
Genome-Wide Association Study in Rice Revealed a Novel Gene in Determining Plant Height and Stem Development, by Encoding a WRKY Transcription Factor

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
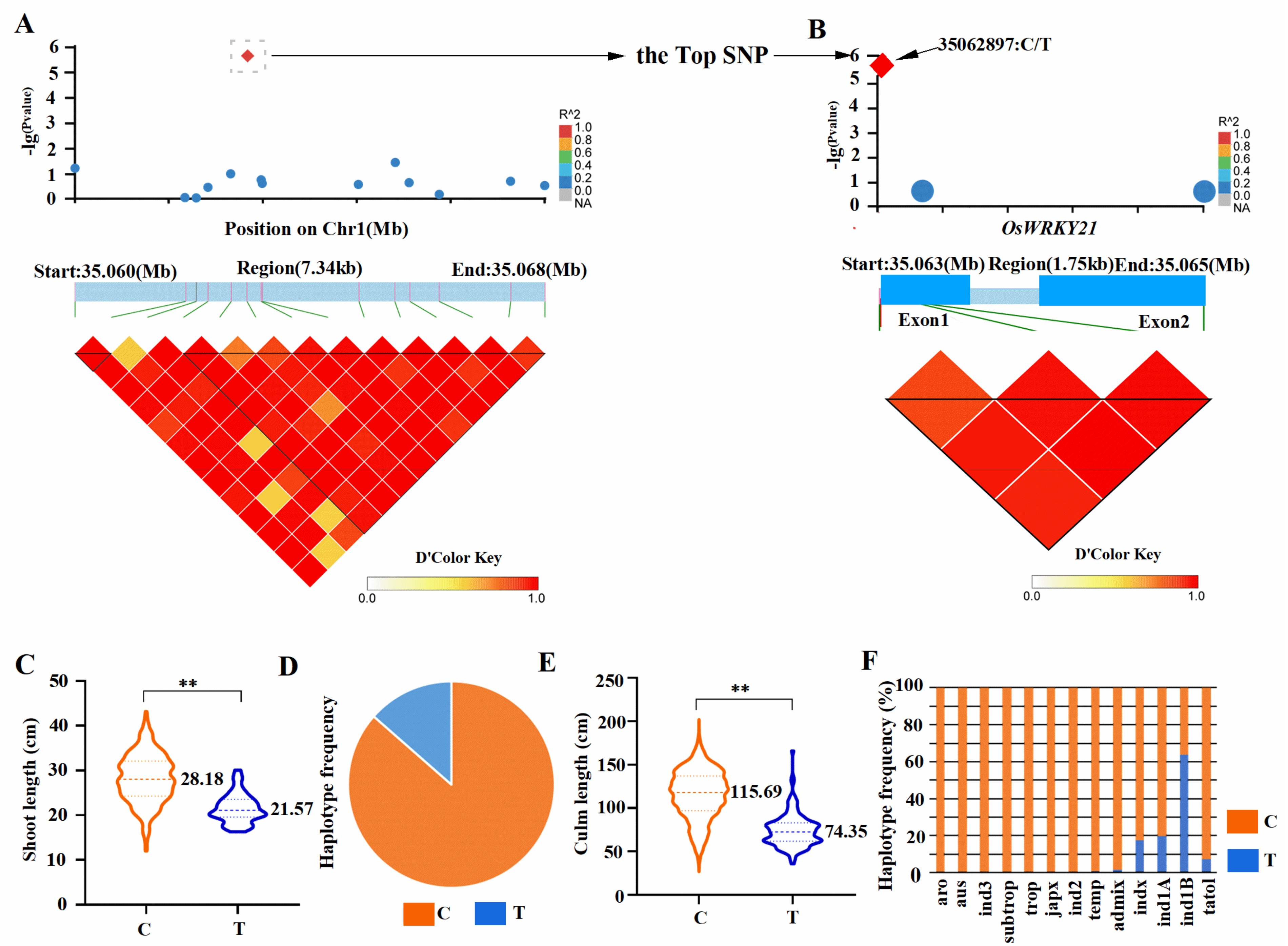
2.1. Genome-Wide Association Study (GWAS) Identified the OsWRKY21 as a Candidate Gene for Plant Height in Rice
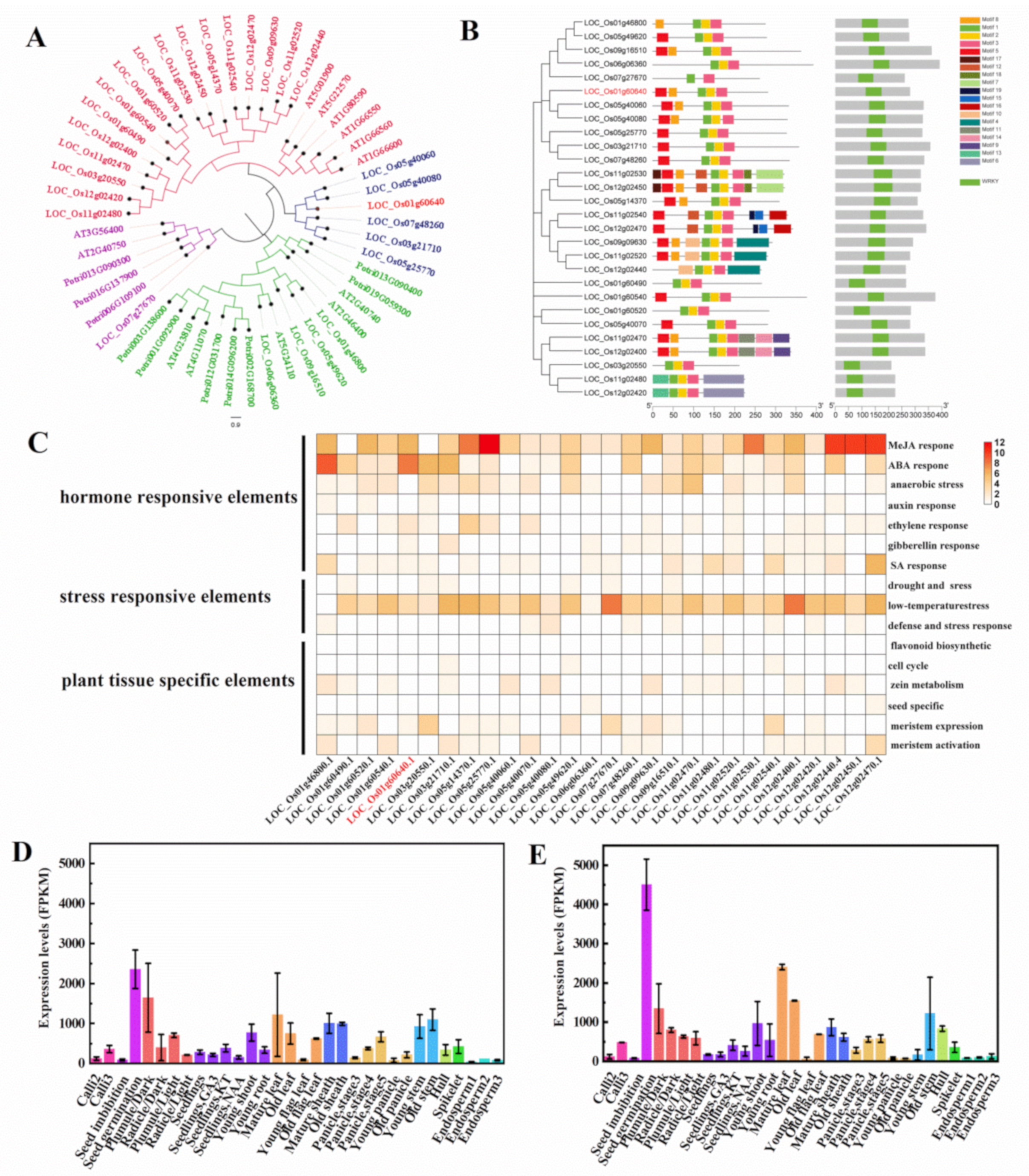
2.2. Bioinformatics and Expression Analysis of OsWRKY21 during the Development of Rice
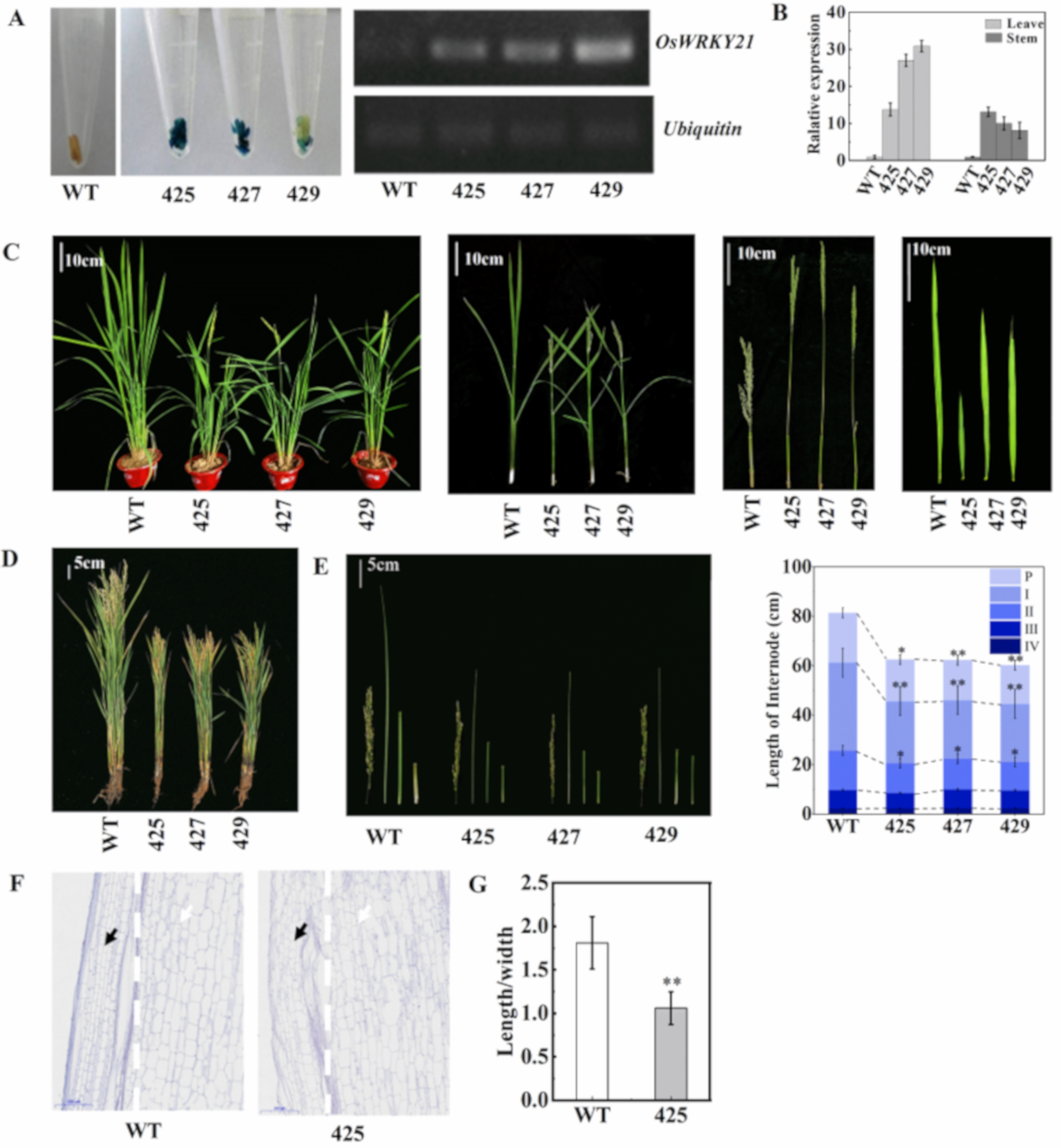
2.3. OsWRKY21 Overexpression Resulted in Semi-Dwarf Phenotype While the Knockout Mutant Plants by CRISPR/Cas9 Technology Yielded the Opposite, Which Confirms the GWAS Results
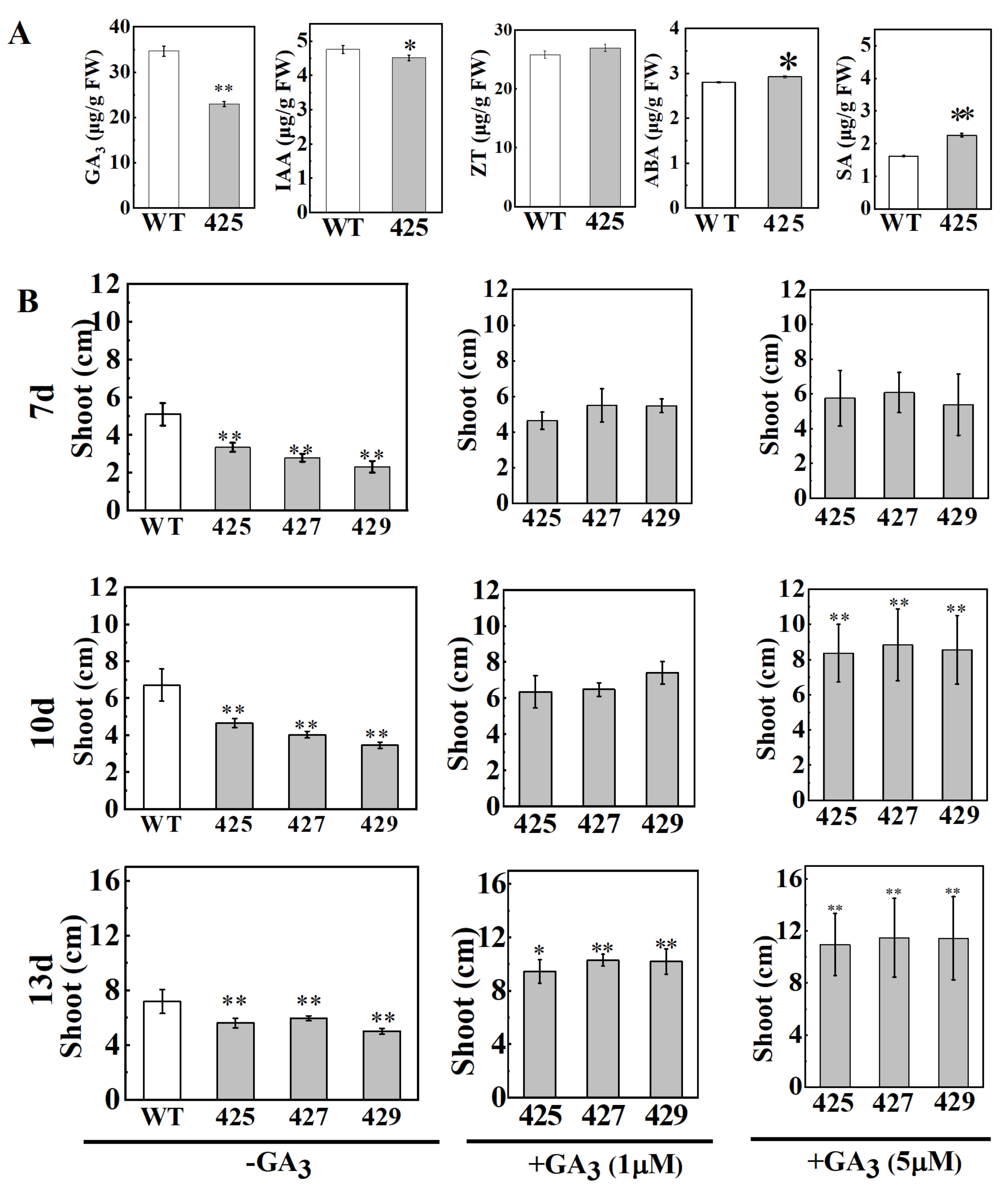
2.4. OsWRKY21 Suppressed IAA and GA Levels and the Dwarf Phenotype Could Be Rescued by Exogenous GA Treatment
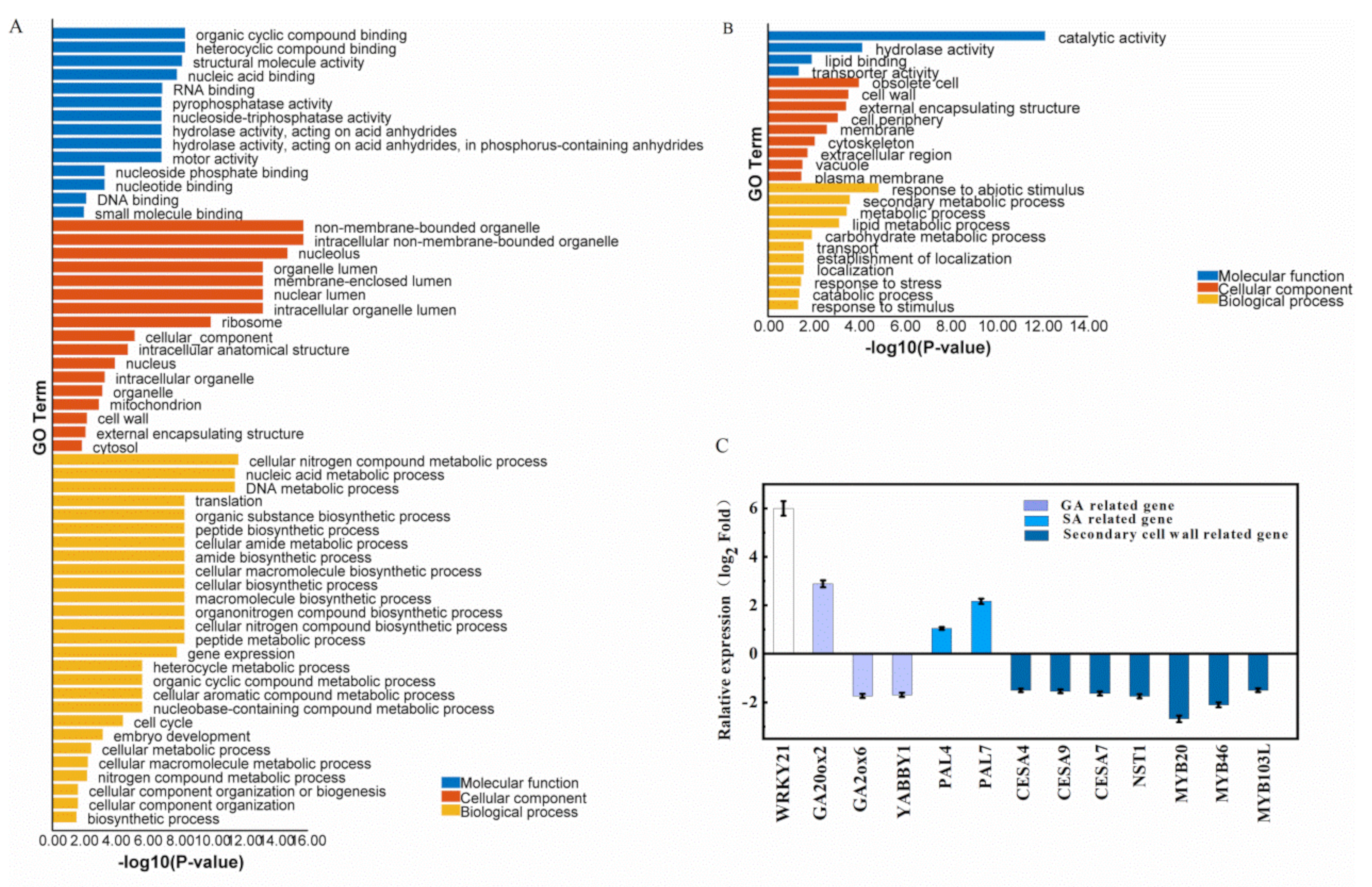
2.5. Overexpression of OsWRKY21 Altered the Expression Levels of the Genes for Phytohormones Metabolic Pathway and Secondary Cell Wall Biosynthesis
3. Discussion
3.1. OsWRKY21 Shows Multiple Functions in Growth/Development and Stress Responses in Rice
3.2. Semi-Dwarf Phenotype of OsWRKY21 Overexpression Is Attributed to the Integral Effects of Phytohormones in Rice
4. Materials and Methods
4.1. Population Structure and GWAS
4.2. Bioinformatics Analysis
4.3. Expression Analysis of OsWRKY21 in Rice
4.4. Plasmid Vector Construction and Rice Transformation
4.5. Plant Materials, Growth Conditions, and GA3 Treatment
4.6. RNA Extraction and qRT-PCR
4.7. Measurement of Endogenous Phytohormones IAA and GA3
4.8. Histological Analysis
4.9. Analysis of the Sequencing Data
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yu, J.; Hu, S.; Wang, J.; Wong, G.K.-S.; Li, S.; Liu, B.; Deng, Y.; Dai, L.; Zhou, Y.; Zhang, X. A draft sequence of the rice genome (oryza sativa l. Ssp. Indica). Science 2002, 296, 79–92. [Google Scholar] [CrossRef]
- Khush, G. Green revolution: The way forward. Nat. Rev. Genet. 2001, 2, 815–822. [Google Scholar] [CrossRef] [PubMed]
- Hedden, P. The genes of the green revolution. TRENDS Genet. 2003, 19, 5–9. [Google Scholar] [CrossRef]
- Itoh, H.; Ueguchi-Tanaka, M.; Sentoku, N.; Kitano, H.; Matsuoka, M.; Kobayashi, M. Cloning and functional analysis of two gibberellin 3β-hydroxylase genes that are differently expressed during the growth of rice. Proc. Natl. Acad. Sci. USA 2001, 98, 8909–8914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakamoto, T.; Matsuoka, M. Generating high-yielding varieties by genetic manipulation of plant architecture. Curr. Opin. Biotechnol. 2004, 15, 144–147. [Google Scholar] [CrossRef]
- Asano, K.; Hirano, K.; Ueguchi-Tanaka, M.; Angeles-Shim, R.B.; Komura, T.; Satoh, H.; Kitano, H.; Matsuoka, M.; Ashikari, M. Isolation and characterization of dominant dwarf mutants, slr1-d, in rice. Mol. Genet. Genom. 2009, 281, 223–231. [Google Scholar] [CrossRef]
- Li, J.; Jiang, J.; Qian, Q.; Xu, Y.; Zhang, C.; Xiao, J.; Du, C.; Luo, W.; Zou, G.; Chen, M. Mutation of rice bc12/gdd1, which encodes a kinesin-like protein that binds to a ga biosynthesis gene promoter, leads to dwarfism with impaired cell elongation. Plant Cell 2011, 23, 628–640. [Google Scholar] [CrossRef] [Green Version]
- Rushton, P.J.; Somssich, I.E.; Ringler, P.; Shen, Q.J. Wrky transcription factors. Trends Plant Sci. 2010, 15, 247–258. [Google Scholar] [CrossRef]
- Eulgem, T.; Rushton, P.J.; Robatzek, S.; Somssich, I.E. The wrky superfamily of plant transcription factors. Trends Plant Sci. 2000, 5, 199–206. [Google Scholar] [CrossRef]
- Sun, Y.; Yu, D. Activated expression of atwrky53 negatively regulates drought tolerance by mediating stomatal movement. Plant Cell Rep. 2015, 34, 1295–1306. [Google Scholar] [CrossRef]
- Shen, H.; Liu, C.; Zhang, Y.; Meng, X.; Zhou, X.; Chu, C.; Wang, X. Oswrky30 is activated by map kinases to confer drought tolerance in rice. Plant Mol. Biol. 2012, 80, 241–253. [Google Scholar] [CrossRef]
- Liu, X.; Bai, X.; Wang, X.; Chu, C. Oswrky71, a rice transcription factor, is involved in rice defense response. J. Plant Physiol. 2007, 164, 969–979. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Hu, Y.; Tang, X.; Zhou, P.; Deng, X.; Wang, H.; Guo, Z. Constitutive expression of rice wrky30 gene increases the endogenous jasmonic acid accumulation, pr gene expression and resistance to fungal pathogens in rice. Planta 2012, 236, 1485–1498. [Google Scholar] [CrossRef]
- Han, M.; Kim, C.-Y.; Lee, J.; Lee, S.-K.; Jeon, J.-S. Oswrky42 represses osmt1d and induces reactive oxygen species and leaf senescence in rice. Mol. Cells 2014, 37, 532. [Google Scholar] [CrossRef]
- Chen, L.; Song, Y.; Li, S.; Zhang, L.; Zou, C.; Yu, D. The role of wrky transcription factors in plant abiotic stresses. Biochim. Biophys. Acta Gene Regul. Mech. 2012, 1819, 120–128. [Google Scholar] [CrossRef]
- Li, W.; Wang, H.; Yu, D. Arabidopsis wrky transcription factors wrky12 and wrky13 oppositely regulate flowering under short-day conditions. Mol. Plant 2016, 9, 1492–1503. [Google Scholar] [CrossRef] [PubMed]
- Garcia, D.; Gerald, J.N.F.; Berger, F. Maternal control of integument cell elongation and zygotic control of endosperm growth are coordinated to determine seed size in arabidopsis. Plant Cell 2005, 17, 52–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, M.; Dennis, E.S.; Berger, F.; Peacock, W.J.; Chaudhury, A. Miniseed3 (mini3), a wrky family gene, and haiku2 (iku2), a leucine-rich repeat (lrr) kinase gene, are regulators of seed size in arabidopsis. Proc. Natl. Acad. Sci. USA 2005, 102, 17531–17536. [Google Scholar] [CrossRef] [Green Version]
- Ramamoorthy, R.; Jiang, S.-Y.; Kumar, N.; Venkatesh, P.N.; Ramachandran, S. A comprehensive transcriptional profiling of the wrky gene family in rice under various abiotic and phytohormone treatments. Plant Cell Physiol. 2008, 49, 865–879. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Zhang, Z.-L.; Zou, X.; Huang, J.; Ruas, P.; Thompson, D.; Shen, Q.J. Annotations and functional analyses of the rice wrky gene superfamily reveal positive and negative regulators of abscisic acid signaling in aleurone cells. Plant Physiol. 2005, 137, 176–189. [Google Scholar] [CrossRef] [Green Version]
- Ryu, H.-S.; Han, M.; Lee, S.-K.; Cho, J.-I.; Ryoo, N.; Heu, S.; Lee, Y.-H.; Bhoo, S.H.; Wang, G.-L.; Hahn, T.-R. A comprehensive expression analysis of the wrky gene superfamily in rice plants during defense response. Plant Cell Rep. 2006, 25, 836–847. [Google Scholar] [CrossRef] [PubMed]
- Qiu, D.; Xiao, J.; Ding, X.; Xiong, M.; Cai, M.; Cao, Y.; Li, X.; Xu, C.; Wang, S. Oswrky13 mediates rice disease resistance by regulating defense-related genes in salicylate-and jasmonate-dependent signaling. Mol. Plant Microbe Interact. 2007, 20, 492–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimono, M.; Sugano, S.; Nakayama, A.; Jiang, C.-J.; Ono, K.; Toki, S.; Takatsuji, H. Rice wrky45 plays a crucial role in benzothiadiazole-inducible blast resistance. Plant Cell 2007, 19, 2064–2076. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.-L.; Xie, Z.; Zou, X.; Casaretto, J.; Ho, T.-h.D.; Shen, Q.J. A rice wrky gene encodes a transcriptional repressor of the gibberellin signaling pathway in aleurone cells. Plant Physiol. 2004, 134, 1500–1513. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Xu, Y.; Lu, Y.; Yu, H.; Gu, M.; Liu, Q. The wrky transcription factor oswrky78 regulates stem elongation and seed development in rice. Planta 2011, 234, 541–554. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Chen, X.; Xie, K.; Xing, Q.; Wu, Y.; Li, J.; Du, C.; Sun, Z.; Guo, Z. Dlf1, a wrky transcription factor, is involved in the control of flowering time and plant height in rice. PLoS ONE 2014, 9, e102529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lan, J.; Lin, Q.; Zhou, C.; Ren, Y.; Liu, X.; Miao, R.; Jing, R.; Mou, C.; Nguyen, T.; Zhu, X. Small grain and semi-dwarf 3, a wrky transcription factor, negatively regulates plant height and grain size by stabilizing slr1 expression in rice. Plant Mol. Biol. 2020, 104, 429–450. [Google Scholar] [CrossRef]
- Ross, C.A.; Liu, Y.; Shen, Q.J. The wrky gene family in rice (oryza sativa). J. Integr. Plant Biol. 2007, 49, 827–842. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, L. The wrky transcription factor superfamily: Its origin in eukaryotes and expansion in plants. BMC Evol. Biol. 2005, 5, 1. [Google Scholar]
- Biłas, R.; Szafran, K.; Hnatuszko-Konka, K.; Kononowicz, A.K. Cis-regulatory elements used to control gene expression in plants. Plant Cell Tissue Organ Cult. 2016, 127, 269–287. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Xie, W.; Chen, Y.; Tang, W.; Yang, J.; Ye, R.; Liu, L.; Lin, Y.; Xu, C.; Xiao, J. A dynamic gene expression atlas covering the entire life cycle of rice. Plant J. 2010, 61, 752–766. [Google Scholar] [CrossRef]
- Dai, M.; Zhao, Y.; Ma, Q.; Hu, Y.; Hedden, P.; Zhang, Q.; Zhou, D.-X. The rice yabby1 gene is involved in the feedback regulation of gibberellin metabolism. Plant Physiol. 2007, 144, 121–133. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Ma, Y.; Li, J. The rice yabby4 gene regulates plant growth and development through modulating the gibberellin pathway. J. Exp. Bot. 2016, 67, 5545–5556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Guo, K.; Li, Y.; Tu, Y.; Hu, H.; Wang, B.; Cui, X.; Peng, L. Expression profiling and integrative analysis of the cesa/csl superfamily in rice. BMC Plant Biol. 2010, 10, 282. [Google Scholar] [CrossRef] [Green Version]
- Wan, H.; Chen, L.; Guo, J.; Li, Q.; Wen, J.; Yi, B.; Ma, C.; Tu, J.; Fu, T.; Shen, J. Genome-wide association study reveals the genetic architecture underlying salt tolerance-related traits in rapeseed (brassica napus l.). Front. Plant Sci. 2017, 8, 593. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Liu, Y.; Xu, H.; Tang, K.; Wu, H.; Lu, L.; Wang, Z.; Chen, Z.; Xu, J.; Zhu, Y. Genome-wide association studies of hiv-1 host control in ethnically diverse chinese populations. Sci. Rep. 2015, 5, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Cao, K.; Zhou, Z.; Wang, Q.; Guo, J.; Zhao, P.; Zhu, G.; Fang, W.; Chen, C.; Wang, X.; Wang, X. Genome-wide association study of 12 agronomic traits in peach. Nat. Commun. 2016, 7, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Li, D.; Liu, X.; Ji, C.; Hao, L.; Zhao, X.; Li, X.; Chen, C.; Cheng, Z.; Zhu, L. Osmyb103l, an r2r3-myb transcription factor, influences leaf rolling and mechanical strength in rice (oryza satival.). BMC Plant Biol. 2014, 14, 158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, Y.; Liu, B.; Zhao, M.; Wu, K.; Cheng, W.; Chen, X.; Liu, Q.; Liu, Z.; Fu, X.; Wu, Y. Cef1/osmyb103l is involved in ga-mediated regulation of secondary wall biosynthesis in rice. Plant Mol. Biol. 2015, 89, 385–401. [Google Scholar] [CrossRef]
- Tanaka, K.; Murata, K.; Yamazaki, M.; Onosato, K.; Miyao, A.; Hirochika, H. Three distinct rice cellulose synthase catalytic subunit genes required for cellulose synthesis in the secondary wall. Plant Physiol. 2003, 133, 73–83. [Google Scholar] [CrossRef] [Green Version]
- Yan, C.; Yan, S.; Zeng, X.; Zhang, Z.; Gu, M. Fine mapping and isolation of bc7 (t), allelic to oscesa4. J. Genet. Genom. 2007, 34, 1019–1027. [Google Scholar] [CrossRef]
- Zhang, J.; Gu, M.; Liang, R.; Shi, X.; Chen, L.; Hu, X.; Wang, S.; Dai, X.; Qu, H.; Li, H. Oswrky21 and oswrky108 function redundantly to promote phosphate accumulation through maintaining the constitutive expression of ospht1; 1 under phosphate-replete conditions. N. Phytol. 2021, 229, 1598. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-Y.; Wang, J.; Zeigler, R.S. The 3,000 rice genomes project: New opportunities and challenges for future rice research. Gigascience 2014, 3. [Google Scholar] [CrossRef] [Green Version]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; De Bakker, P.I.; Daly, M.J. Plink: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Liu, X.; Wang, J.; Li, M.; Wang, Q.; Tian, F.; Su, Z.; Pan, Y.; Liu, D.; Lipka, A.E. Gapit version 2: An enhanced integrated tool for genomic association and prediction. Plant Genome 2016, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, L.; Zhang, H.; Tang, Z.; Xu, J.; Yin, D.; Zhang, Z.; Yuan, X.; Zhu, M.; Zhao, S.; Li, X. Rmvp: A memory-efficient, visualization-enhanced, and parallel-accelerated tool for genome-wide association study. Genom. Proteom. Bioinform. 2021. [Google Scholar] [CrossRef]
- Kumar, S.; Nei, M.; Dudley, J.; Tamura, K. Mega: A biologist-centric software for evolutionary analysis of DNA and protein sequences. Brief. Bioinform. 2008, 9, 299–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. Meme suite: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, W202–W208. [Google Scholar] [CrossRef]
- Krogh, A.; Larsson, B.; Von Heijne, G.; Sonnhammer, E.L. Predicting transmembrane protein topology with a hidden markov model: Application to complete genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef] [Green Version]
- Horton, P.; Park, K.-J.; Obayashi, T.; Fujita, N.; Harada, H.; Adams-Collier, C.; Nakai, K. Wolf psort: Protein localization predictor. Nucleic Acids Res. 2007, 35, W585–W587. [Google Scholar] [CrossRef] [Green Version]
- Lescot, M.; Déhais, P.; Thijs, G.; Marchal, K.; Moreau, Y.; Van de Peer, Y.; Rouzé, P.; Rombauts, S. Plantcare, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 2002, 30, 325–327. [Google Scholar] [CrossRef]
- Lin, Y.J.; Zhang, Q. Optimising the tissue culture conditions for high efficiency transformation of indica rice. Plant Cell Rep. 2005, 23, 540–547. [Google Scholar] [CrossRef] [PubMed]
- Xie, K.; Minkenberg, B.; Yang, Y. Boosting crispr/cas9 multiplex editing capability with the endogenous trna-processing system. Proc. Natl. Acad. Sci. USA 2015, 112, 3570–3575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative pcr and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. Hisat: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Smyth, G.K.; Shi, W. Featurecounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Anders, S. Analysing rna-seq data with the deseq package. Mol. Biol. 2010, 43, 1–17. [Google Scholar]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Xia, R. Tbtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wei, X.; Zhou, H.; Xie, D.; Li, J.; Yang, M.; Chang, T.; Wang, D.; Hu, L.; Xie, G.; Wang, J.; et al. Genome-Wide Association Study in Rice Revealed a Novel Gene in Determining Plant Height and Stem Development, by Encoding a WRKY Transcription Factor. Int. J. Mol. Sci. 2021, 22, 8192. https://doi.org/10.3390/ijms22158192
Wei X, Zhou H, Xie D, Li J, Yang M, Chang T, Wang D, Hu L, Xie G, Wang J, et al. Genome-Wide Association Study in Rice Revealed a Novel Gene in Determining Plant Height and Stem Development, by Encoding a WRKY Transcription Factor. International Journal of Molecular Sciences. 2021; 22(15):8192. https://doi.org/10.3390/ijms22158192
Chicago/Turabian StyleWei, Xiaoshuang, Hailian Zhou, Deying Xie, Jianguo Li, Mingchong Yang, Tianli Chang, Dongxin Wang, Lihua Hu, Guosheng Xie, Jihong Wang, and et al. 2021. "Genome-Wide Association Study in Rice Revealed a Novel Gene in Determining Plant Height and Stem Development, by Encoding a WRKY Transcription Factor" International Journal of Molecular Sciences 22, no. 15: 8192. https://doi.org/10.3390/ijms22158192