That Modulate Pain and Inflammation. Nature 2013, 501, 52–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evers, A.W.M.; Schut, C.; Gieler, U.; Spillekom-van Koulil, S.; van Beugen, S. Itch Management: Psychotherapeutic Approach. Curr. Probl. Dermatol. 2016, 50, 64–70. [Google Scholar] [CrossRef]
- Mijouin, L.; Hillion, M.; Ramdani, Y.; Jaouen, T.; Duclairoir-Poc, C.; Follet-Gueye, M.-L.; Lati, E.; Yvergnaux, F.; Driouich, A.; Lefeuvre, L.; et al. Effects of a Skin Neuropeptide (Substance p) on Cutaneous Microflora. PLoS ONE 2013, 8, e78773. [Google Scholar] [CrossRef]
- N’Diaye, A.; Gannesen, A.; Borrel, V.; Maillot, O.; Enaut, J.; Racine, P.-J.; Plakunov, V.; Chevalier, S.; Lesouhaitier, O.; Feuilloley, M.G.J. Substance P and Calcitonin Gene-Related Peptide: Key Regulators of Cutaneous Microbiota Homeostasis. Front. Endocrinol. 2017, 8, 15. [Google Scholar] [CrossRef] [Green Version]
- Park, K.-H.; Greenwood-Quaintance, K.E.; Uhl, J.R.; Cunningham, S.A.; Chia, N.; Jeraldo, P.R.; Sampathkumar, P.; Nelson, H.; Patel, R. Molecular Epidemiology of Staphylococcus aureus Bacteremia in a Single Large Minnesota Medical Center in 2015 as Assessed Using MLST, Core Genome MLST and Spa Typing. PLoS ONE 2017, 12, e0179003. [Google Scholar] [CrossRef]
- Harkins, C.P.; Pettigrew, K.A.; Oravcová, K.; Gardner, J.; Hearn, R.M.R.; Rice, D.; Mather, A.E.; Parkhill, J.; Brown, S.J.; Proby, C.M.; et al. The Microevolution and Epidemiology of Staphylococcus aureus Colonization during Atopic Eczema Disease Flare. J. Investig. Dermatol. 2018, 138, 336–343. [Google Scholar] [CrossRef] [Green Version]
- Fleury, O.M.; McAleer, M.A.; Feuillie, C.; Formosa-Dague, C.; Sansevere, E.; Bennett, D.E.; Towell, A.M.; McLean, W.H.I.; Kezic, S.; Robinson, D.A.; et al. Clumping Factor B Promotes Adherence of Staphylococcus aureus to Corneocytes in Atopic Dermatitis. Infect. Immun. 2017, 85, e00994-16. [Google Scholar] [CrossRef] [Green Version]
- Lone, A.G.; Atci, E.; Renslow, R.; Beyenal, H.; Noh, S.; Fransson, B.; Abu-Lail, N.; Park, J.-J.; Gang, D.R.; Call, D.R. Staphylococcus aureus Induces Hypoxia and Cellular Damage in Porcine Dermal Explants. Infect. Immun. 2015, 83, 2531–2541. [Google Scholar] [CrossRef] [Green Version]
- Di Domenico, E.G.; Cavallo, I.; Bordignon, V.; Prignano, G.; Sperduti, I.; Gurtner, A.; Trento, E.; Toma, L.; Pimpinelli, F.; Capitanio, B.; et al. Inflammatory Cytokines and Biofilm Production Sustain Staphylococcus aureus Outgrowth and Persistence: A Pivotal Interplay in the Pathogenesis of Atopic Dermatitis. Sci. Rep. 2018, 8, 9573. [Google Scholar] [CrossRef]
- Tankersley, A.; Frank, M.B.; Bebak, M.; Brennan, R. Early Effects of Staphylococcus aureus Biofilm Secreted Products on Inflammatory Responses of Human Epithelial Keratinocytes. J. Inflamm. Lond. Engl. 2014, 11, 17. [Google Scholar] [CrossRef] [Green Version]
- Allen, H.B.; Vaze, N.D.; Choi, C.; Hailu, T.; Tulbert, B.H.; Cusack, C.A.; Joshi, S.G. The Presence and Impact of Biofilm-Producing Staphylococci in Atopic Dermatitis. JAMA Dermatol. 2014, 150, 260–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, X.Q.; Tang, Y.; Ju, Y.; Zhang, X.Y.; Yan, J.J.; Wang, C.M.; Yang, Y.; Zhu, C.; Tang, Z.X.; Zhou, Y.; et al. Scratching Damages Tight Junctions through the Akt–Claudin 1 Axis in Atopic Dermatitis. Clin. Exp. Dermatol. 2021, 46, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Watters, C.; Fleming, D.; Bishop, D.; Rumbaugh, K.P. Host Responses to Biofilm. Prog. Mol. Biol. Transl. Sci. 2016, 142, 193–239. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, T.; Stevens, M.L.; Baatyrbek Kyzy, A.; Alarcon, R.; He, H.; Kroner, J.W.; Spagna, D.; Grashel, B.; Sidler, E.; Martin, L.J.; et al. Biofilm Propensity of Staphylococcus aureus Skin Isolates Is Associated with Increased Atopic Dermatitis Severity and Barrier Dysfunction in the MPAACH Pediatric Cohort. Allergy 2021, 76, 302–313. [Google Scholar] [CrossRef]
- Sonesson, A.; Przybyszewska, K.; Eriksson, S.; Mörgelin, M.; Kjellström, S.; Davies, J.; Potempa, J.; Schmidtchen, A. Identification of Bacterial Biofilm and the Staphylococcus aureus Derived Protease, Staphopain, on the Skin Surface of Patients with Atopic Dermatitis. Sci. Rep. 2017, 7, 8689. [Google Scholar] [CrossRef]
- Takai, T. TSLP Expression: Cellular Sources, Triggers, and Regulatory Mechanisms. Allergol. Int. Off. J. Jpn. Soc. Allergol. 2012, 61, 3–17. [Google Scholar] [CrossRef] [Green Version]
- Panchatcharam, B.S.; Cooksley, C.M.; Ramezanpour, M.; Vediappan, R.S.; Bassiouni, A.; Wormald, P.J.; Psaltis, A.J.; Vreugde, S. Staphylococcus aureus Biofilm Exoproteins Are Cytotoxic to Human Nasal Epithelial Barrier in Chronic Rhinosinusitis. Int. Forum Allergy Rhinol. 2020, 10, 871–883. [Google Scholar] [CrossRef]
- Błażewicz, I.; Jaśkiewicz, M.; Piechowicz, L.; Neubauer, D.; Nowicki, R.J.; Kamysz, W.; Barańska-Rybak, W. Activity of Antimicrobial Peptides and Conventional Antibiotics against Superantigen Positive Staphylococcus aureus Isolated from Patients with Atopic Dermatitis. Postepy Dermatol. Alergol. 2018, 35, 74–82. [Google Scholar] [CrossRef] [Green Version]
- Harkins, C.P.; McAleer, M.A.; Bennett, D.; McHugh, M.; Fleury, O.M.; Pettigrew, K.A.; Oravcová, K.; Parkhill, J.; Proby, C.M.; Dawe, R.S.; et al. The Widespread Use of Topical Antimicrobials Enriches for Resistance in Staphylococcus aureus Isolated from Patients with Atopic Dermatitis. Br. J. Dermatol. 2018, 179, 951–958. [Google Scholar] [CrossRef] [Green Version]
- Majewski, S.; Bhattacharya, T.; Asztalos, M.; Bohaty, B.; Durham, K.C.; West, D.P.; Hebert, A.A.; Paller, A.S. Sodium Hypochlorite Body Wash in the Management of Staphylococcus aureus–Colonized Moderate-to-severe Atopic Dermatitis in Infants, Children, and Adolescents. Pediatr. Dermatol. 2019, 36, 442–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maarouf, M.; Shi, V.Y. Bleach for Atopic Dermatitis. Dermatitis 2018, 29, 120–126. [Google Scholar] [CrossRef]
- Eriksson, S.; van der Plas, M.J.A.; Mörgelin, M.; Sonesson, A. Antibacterial and Antibiofilm Effects of Sodium Hypochlorite against Staphylococcus aureus Isolates Derived from Patients with Atopic Dermatitis. Br. J. Dermatol. 2017, 177, 513–521. [Google Scholar] [CrossRef]
- Nakatsuji, T.; Chen, T.H.; Narala, S.; Chun, K.A.; Two, A.M.; Yun, T.; Shafiq, F.; Kotol, P.F.; Bouslimani, A.; Melnik, A.V.; et al. Antimicrobials from Human Skin Commensal Bacteria Protect against Staphylococcus aureus and Are Deficient in Atopic Dermatitis. Sci. Transl. Med. 2017, 9, eaah4680. [Google Scholar] [CrossRef] [Green Version]
- Myles, I.A.; Earland, N.J.; Anderson, E.D.; Moore, I.N.; Kieh, M.D.; Williams, K.W.; Saleem, A.; Fontecilla, N.M.; Welch, P.A.; Darnell, D.A.; et al. First-in-Human Topical Microbiome Transplantation with Roseomonas Mucosa for Atopic Dermatitis. JCI Insight 2018, 3, 120608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimamori, Y.; Mitsunaka, S.; Yamashita, H.; Suzuki, T.; Kitao, T.; Kubori, T.; Nagai, H.; Takeda, S.; Ando, H. Staphylococcal Phage in Combination with Staphylococcus Epidermidis as a Potential Treatment for Staphylococcus aureus-Associated Atopic Dermatitis and Suppressor of Phage-Resistant Mutants. Viruses 2020, 13, 7. [Google Scholar] [CrossRef] [PubMed]
- Totté, J.; de Wit, J.; Pardo, L.; Schuren, F.; van Doorn, M.; Pasmans, S. Targeted Anti-Staphylococcal Therapy with Endolysins in Atopic Dermatitis and the Effect on Steroid Use, Disease Severity and the Microbiome: Study Protocol for a Randomized Controlled Trial (MAAS Trial). Trials 2017, 18, 404. [Google Scholar] [CrossRef] [Green Version]
- Dawgul, M.; Baranska-Rybak, W.; Piechowicz, L.; Bauer, M.; Neubauer, D.; Nowicki, R.; Kamysz, W. The Antistaphylococcal Activity of Citropin 1.1 and Temporin A against Planktonic Cells and Biofilms Formed by Isolates from Patients with Atopic Dermatitis: An Assessment of Their Potential to Induce Microbial Resistance Compared to Conventional Antimicrobials. Pharmaceuticals 2016, 9, 30. [Google Scholar] [CrossRef]
- Niemeyer-van der Kolk, T.; van der Wall, H.; Hogendoorn, G.K.; Rijneveld, R.; Luijten, S.; van Alewijk, D.C.J.G.; van den Munckhof, E.H.A.; de Kam, M.L.; Feiss, G.L.; Prens, E.P.; et al. Pharmacodynamic Effects of Topical Omiganan in Patients With Mild to Moderate Atopic Dermatitis in a Randomized, Placebo-Controlled, Phase II Trial. Clin. Transl. Sci. 2020, 13, 994–1003. [Google Scholar] [CrossRef]
- Olesen, C.M.; Ingham, A.C.; Thomsen, S.F.; Clausen, M.-L.; Andersen, P.S.; Edslev, S.M.; Yüksel, Y.T.; Guttman-Yassky, E.; Agner, T. Changes in Skin and Nasal Microbiome and Staphylococcal Species Following Treatment of Atopic Dermatitis with Dupilumab. Microorganisms 2021, 9, 1487. [Google Scholar] [CrossRef]
- Lossius, A.H.; Sundnes, O.; Ingham, A.C.; Edslev, S.M.; Bjørnholt, J.V.; Lilje, B.; Bradley, M.; Asad, S.; Haraldsen, G.; Skytt-Andersen, P.; et al. Shifts in the Skin Microbiota after UVB Treatment in Adult Atopic Dermatitis. Dermatology 2021, 1–12. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blicharz, L.; Rudnicka, L.; Czuwara, J.; Waśkiel-Burnat, A.; Goldust, M.; Olszewska, M.; Samochocki, Z. The Influence of Microbiome Dysbiosis and Bacterial Biofilms on Epidermal Barrier Function in Atopic Dermatitis—An Update. Int. J. Mol. Sci. 2021, 22, 8403. https://doi.org/10.3390/ijms22168403
Blicharz L, Rudnicka L, Czuwara J, Waśkiel-Burnat A, Goldust M, Olszewska M, Samochocki Z. The Influence of Microbiome Dysbiosis and Bacterial Biofilms on Epidermal Barrier Function in Atopic Dermatitis—An Update. International Journal of Molecular Sciences. 2021; 22(16):8403. https://doi.org/10.3390/ijms22168403
Chicago/Turabian StyleBlicharz, Leszek, Lidia Rudnicka, Joanna Czuwara, Anna Waśkiel-Burnat, Mohamad Goldust, Małgorzata Olszewska, and Zbigniew Samochocki. 2021. "The Influence of Microbiome Dysbiosis and Bacterial Biofilms on Epidermal Barrier Function in Atopic Dermatitis—An Update" International Journal of Molecular Sciences 22, no. 16: 8403. https://doi.org/10.3390/ijms22168403