
Application of Patient-Specific iPSCs for Modelling and Treatment of X-Linked Cardiomyopathies

 ,
,
Abstract
:1. Introduction
2. The Advantages of an iPSC-Based Model
3. The Procedures for Producing iPSCs
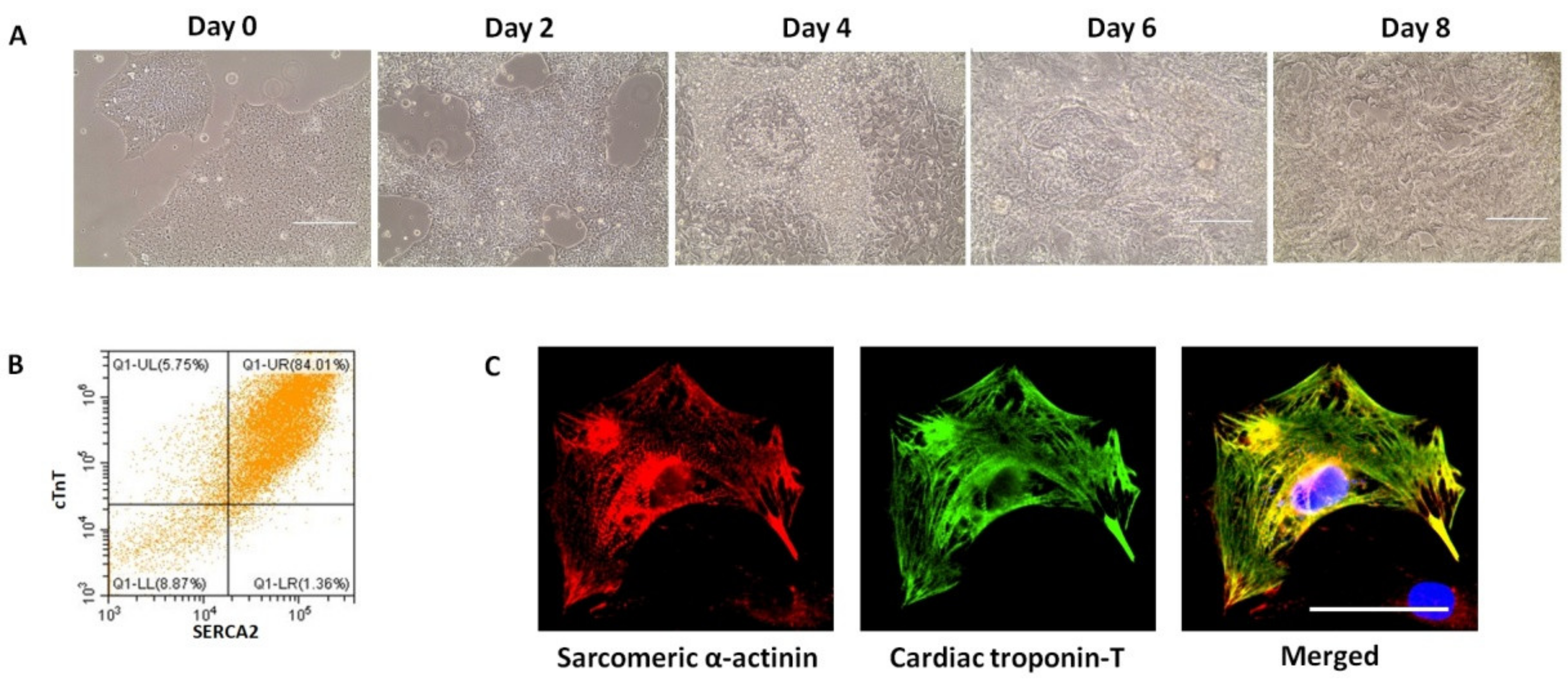
4. Differentiation of Established iPSC Lines to Cardiomyocytes
5. Functional Characterisation of Cardiomyocytes Derived from iPSCs
5.1. Characterisation of Structural Properties
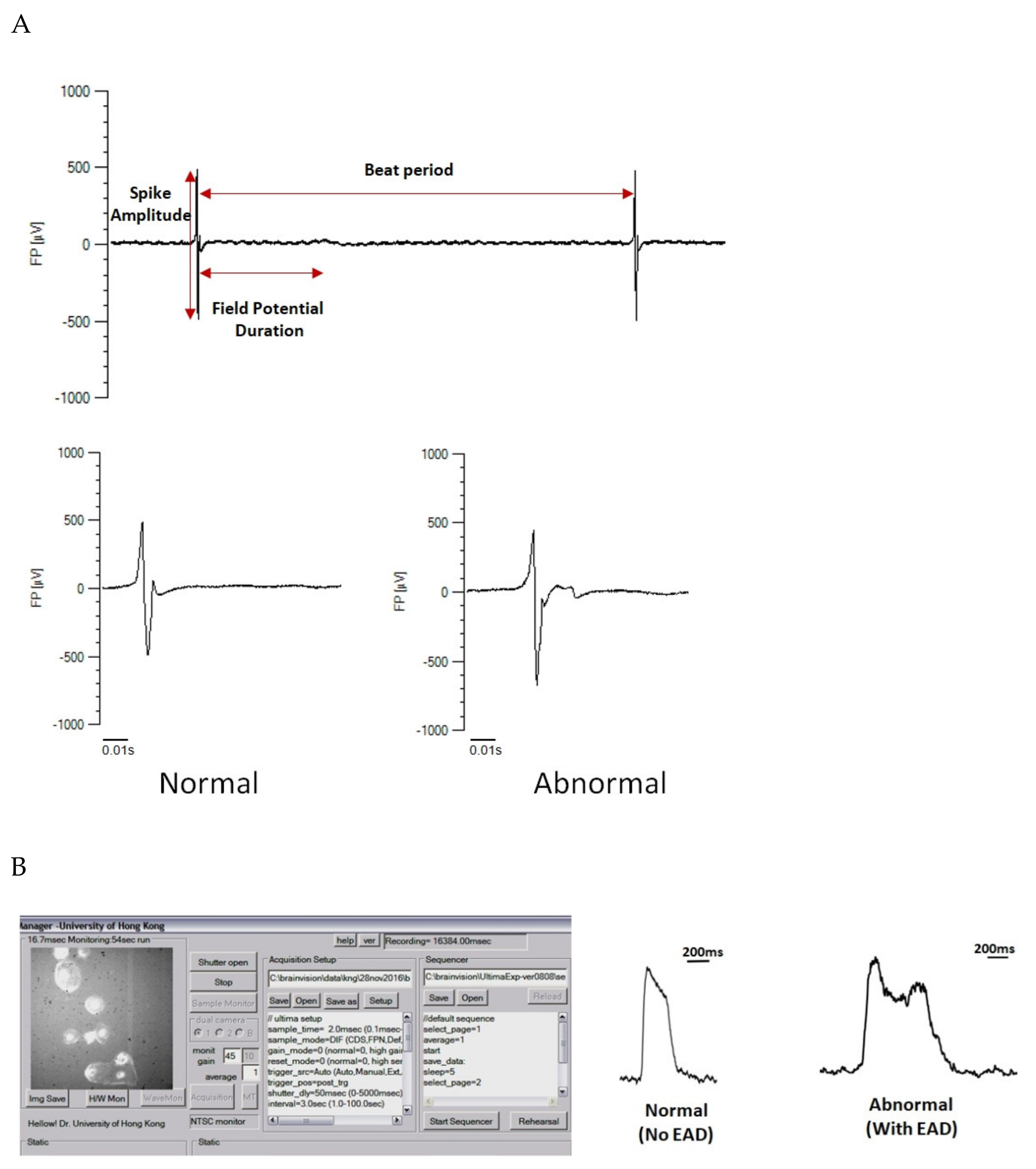
5.2. Characterisation of Electrophysiological Properties
5.3. Characterisation of the Calcium Handling Properties
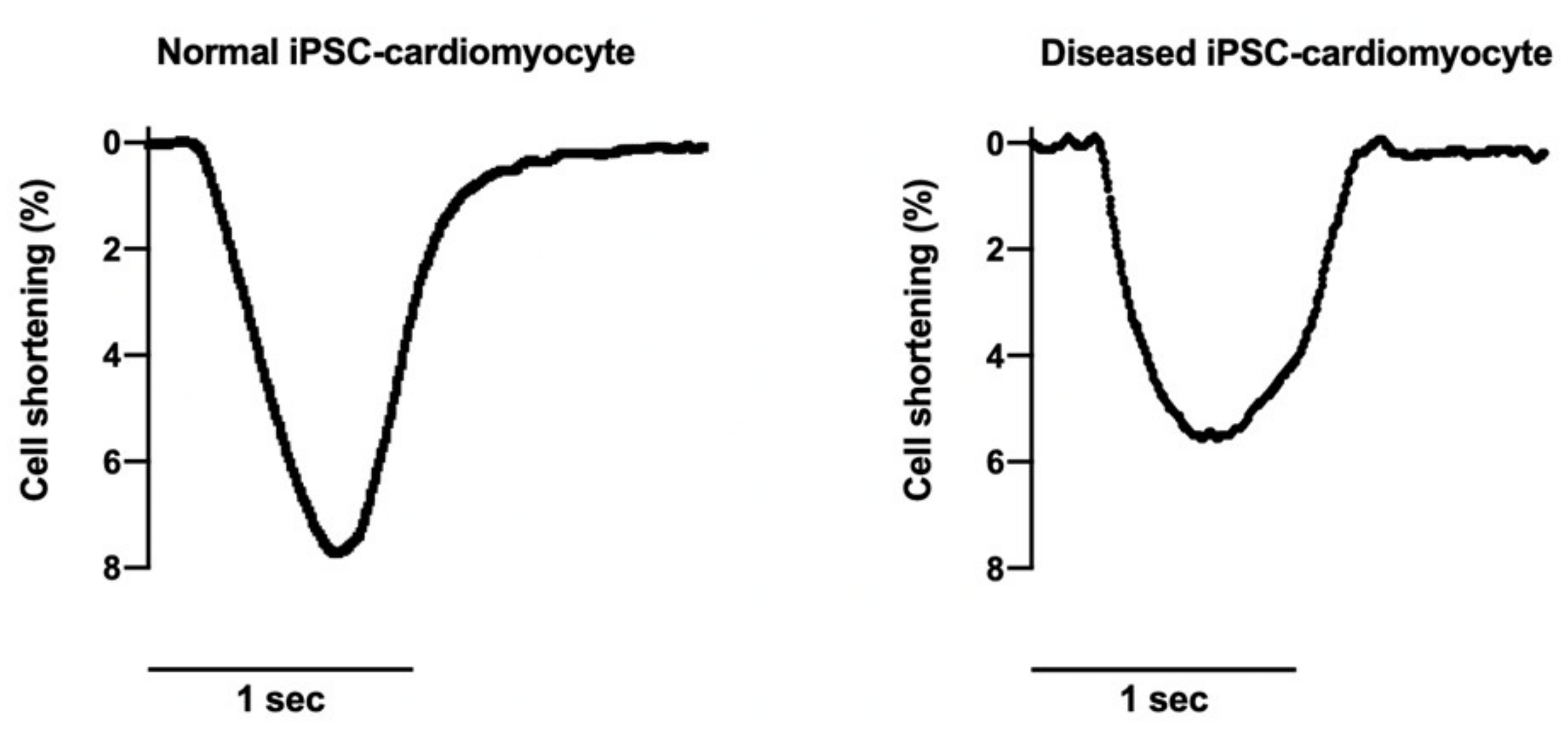
5.4. Characterisations of Contraction Properties
6. Strategies for Using iPSC-Based Models for Disease Modelling and Drug Testing
6.1. Duchenne Muscular Dystrophy
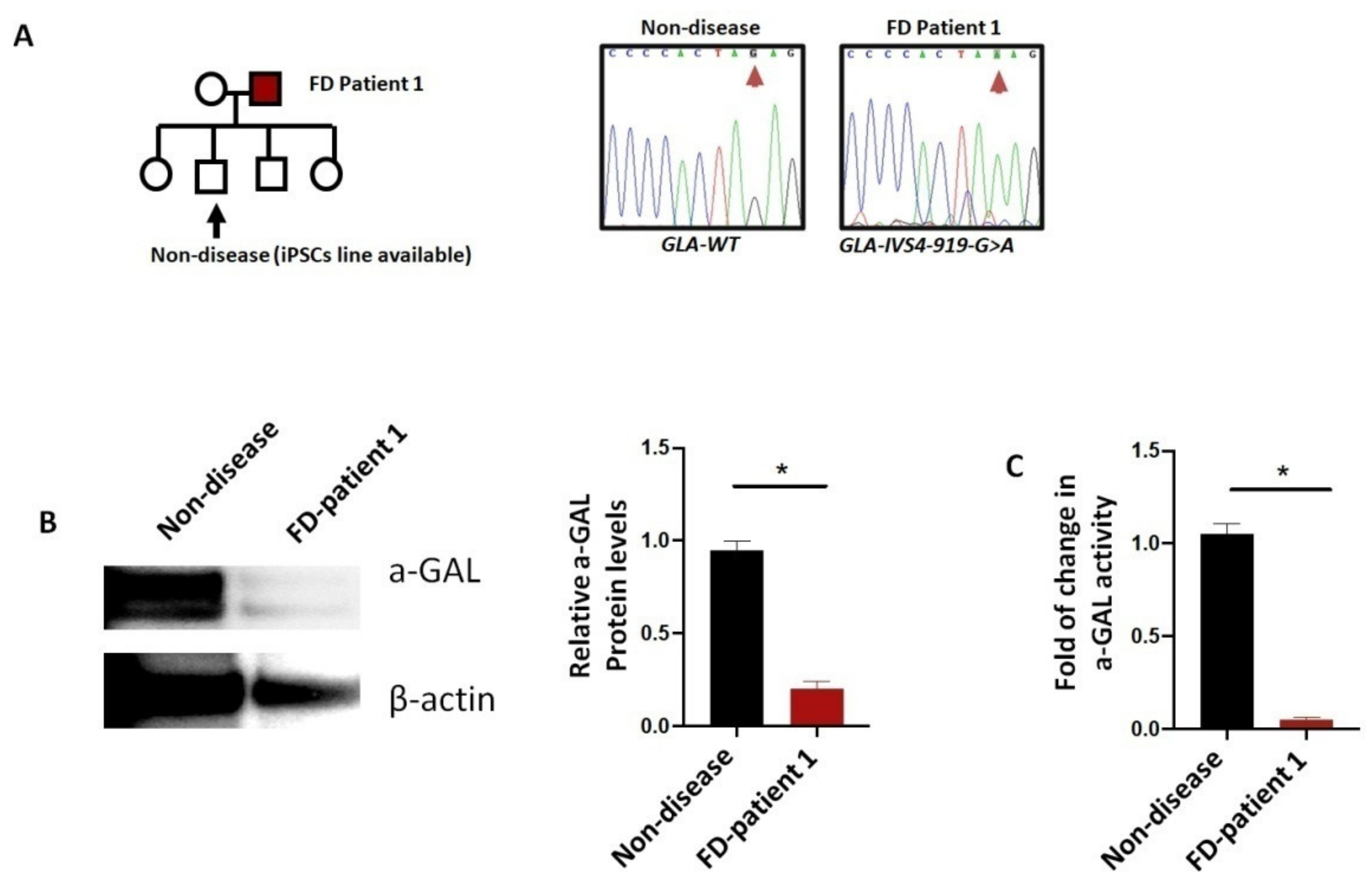
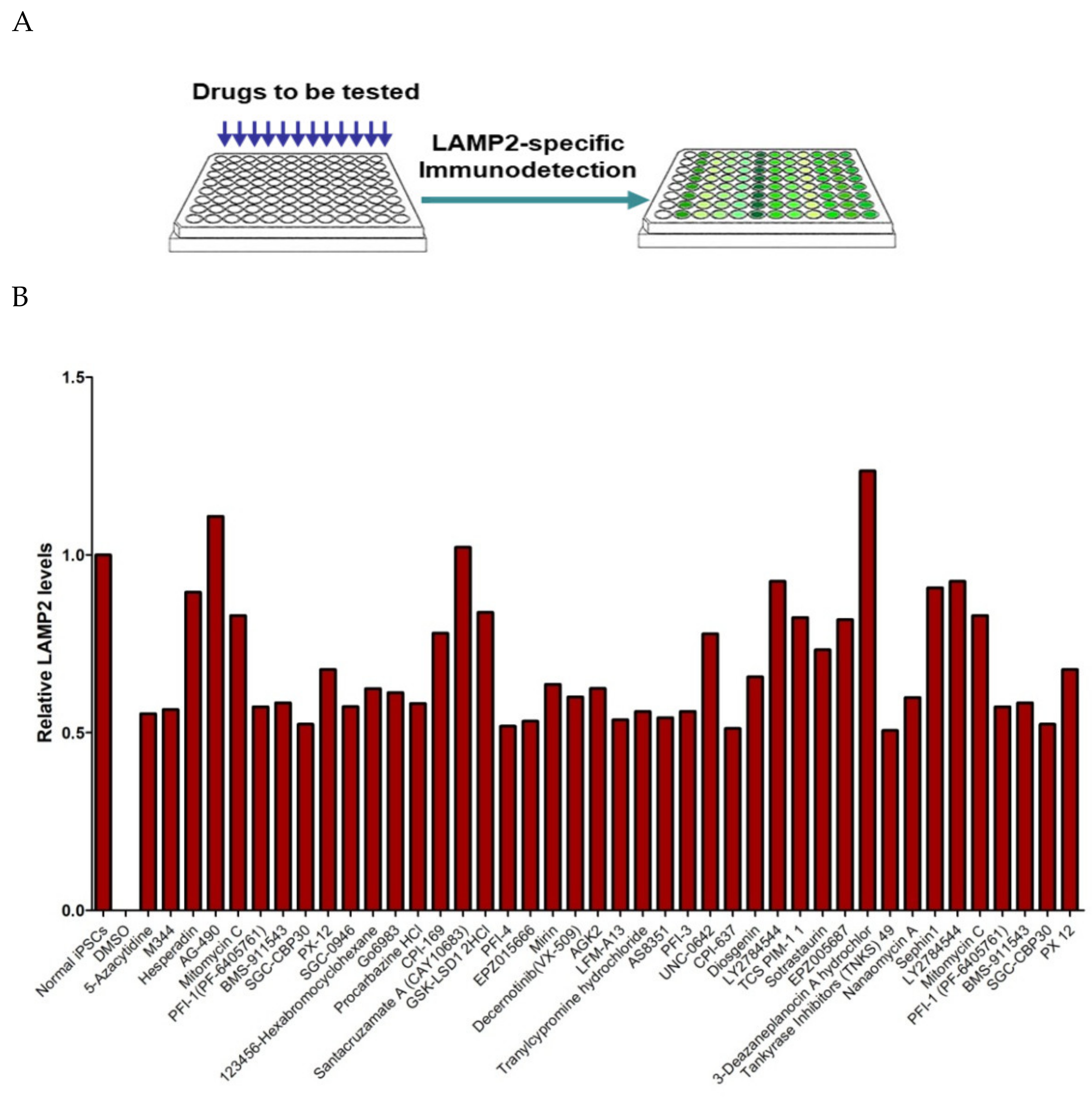
6.2. Fabry Disease
6.3. Rett Syndrome
6.4. Danon Disease
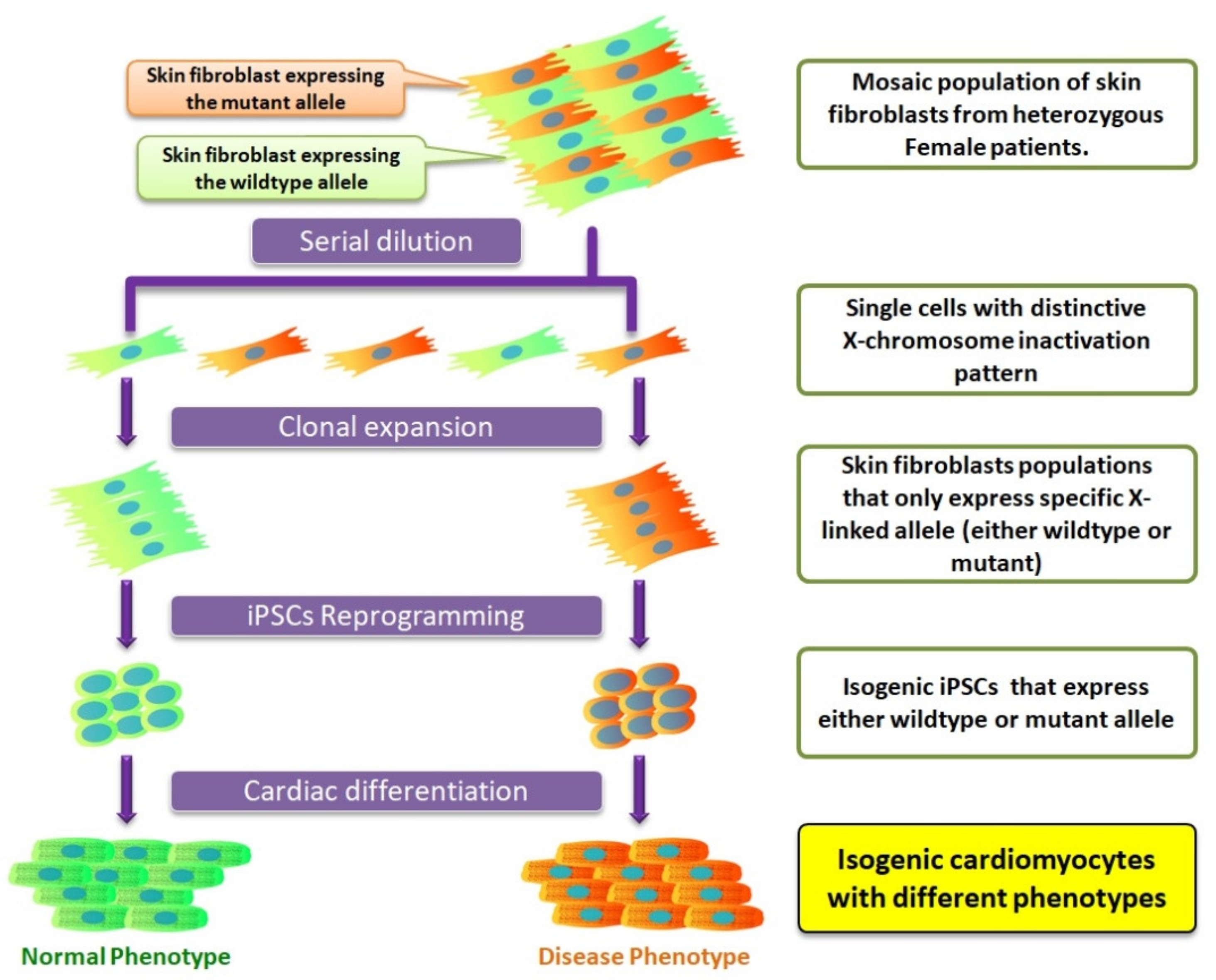
7. Isogenic iPSCs as a New Platform to Study X-Chromosome Inactivation
8. Limitations
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Milani-Nejad, N.; Janssen, P.M. Small and large animal models in cardiac contraction research: Advantages and disadvantages. Pharm. 2014, 141, 235–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nerbonne, J.M. Studying cardiac arrhythmias in the mouse--a reasonable model for probing mechanisms? Trends Cardiovasc. Med. 2004, 14, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Sutanto, H.; Laudy, L.; Clerx, M.; Dobrev, D.; Crijns, H.; Heijman, J. Maastricht antiarrhythmic drug evaluator (manta): A computational tool for better understanding of antiarrhythmic drugs. Pharm. Res. 2019, 148, 104444. [Google Scholar] [CrossRef] [PubMed]
- Alpert, N.R.; Brosseau, C.; Federico, A.; Krenz, M.; Robbins, J.; Warshaw, D.M. Molecular mechanics of mouse cardiac myosin isoforms. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H1446–H1454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakagawa, M.; Koyanagi, M.; Tanabe, K.; Takahashi, K.; Ichisaka, T.; Aoi, T.; Okita, K.; Mochiduki, Y.; Takizawa, N.; Yamanaka, S. Generation of induced pluripotent stem cells without myc from mouse and human fibroblasts. Nat. Biotechnol. 2008, 26, 101–106. [Google Scholar] [CrossRef]
- Farini, A.; Gowran, A.; Bella, P.; Sitzia, C.; Scopece, A.; Castiglioni, E.; Rovina, D.; Nigro, P.; Villa, C.; Fortunato, F.; et al. Fibrosis rescue improves cardiac function in dystrophin-deficient mice and duchenne patient-specific cardiomyocytes by immunoproteasome modulation. Am. J. Pathol. 2019, 189, 339–353. [Google Scholar] [CrossRef] [Green Version]
- Peper, J.; Kownatzki-Danger, D.; Weninger, G.; Seibertz, F.; Pronto, J.R.D.; Sutanto, H.; Pacheu-Grau, D.; Hindmarsh, R.; Brandenburg, S.; Kohl, T.; et al. Caveolin3 stabilizes mct1-mediated lactate/proton transport in cardiomyocytes. Circ. Res. 2021, 128, e102–e120. [Google Scholar] [CrossRef]
- Brambatti, M.; Caspi, O.; Maolo, A.; Koshi, E.; Greenberg, B.; Taylor, M.R.G.; Adler, E.D. Danon disease: Gender differences in presentation and outcomes. Int. J. Cardiol. 2019, 286, 92–98. [Google Scholar] [CrossRef]
- Jefferies, J.L.; Eidem, B.W.; Belmont, J.W.; Craigen, W.J.; Ware, S.M.; Fernbach, S.D.; Neish, S.R.; Smith, E.O.; Towbin, J.A. Genetic predictors and remodeling of dilated cardiomyopathy in muscular dystrophy. Circulation 2005, 112, 2799–2804. [Google Scholar] [CrossRef] [Green Version]
- Rivera, S.R.; Jhamb, S.K.; Abdel-Hamid, H.Z.; Acsadi, G.; Brandsema, J.; Ciafaloni, E.; Darras, B.T.; Iannaccone, S.T.; Konersman, C.G.; Kuntz, N.L.; et al. Medical management of muscle weakness in duchenne muscular dystrophy. PLoS ONE 2020, 15, e0240687. [Google Scholar] [CrossRef]
- Eiholzer, U.; Boltshauser, E.; Frey, D.; Molinari, L.; Zachmann, M. Short stature: A common feature in duchenne muscular dystrophy. Eur. J. Pediatr. 1988, 147, 602–605. [Google Scholar] [CrossRef] [PubMed]
- Ommen, S.R.; Nishimura, R.A.; Edwards, W.D. Fabry disease: A mimic for obstructive hypertrophic cardiomyopathy? Heart 2003, 89, 929–930. [Google Scholar] [CrossRef] [Green Version]
- Bokhari, S.R.A.; Zulfiqar, H.; Hariz, A. Fabry disease; Statpearls: Treasure Island, FL, USA, 2021. [Google Scholar]
- Kyndt, F.; Gueffet, J.P.; Probst, V.; Jaafar, P.; Legendre, A.; Le Bouffant, F.; Toquet, C.; Roy, E.; McGregor, L.; Lynch, S.A.; et al. Mutations in the gene encoding filamin a as a cause for familial cardiac valvular dystrophy. Circulation 2007, 115, 40–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Kogelenberg, M.; Clark, A.R.; Jenkins, Z.; Morgan, T.; Anandan, A.; Sawyer, G.M.; Edwards, M.; Dudding, T.; Homfray, T.; Castle, B.; et al. Diverse phenotypic consequences of mutations affecting the c-terminus of flna. J. Mol. Med. 2015, 93, 773–782. [Google Scholar] [CrossRef]
- Monteleone, P.L.; Fagan, L.F. Possible x-linked congenital heart disease. Circulation 1969, 39, 611–614. [Google Scholar] [CrossRef] [Green Version]
- Ritelli, M.; Morlino, S.; Giacopuzzi, E.; Carini, G.; Cinquina, V.; Chiarelli, N.; Majore, S.; Colombi, M.; Castori, M. Ehlers-danlos syndrome with lethal cardiac valvular dystrophy in males carrying a novel splice mutation in flna. Am. J. Med. Genet. A 2017, 173, 169–176. [Google Scholar] [CrossRef]
- Arad, M.; Maron, B.J.; Gorham, J.M.; Johnson, W.H., Jr.; Saul, J.P.; Perez-Atayde, A.R.; Spirito, P.; Wright, G.B.; Kanter, R.J.; Seidman, C.E.; et al. Glycogen storage diseases presenting as hypertrophic cardiomyopathy. N. Engl. J. Med. 2005, 352, 362–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prall, F.R.; Drack, A.; Taylor, M.; Ku, L.; Olson, J.L.; Gregory, D.; Mestroni, L.; Mandava, N. Ophthalmic manifestations of danon disease. Ophthalmology 2006, 113, 1010–1013. [Google Scholar] [CrossRef] [PubMed]
- Yardeni, M.; Weisman, O.; Mandel, H.; Weinberger, R.; Quarta, G.; Salazar-Mendiguchia, J.; Garcia-Pavia, P.; Lobato-Rodriguez, M.J.; Simon, L.F.; Dov, F.; et al. Psychiatric and cognitive characteristics of individuals with danon disease (lamp2 gene mutation). Am. J. Med. Genet. A 2017, 173, 2461–2466. [Google Scholar] [CrossRef]
- Gourzi, P.; Pantou, M.P.; Gkouziouta, A.; Kaklamanis, L.; Tsiapras, D.; Zygouri, C.; Constantoulakis, P.; Adamopoulos, S.; Degiannis, D. A new phenotype of severe dilated cardiomyopathy associated with a mutation in the lamp2 gene previously known to cause hypertrophic cardiomyopathy in the context of danon disease. Eur. J. Med. Genet. 2019, 62, 77–80. [Google Scholar] [CrossRef]
- Codron, P.; Pautot, V.; Tassin, A.; Sternberg, D.; Letournel, F.; Richard, P.; Nadaj-Pakleza, A. Abundant electrical myotonia and left ventricular noncompaction: Unusual features of danon disease due to a novel mutation in lamp2 gene. Rev. Neurol. 2019, 175, 201–203. [Google Scholar] [CrossRef] [PubMed]
- Amir, R.E.; Van den Veyver, I.B.; Wan, M.; Tran, C.Q.; Francke, U.; Zoghbi, H.Y. Rett syndrome is caused by mutations in x-linked mecp2, encoding methyl-cpg-binding protein 2. Nat. Genet. 1999, 23, 185–188. [Google Scholar] [CrossRef]
- Hagberg, B.; Aicardi, J.; Dias, K.; Ramos, O. A progressive syndrome of autism, dementia, ataxia, and loss of purposeful hand use in girls: Rett’s syndrome: Report of 35 cases. Ann. Neurol. 1983, 14, 471–479. [Google Scholar] [CrossRef]
- Dolce, A.; Ben-Zeev, B.; Naidu, S.; Kossoff, E.H. Rett syndrome and epilepsy: An update for child neurologists. Pediatr. Neurol. 2013, 48, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Leonard, H.; Thomson, M.R.; Glasson, E.J.; Fyfe, S.; Leonard, S.; Bower, C.; Christodoulou, J.; Ellaway, C. A population-based approach to the investigation of osteopenia in rett syndrome. Dev. Med. Child. Neurol. 1999, 41, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, P.B.; Pierson, C.R.; Joshi, M.; Liu, X.; Ravenscroft, G.; Moghadaszadeh, B.; Talabere, T.; Viola, M.; Swanson, L.C.; Haliloglu, G.; et al. Speg interacts with myotubularin, and its deficiency causes centronuclear myopathy with dilated cardiomyopathy. Am. J. Hum. Genet. 2014, 95, 218–226. [Google Scholar] [CrossRef] [Green Version]
- Dowling, J.J.; Lawlor, M.W.; Das, S. X-linked myotubular myopathy. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Zakrzewski, W.; Dobrzynski, M.; Szymonowicz, M.; Rybak, Z. Stem cells: Past, present, and future. Stem. Cell Res. Ther. 2019, 10, 68. [Google Scholar] [CrossRef]
- Hu, S.; Zhao, M.T.; Jahanbani, F.; Shao, N.Y.; Lee, W.H.; Chen, H.; Snyder, M.P.; Wu, J.C. Effects of cellular origin on differentiation of human induced pluripotent stem cell-derived endothelial cells. JCI Insight 2016, 1. [Google Scholar] [CrossRef]
- Ng, K.M.; Mok, P.Y.; Butler, A.W.; Ho, J.C.; Choi, S.W.; Lee, Y.K.; Lai, W.H.; Au, K.W.; Lau, Y.M.; Wong, L.Y.; et al. Amelioration of x-linked related autophagy failure in danon disease with DNA methylation inhibitor. Circulation 2016, 134, 1373–1389. [Google Scholar] [CrossRef]
- Ghanbari, H.A.; McCarl, R.L. A comparative study of myosin and its subunits in adult and neonatal-rat hearts and in rat heart cells from young and old cultures. Biochem. J. 1980, 191, 627–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peter, A.K.; Bjerke, M.A.; Leinwand, L.A. Biology of the cardiac myocyte in heart disease. Mol. Biol. Cell 2016, 27, 2149–2160. [Google Scholar] [CrossRef] [PubMed]
- Xue, H.; Wu, J.; Li, S.; Rao, M.S.; Liu, Y. Genetic modification in human pluripotent stem cells by homologous recombination and crispr/cas9 system. Methods Mol. Biol. 2016, 1307, 173–190. [Google Scholar] [PubMed]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the crispr-cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Masi, C.; Spitalieri, P.; Murdocca, M.; Novelli, G.; Sangiuolo, F. Application of crispr/cas9 to human-induced pluripotent stem cells: From gene editing to drug discovery. Hum. Genom. 2020, 14, 25. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhang, Y.; Gao, F.; Han, S.; Cheah, K.S.; Tse, H.F.; Lian, Q. Crispr/cas9 genome-editing system in human stem cells: Current status and future prospects. Mol. Ther. Nucleic. Acids 2017, 9, 230–241. [Google Scholar] [CrossRef] [Green Version]
- Sterneckert, J.L.; Reinhardt, P.; Scholer, H.R. Investigating human disease using stem cell models. Nat. Rev. Genet. 2014, 15, 625–639. [Google Scholar] [CrossRef] [PubMed]
- Hockemeyer, D.; Jaenisch, R. Induced pluripotent stem cells meet genome editing. Cell Stem Cell 2016, 18, 573–586. [Google Scholar] [CrossRef] [Green Version]
- McCauley, H.A.; Wells, J.M. Pluripotent stem cell-derived organoids: Using principles of developmental biology to grow human tissues in a dish. Development 2017, 144, 958–962. [Google Scholar] [CrossRef] [Green Version]
- Machiraju, P.; Greenway, S.C. Current methods for the maturation of induced pluripotent stem cell-derived cardiomyocytes. World J. Stem Cells 2019, 11, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Wang, Y.; Zhou, T.; Wong, L.Y.; Tian, X.Y.; Hong, X.; Lai, W.H.; Au, K.W.; Wei, R.; Liu, Y.; et al. Generation of human liver chimeric mice with hepatocytes from familial hypercholesterolemia induced pluripotent stem cells. Stem Cell Rep. 2017, 8, 605–618. [Google Scholar] [CrossRef] [Green Version]
- Vitrac, A.; Pons, S.; Balkota, M.; Lemiere, N.; Rais, C.; Bourgeois, J.P.; Maskos, U.; Bourgeron, T.; Cloez-Tayarani, I. A chimeric mouse model to study human ipsc-derived neurons: The case of a truncating shank3 mutation. Sci. Rep. 2020, 10, 13315. [Google Scholar] [CrossRef]
- Yu, J.; Hu, K.; Smuga-Otto, K.; Tian, S.; Stewart, R.; Slukvin, I.I.; Thomson, J.A. Human induced pluripotent stem cells free of vector and transgene sequences. Science 2009, 324, 797–801. [Google Scholar] [CrossRef] [Green Version]
- El Hokayem, J.; Cukier, H.N.; Dykxhoorn, D.M. Blood derived induced pluripotent stem cells (ipscs): Benefits, challenges and the road ahead. J. Alzheimers Dis. Parkinsonism 2016, 6. [Google Scholar] [CrossRef] [Green Version]
- Spitzhorn, L.-S.; Megges, M.; Wruck, W.; Rahman, M.S.; Otte, J.; Degistirici, Ö.; Meisel, R.; Sorg, R.V.; Oreffo, R.O.C.; Adjaye, J. Human ipsc-derived mscs (imscs) from aged individuals acquire a rejuvenation signature. Stem Cell Res. Ther. 2019, 10, 100. [Google Scholar] [CrossRef] [Green Version]
- Zhou, T.; Benda, C.; Duzinger, S.; Huang, Y.; Li, X.; Li, Y.; Guo, X.; Cao, G.; Chen, S.; Hao, L.; et al. Generation of induced pluripotent stem cells from urine. J. Am. Soc. Nephrol. 2011, 22, 1221–1228. [Google Scholar] [CrossRef] [PubMed]
- Sommer, C.A.; Stadtfeld, M.; Murphy, G.J.; Hochedlinger, K.; Kotton, D.N.; Mostoslavsky, G. Induced pluripotent stem cell generation using a single lentiviral stem cell cassette. Stem Cells 2009, 27, 543–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamasaki, S.; Taguchi, Y.; Shimamoto, A.; Mukasa, H.; Tahara, H.; Okamoto, T. Generation of human induced pluripotent stem (ips) cells in serum- and feeder-free defined culture and tgf-beta1 regulation of pluripotency. PLoS ONE 2014, 9, e87151. [Google Scholar] [CrossRef]
- Ghaedi, M.; Niklason, L.E. Human pluripotent stem cells (ipsc) generation, culture, and differentiation to lung progenitor cells. Methods Mol. Biol. 2019, 1576, 55–92. [Google Scholar] [PubMed]
- Kehat, I.; Kenyagin-Karsenti, D.; Snir, M.; Segev, H.; Amit, M.; Gepstein, A.; Livne, E.; Binah, O.; Itskovitz-Eldor, J.; Gepstein, L. Human embryonic stem cells can differentiate into myocytes with structural and functional properties of cardiomyocytes. J. Clin. Invest. 2001, 108, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Segev, H.; Kenyagin-Karsenti, D.; Fishman, B.; Gerecht-Nir, S.; Ziskind, A.; Amit, M.; Coleman, R.; Itskovitz-Eldor, J. Molecular analysis of cardiomyocytes derived from human embryonic stem cells. Dev. Growth Differ. 2005, 47, 295–306. [Google Scholar] [CrossRef]
- Blazeski, A.; Zhu, R.; Hunter, D.W.; Weinberg, S.H.; Boheler, K.R.; Zambidis, E.T.; Tung, L. Electrophysiological and contractile function of cardiomyocytes derived from human embryonic stem cells. Prog. Biophys. Mol. Biol. 2012, 110, 178–195. [Google Scholar] [CrossRef] [Green Version]
- Kattman, S.J.; Witty, A.D.; Gagliardi, M.; Dubois, N.C.; Niapour, M.; Hotta, A.; Ellis, J.; Keller, G. Stage-specific optimization of activin/nodal and bmp signaling promotes cardiac differentiation of mouse and human pluripotent stem cell lines. Cell Stem Cell 2011, 8, 228–240. [Google Scholar] [CrossRef] [Green Version]
- Burridge, P.W.; Thompson, S.; Millrod, M.A.; Weinberg, S.; Yuan, X.; Peters, A.; Mahairaki, V.; Koliatsos, V.E.; Tung, L.; Zambidis, E.T. A universal system for highly efficient cardiac differentiation of human induced pluripotent stem cells that eliminates interline variability. PLoS ONE 2011, 6, e18293. [Google Scholar] [CrossRef]
- Zhu, W.Z.; Van Biber, B.; Laflamme, M.A. Methods for the derivation and use of cardiomyocytes from human pluripotent stem cells. Methods Mol. Biol. 2011, 767, 419–431. [Google Scholar]
- Jha, R.; Xu, R.H.; Xu, C. Efficient differentiation of cardiomyocytes from human pluripotent stem cells with growth factors. Methods Mol. Biol. 2015, 1299, 115–131. [Google Scholar]
- Yang, L.; Soonpaa, M.H.; Adler, E.D.; Roepke, T.K.; Kattman, S.J.; Kennedy, M.; Henckaerts, E.; Bonham, K.; Abbott, G.W.; Linden, R.M.; et al. Human cardiovascular progenitor cells develop from a kdr+ embryonic-stem-cell-derived population. Nature 2008, 453, 524–528. [Google Scholar] [CrossRef]
- Willems, E.; Cabral-Teixeira, J.; Schade, D.; Cai, W.; Reeves, P.; Bushway, P.J.; Lanier, M.; Walsh, C.; Kirchhausen, T.; Izpisua Belmonte, J.C.; et al. Small molecule-mediated tgf-beta type ii receptor degradation promotes cardiomyogenesis in embryonic stem cells. Cell Stem Cell 2012, 11, 242–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, Y.; Lee, M.Y.; Schliffke, S.; Paavola, J.; Amos, P.J.; Ge, X.; Ye, M.; Zhu, S.; Senyei, G.; Lum, L.; et al. Small molecule wnt inhibitors enhance the efficiency of bmp-4-directed cardiac differentiation of human pluripotent stem cells. J. Mol. Cell Cardiol. 2011, 51, 280–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamad, S.; Derichsweiler, D.; Papadopoulos, S.; Nguemo, F.; Saric, T.; Sachinidis, A.; Brockmeier, K.; Hescheler, J.; Boukens, B.J.; Pfannkuche, K. Generation of human induced pluripotent stem cell-derived cardiomyocytes in 2d monolayer and scalable 3d suspension bioreactor cultures with reduced batch-to-batch variations. Theranostics 2019, 9, 7222–7238. [Google Scholar] [CrossRef] [PubMed]
- Burridge, P.W.; Matsa, E.; Shukla, P.; Lin, Z.C.; Churko, J.M.; Ebert, A.D.; Lan, F.; Diecke, S.; Huber, B.; Mordwinkin, N.M.; et al. Chemically defined generation of human cardiomyocytes. Nat. Methods 2014, 11, 855–860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Valk, J.; Brunner, D.; De Smet, K.; Fex Svenningsen, A.; Honegger, P.; Knudsen, L.E.; Lindl, T.; Noraberg, J.; Price, A.; Scarino, M.L.; et al. Optimization of chemically defined cell culture media--replacing fetal bovine serum in mammalian in vitro methods. Toxicol Vitr. 2010, 24, 1053–1063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dambrot, C.; Braam, S.R.; Tertoolen, L.G.; Birket, M.; Atsma, D.E.; Mummery, C.L. Serum supplemented culture medium masks hypertrophic phenotypes in human pluripotent stem cell derived cardiomyocytes. J. Cell Mol. Med. 2014, 18, 1509–1518. [Google Scholar] [CrossRef] [PubMed]
- Tohyama, S.; Hattori, F.; Sano, M.; Hishiki, T.; Nagahata, Y.; Matsuura, T.; Hashimoto, H.; Suzuki, T.; Yamashita, H.; Satoh, Y.; et al. Distinct metabolic flow enables large-scale purification of mouse and human pluripotent stem cell-derived cardiomyocytes. Cell Stem Cell 2013, 12, 127–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elliott, D.A.; Braam, S.R.; Koutsis, K.; Ng, E.S.; Jenny, R.; Lagerqvist, E.L.; Biben, C.; Hatzistavrou, T.; Hirst, C.E.; Yu, Q.C.; et al. Nkx2-5(egfp/w) hescs for isolation of human cardiac progenitors and cardiomyocytes. Nat. Methods 2011, 8, 1037–1040. [Google Scholar] [CrossRef]
- Uosaki, H.; Fukushima, H.; Takeuchi, A.; Matsuoka, S.; Nakatsuji, N.; Yamanaka, S.; Yamashita, J.K. Efficient and scalable purification of cardiomyocytes from human embryonic and induced pluripotent stem cells by vcam1 surface expression. PLoS ONE 2011, 6, e23657. [Google Scholar] [CrossRef] [PubMed]
- Dubois, N.C.; Craft, A.M.; Sharma, P.; Elliott, D.A.; Stanley, E.G.; Elefanty, A.G.; Gramolini, A.; Keller, G. Sirpa is a specific cell-surface marker for isolating cardiomyocytes derived from human pluripotent stem cells. Nat. Biotechnol. 2011, 29, 1011–1018. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.T.; Shao, N.Y.; Garg, V. Subtype-specific cardiomyocytes for precision medicine: Where are we now? Stem Cells 2020, 38, 822–833. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Jiang, J.; Han, P.; Yuan, Q.; Zhang, J.; Zhang, X.; Xu, Y.; Cao, H.; Meng, Q.; Chen, L.; et al. Direct differentiation of atrial and ventricular myocytes from human embryonic stem cells by alternating retinoid signals. Cell Res. 2011, 21, 579–587. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Protze, S.I.; Laksman, Z.; Backx, P.H.; Keller, G.M. Human pluripotent stem cell-derived atrial and ventricular cardiomyocytes develop from distinct mesoderm populations. Cell Stem Cell 2017, 21, 179–194 e174. [Google Scholar] [CrossRef]
- Kleinsorge, M.; Cyganek, L. Subtype-directed differentiation of human ipscs into atrial and ventricular cardiomyocytes. STAR Protoc. 2020, 1, 100026. [Google Scholar] [CrossRef]
- Fu, J.D.; Rushing, S.N.; Lieu, D.K.; Chan, C.W.; Kong, C.W.; Geng, L.; Wilson, K.D.; Chiamvimonvat, N.; Boheler, K.R.; Wu, J.C.; et al. Distinct roles of microrna-1 and -499 in ventricular specification and functional maturation of human embryonic stem cell-derived cardiomyocytes. PLoS ONE 2011, 6, e27417. [Google Scholar] [CrossRef] [Green Version]
- Zhu, W.Z.; Xie, Y.; Moyes, K.W.; Gold, J.D.; Askari, B.; Laflamme, M.A. Neuregulin/erbb signaling regulates cardiac subtype specification in differentiating human embryonic stem cells. Circ. Res. 2010, 107, 776–786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yechikov, S.; Kao, H.K.J.; Chang, C.W.; Pretto, D.; Zhang, X.D.; Sun, Y.H.; Smithers, R.; Sirish, P.; Nolta, J.A.; Chan, J.W.; et al. Nodal inhibition promotes differentiation of pacemaker-like cardiomyocytes from human induced pluripotent stem cells. Stem Cell Res. 2020, 49, 102043. [Google Scholar] [CrossRef]
- Fukushima, H.; Yoshioka, M.; Kawatou, M.; Lopez-Davila, V.; Takeda, M.; Kanda, Y.; Sekino, Y.; Yoshida, Y.; Yamashita, J.K. Specific induction and long-term maintenance of high purity ventricular cardiomyocytes from human induced pluripotent stem cells. PLoS ONE 2020, 15, e0241287. [Google Scholar] [CrossRef]
- Schweizer, P.A.; Darche, F.F.; Ullrich, N.D.; Geschwill, P.; Greber, B.; Rivinius, R.; Seyler, C.; Muller-Decker, K.; Draguhn, A.; Utikal, J.; et al. Subtype-specific differentiation of cardiac pacemaker cell clusters from human induced pluripotent stem cells. Stem Cell Res. Ther. 2017, 8, 229. [Google Scholar] [CrossRef] [Green Version]
- Towbin, J.A. The role of cytoskeletal proteins in cardiomyopathies. Curr. Opin. Cell Biol. 1998, 10, 131–139. [Google Scholar] [CrossRef]
- Mudhar, H.S.; Wagner, B.E.; Suvarna, S.K. Electron microscopy of myocardial tissue. A nine year review. J. Clin. Pathol. 2001, 54, 321–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, B.; Li, Y.; Han, L.; Kaplan, A.D.; Ao, Y.; Kalra, S.; Bett, G.C.; Rasmusson, R.L.; Denning, C.; Yang, L. Modeling and study of the mechanism of dilated cardiomyopathy using induced pluripotent stem cells derived from individuals with duchenne muscular dystrophy. Dis. Models Mech. 2015, 8, 457–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verkerk, A.O.; Veerman, C.C.; Zegers, J.G.; Mengarelli, I.; Bezzina, C.R.; Wilders, R. Patch-clamp recording from human induced pluripotent stem cell-derived cardiomyocytes: Improving action potential characteristics through dynamic clamp. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kornreich, B.G. The patch clamp technique: Principles and technical considerations. J. Vet. Cardiol. 2007, 9, 25–37. [Google Scholar] [CrossRef]
- Emmenegger, V.; Obien, M.E.J.; Franke, F.; Hierlemann, A. Technologies to study action potential propagation with a focus on hd-meas. Front. Cell Neurosci. 2019, 13, 159. [Google Scholar] [CrossRef]
- Spira, M.E.; Hai, A. Multi-electrode array technologies for neuroscience and cardiology. Nat. Nanotechnol. 2013, 8, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Attin, M.; Clusin, W.T. Basic concepts of optical mapping techniques in cardiac electrophysiology. Biol. Res. Nurs. 2009, 11, 195–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lullmann, H.; Ziegler, A. Calcium, cell membrane, and excitation-contraction coupling. J. Cardiovasc. Pharmacol. 1987, 10 (Suppl. 1), S2-8. [Google Scholar] [CrossRef]
- Sedwick, C. How a mutation undermines cardiac function. J. Gen. Physiol. 2019, 151, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zatti, S.; Martewicz, S.; Serena, E.; Uno, N.; Giobbe, G.; Kazuki, Y.; Oshimura, M.; Elvassore, N. Complete restoration of multiple dystrophin isoforms in genetically corrected duchenne muscular dystrophy patient-derived cardiomyocytes. Mol. Therapy. Methods Clin. Dev. 2014, 1, 1. [Google Scholar] [CrossRef]
- Csobonyeiova, M.; Polak, S.; Danisovic, L. Toxicity testing and drug screening using ipsc-derived hepatocytes, cardiomyocytes, and neural cells. Can J. Physiol. Pharmacol. 2016, 94, 687–694. [Google Scholar] [CrossRef] [Green Version]
- Wallace, E.; Howard, L.; Liu, M.; O’Brien, T.; Ward, D.; Shen, S.; Prendiville, T. Long qt syndrome: Genetics and future perspective. Pediatr. Cardiol. 2019, 40, 1419–1430. [Google Scholar] [CrossRef] [Green Version]
- Sendfeld, F.; Selga, E.; Scornik, F.S.; Perez, G.J.; Mills, N.L.; Brugada, R. Experimental models of brugada syndrome. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef] [Green Version]
- Sun, N.; Yazawa, M.; Liu, J.; Han, L.; Sanchez-Freire, V.; Abilez, O.J.; Navarrete, E.G.; Hu, S.; Wang, L.; Lee, A.; et al. Patient-specific induced pluripotent stem cells as a model for familial dilated cardiomyopathy. Sci. Transl. Med. 2012, 4, 130ra147. [Google Scholar] [CrossRef] [Green Version]
- Pfeiffer-Kaushik, E.R.; Smith, G.L.; Cai, B.; Dempsey, G.T.; Hortigon-Vinagre, M.P.; Zamora, V.; Feng, S.; Ingermanson, R.; Zhu, R.; Hariharan, V.; et al. Electrophysiological characterization of drug response in hsc-derived cardiomyocytes using voltage-sensitive optical platforms. J. Pharmacol. Toxicol. Methods 2019, 99, 106612. [Google Scholar] [CrossRef] [PubMed]
- Izumi-Nakaseko, H.; Hagiwara-Nagasawa, M.; Naito, A.T.; Goto, A.; Chiba, K.; Sekino, Y.; Kanda, Y.; Sugiyama, A. Application of human induced pluripotent stem cell-derived cardiomyocytes sheets with microelectrode array system to estimate antiarrhythmic properties of multi-ion channel blockers. J. Pharmacol. Sci. 2018, 137, 372–378. [Google Scholar] [CrossRef] [PubMed]
- Kussauer, S.; David, R.; Lemcke, H. Hipscs derived cardiac cells for drug and toxicity screening and disease modeling: What micro- electrode-array analyses can tell us. Cells 2019, 8. [Google Scholar] [CrossRef] [Green Version]
- Huo, J.; Wei, F.; Cai, C.; Lyn-Cook, B.; Pang, L. Sex-related differences in drug-induced qt prolongation and torsades de pointes: A new model system with human ipsc-cms. Toxicol Sci. 2019, 167, 360–374. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.Z.; Termglinchan, V.; Shao, N.Y.; Itzhaki, I.; Liu, C.; Ma, N.; Tian, L.; Wang, V.Y.; Chang, A.C.Y.; Guo, H.; et al. A human ipsc double-reporter system enables purification of cardiac lineage subpopulations with distinct function and drug response profiles. Cell Stem Cell 2019, 24, 802–811 e805. [Google Scholar] [CrossRef]
- Devalla, H.D.; Schwach, V.; Ford, J.W.; Milnes, J.T.; El-Haou, S.; Jackson, C.; Gkatzis, K.; Elliott, D.A.; Chuva de Sousa Lopes, S.M.; Mummery, C.L.; et al. Atrial-like cardiomyocytes from human pluripotent stem cells are a robust preclinical model for assessing atrial-selective pharmacology. EMBO Mol. Med. 2015, 7, 394–410. [Google Scholar] [CrossRef]
- Lemme, M.; Ulmer, B.M.; Lemoine, M.D.; Zech, A.T.L.; Flenner, F.; Ravens, U.; Reichenspurner, H.; Rol-Garcia, M.; Smith, G.; Hansen, A.; et al. Atrial-like engineered heart tissue: An in vitro model of the human atrium. Stem Cell Rep. 2018, 11, 1378–1390. [Google Scholar] [CrossRef] [Green Version]
- Geng, L.; Kong, C.W.; Wong, A.O.T.; Shum, A.M.; Chow, M.Z.Y.; Che, H.; Zhang, C.; Yau, K.L.; Chan, C.W.; Keung, W.; et al. Probing flecainide block of ina using human pluripotent stem cell-derived ventricular cardiomyocytes adapted to automated patch-clamping and 2d monolayers. Toxicol. Lett. 2018, 294, 61–72. [Google Scholar] [CrossRef]
- Sacchetto, C.; Vitiello, L.; de Windt, L.J.; Rampazzo, A.; Calore, M. Modeling cardiovascular diseases with hipsc-derived cardiomyocytes in 2d and 3d cultures. Int. J. Mol. Sci. 2020, 21. [Google Scholar] [CrossRef]
- Shadrin, I.Y.; Allen, B.W.; Qian, Y.; Jackman, C.P.; Carlson, A.L.; Juhas, M.E.; Bursac, N. Cardiopatch platform enables maturation and scale-up of human pluripotent stem cell-derived engineered heart tissues. Nat. Commun. 2017, 8, 1825. [Google Scholar] [CrossRef] [Green Version]
- Goldfracht, I.; Efraim, Y.; Shinnawi, R.; Kovalev, E.; Huber, I.; Gepstein, A.; Arbel, G.; Shaheen, N.; Tiburcy, M.; Zimmermann, W.H.; et al. Engineered heart tissue models from hipsc-derived cardiomyocytes and cardiac ecm for disease modeling and drug testing applications. Acta Biomater. 2019, 92, 145–159. [Google Scholar] [CrossRef] [PubMed]
- Tiburcy, M.; Hudson, J.E.; Balfanz, P.; Schlick, S.; Meyer, T.; Chang Liao, M.L.; Levent, E.; Raad, F.; Zeidler, S.; Wingender, E.; et al. Defined engineered human myocardium with advanced maturation for applications in heart failure modeling and repair. Circulation 2017, 135, 1832–1847. [Google Scholar] [CrossRef] [PubMed]
- Cashman, T.J.; Josowitz, R.; Johnson, B.V.; Gelb, B.D.; Costa, K.D. Human engineered cardiac tissues created using induced pluripotent stem cells reveal functional characteristics of braf-mediated hypertrophic cardiomyopathy. PLoS ONE 2016, 11, e0146697. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; McCain, M.L.; Yang, L.; He, A.; Pasqualini, F.S.; Agarwal, A.; Yuan, H.; Jiang, D.; Zhang, D.; Zangi, L.; et al. Modeling the mitochondrial cardiomyopathy of barth syndrome with induced pluripotent stem cell and heart-on-chip technologies. Nat. Med. 2014, 20, 616–623. [Google Scholar] [CrossRef]
- Hinson, J.T.; Chopra, A.; Nafissi, N.; Polacheck, W.J.; Benson, C.C.; Swist, S.; Gorham, J.; Yang, L.; Schafer, S.; Sheng, C.C.; et al. Heart disease. Titin mutations in ips cells define sarcomere insufficiency as a cause of dilated cardiomyopathy. Science 2015, 349, 982–986. [Google Scholar] [CrossRef] [Green Version]
- Lemme, M.; Braren, I.; Prondzynski, M.; Aksehirlioglu, B.; Ulmer, B.M.; Schulze, M.L.; Ismaili, D.; Meyer, C.; Hansen, A.; Christ, T.; et al. Chronic intermittent tachypacing by an optogenetic approach induces arrhythmia vulnerability in human engineered heart tissue. Cardiovasc. Res. 2020, 116, 1487–1499. [Google Scholar] [CrossRef] [Green Version]
- Long, C.; Li, H.; Tiburcy, M.; Rodriguez-Caycedo, C.; Kyrychenko, V.; Zhou, H.; Zhang, Y.; Min, Y.L.; Shelton, J.M.; Mammen, P.P.A.; et al. Correction of diverse muscular dystrophy mutations in human engineered heart muscle by single-site genome editing. Sci. Adv. 2018, 4, eaap9004. [Google Scholar] [CrossRef] [Green Version]
- Shiba, Y.; Gomibuchi, T.; Seto, T.; Wada, Y.; Ichimura, H.; Tanaka, Y.; Ogasawara, T.; Okada, K.; Shiba, N.; Sakamoto, K.; et al. Allogeneic transplantation of ips cell-derived cardiomyocytes regenerates primate hearts. Nature 2016, 538, 388–391. [Google Scholar] [CrossRef]
- Laflamme, M.A.; Chen, K.Y.; Naumova, A.V.; Muskheli, V.; Fugate, J.A.; Dupras, S.K.; Reinecke, H.; Xu, C.; Hassanipour, M.; Police, S.; et al. Cardiomyocytes derived from human embryonic stem cells in pro-survival factors enhance function of infarcted rat hearts. Nat. Biotechnol. 2007, 25, 1015–1024. [Google Scholar] [CrossRef]
- Caspi, O.; Huber, I.; Kehat, I.; Habib, M.; Arbel, G.; Gepstein, A.; Yankelson, L.; Aronson, D.; Beyar, R.; Gepstein, L. Transplantation of human embryonic stem cell-derived cardiomyocytes improves myocardial performance in infarcted rat hearts. J. Am. Coll. Cardiol. 2007, 50, 1884–1893. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Gregorich, Z.R.; Zhu, W.; Mattapally, S.; Oduk, Y.; Lou, X.; Kannappan, R.; Borovjagin, A.V.; Walcott, G.P.; Pollard, A.E.; et al. Large cardiac muscle patches engineered from human induced-pluripotent stem cell-derived cardiac cells improve recovery from myocardial infarction in swine. Circulation 2018, 137, 1712–1730. [Google Scholar] [CrossRef]
- Zhao, X.; Chen, H.; Xiao, D.; Yang, H.; Itzhaki, I.; Qin, X.; Chour, T.; Aguirre, A.; Lehmann, K.; Kim, Y.; et al. Comparison of non-human primate versus human induced pluripotent stem cell-derived cardiomyocytes for treatment of myocardial infarction. Stem Cell Rep. 2018, 10, 422–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rojas, S.V.; Kensah, G.; Rotaermel, A.; Baraki, H.; Kutschka, I.; Zweigerdt, R.; Martin, U.; Haverich, A.; Gruh, I.; Martens, A. Transplantation of purified ipsc-derived cardiomyocytes in myocardial infarction. PLoS ONE 2017, 12, e0173222. [Google Scholar] [CrossRef]
- Willi, L.; Agranovich, B.; Abramovich, I.; Freimark, D.; Arad, M.; Gottlieb, E.; Binah, O. Bioenergetic and metabolic impairments in duchenne muscular dystrophy (dmd) patients’ ipsc-derived cardiomyocytes. Eur. Heart J. 2020, 41. [Google Scholar] [CrossRef]
- Tsurumi, F.; Baba, S.; Yoshinaga, D.; Umeda, K.; Hirata, T.; Takita, J.; Heike, T. The intracellular ca2+ concentration is elevated in cardiomyocytes differentiated from hipscs derived from a duchenne muscular dystrophy patient. PLoS ONE 2019, 14, e0213768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mournetas, V.; Massourides, E.; Dupont, J.B.; Kornobis, E.; Polveche, H.; Jarrige, M.; Dorval, A.R.L.; Gosselin, M.R.F.; Manousopoulou, A.; Garbis, S.D.; et al. Myogenesis modelled by human pluripotent stem cells: A multi-omic study of duchenne myopathy early onset. J. Cachexia Sarcopenia Muscle 2021, 12, 209–232. [Google Scholar] [CrossRef]
- Eisen, B.; Ben Jehuda, R.; Cuttitta, A.J.; Mekies, L.N.; Shemer, Y.; Baskin, P.; Reiter, I.; Willi, L.; Freimark, D.; Gherghiceanu, M.; et al. Electrophysiological abnormalities in induced pluripotent stem cell-derived cardiomyocytes generated from duchenne muscular dystrophy patients. J. Cell Mol. Med. 2019, 23, 2125–2135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birket, M.J.; Raibaud, S.; Lettieri, M.; Adamson, A.D.; Letang, V.; Cervello, P.; Redon, N.; Ret, G.; Viale, S.; Wang, B.; et al. A human stem cell model of fabry disease implicates limp-2 accumulation in cardiomyocyte pathology. Stem Cell Rep. 2019, 13, 380–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuramoto, Y.; Naito, A.T.; Tojo, H.; Sakai, T.; Ito, M.; Shibamoto, M.; Nakagawa, A.; Higo, T.; Okada, K.; Yamaguchi, T.; et al. Generation of fabry cardiomyopathy model for drug screening using induced pluripotent stem cell-derived cardiomyocytes from a female fabry patient. J. Mol. Cell Cardiol. 2018, 121, 256–265. [Google Scholar] [CrossRef]
- Chien, Y.; Chien, C.S.; Chiang, H.C.; Huang, W.L.; Chou, S.J.; Chang, W.C.; Chang, Y.L.; Leu, H.B.; Chen, K.H.; Wang, K.L.; et al. Interleukin-18 deteriorates fabry cardiomyopathy and contributes to the development of left ventricular hypertrophy in fabry patients with gla ivs4+919 g>a mutation. Oncotarget 2016, 7, 87161–87179. [Google Scholar] [CrossRef] [PubMed]
- Bienvenu, T.; Philippe, C.; De Roux, N.; Raynaud, M.; Bonnefond, J.P.; Pasquier, L.; Lesca, G.; Mancini, J.; Jonveaux, P.; Moncla, A.; et al. The incidence of rett syndrome in france. Pediatr. Neurol. 2006, 34, 372–375. [Google Scholar] [CrossRef]
- Chahrour, M.; Zoghbi, H.Y. The story of rett syndrome: From clinic to neurobiology. Neuron 2007, 56, 422–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itier, J.M.; Ret, G.; Viale, S.; Sweet, L.; Bangari, D.; Caron, A.; Le-Gall, F.; Benichou, B.; Leonard, J.; Deleuze, J.F.; et al. Effective clearance of gl-3 in a human ipsc-derived cardiomyocyte model of fabry disease. J. Inherit. Metab. Dis. 2014, 37, 1013–1022. [Google Scholar] [CrossRef] [PubMed]
- Hashem, S.I.; Perry, C.N.; Bauer, M.; Han, S.; Clegg, S.D.; Ouyang, K.; Deacon, D.C.; Spinharney, M.; Panopoulos, A.D.; Izpisua Belmonte, J.C.; et al. Brief report: Oxidative stress mediates cardiomyocyte apoptosis in a human model of danon disease and heart failure. Stem Cells 2015, 33, 2343–2350. [Google Scholar] [CrossRef] [Green Version]
- Marchetto, M.C.; Carromeu, C.; Acab, A.; Yu, D.; Yeo, G.W.; Mu, Y.; Chen, G.; Gage, F.H.; Muotri, A.R. A model for neural development and treatment of rett syndrome using human induced pluripotent stem cells. Cell 2010, 143, 527–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, P.; Mesci, P.; Carromeu, C.; McClatchy, D.R.; Schiapparelli, L.; Yates, J.R., 3rd; Muotri, A.R.; Cline, H.T. Exosomes regulate neurogenesis and circuit assembly. Proc. Natl. Acad. Sci. USA 2019, 116, 16086–16094. [Google Scholar] [CrossRef] [Green Version]
- Landucci, E.; Brindisi, M.; Bianciardi, L.; Catania, L.M.; Daga, S.; Croci, S.; Frullanti, E.; Fallerini, C.; Butini, S.; Brogi, S.; et al. Ipsc-derived neurons profiling reveals gabaergic circuit disruption and acetylated alpha-tubulin defect which improves after ihdac6 treatment in rett syndrome. Exp. Cell Res. 2018, 368, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Gomathi, M.; Balachandar, V. Novel therapeutic approaches: Rett syndrome and human induced pluripotent stem cell technology. Stem Cell Investig. 2017, 4, 20. [Google Scholar] [CrossRef] [Green Version]
- Crisafulli, S.; Sultana, J.; Fontana, A.; Salvo, F.; Messina, S.; Trifiro, G. Global epidemiology of duchenne muscular dystrophy: An updated systematic review and meta-analysis. Orphanet J. Rare. Dis. 2020, 15, 141. [Google Scholar] [CrossRef]
- Duan, D.; Goemans, N.; Takeda, S.; Mercuri, E.; Aartsma-Rus, A. Duchenne muscular dystrophy. Nat. Rev. Dis. Primers 2021, 7, 13. [Google Scholar] [CrossRef] [PubMed]
- Monaco, A.P.; Bertelson, C.J.; Liechti-Gallati, S.; Moser, H.; Kunkel, L.M. An explanation for the phenotypic differences between patients bearing partial deletions of the dmd locus. Genomics 1988, 2, 90–95. [Google Scholar] [CrossRef]
- Wolinsky, H.; Goldfischer, S.; Schiller, B.; Kasak, L.E. Lysosomes in aortic smooth muscle cells. Effects of hypertension. Am. J. Pathol. 1973, 73, 727–734. [Google Scholar] [PubMed]
- Kaspar, R.W.; Allen, H.D.; Montanaro, F. Current understanding and management of dilated cardiomyopathy in duchenne and becker muscular dystrophy. J. Am. Acad. Nurse Pract. 2009, 21, 241–249. [Google Scholar] [CrossRef] [Green Version]
- Mendell, J.R.; Shilling, C.; Leslie, N.D.; Flanigan, K.M.; al-Dahhak, R.; Gastier-Foster, J.; Kneile, K.; Dunn, D.M.; Duval, B.; Aoyagi, A.; et al. Evidence-based path to newborn screening for duchenne muscular dystrophy. Ann. Neurol. 2012, 71, 304–313. [Google Scholar] [CrossRef]
- Frankel, K.A.; Rosser, R.J. The pathology of the heart in progressive muscular dystrophy: Epimyocardial fibrosis. Hum. Pathol. 1976, 7, 375–386. [Google Scholar] [CrossRef]
- Japp, A.G.; Gulati, A.; Cook, S.A.; Cowie, M.R.; Prasad, S.K. The diagnosis and evaluation of dilated cardiomyopathy. J. Am. Coll. Cardiol. 2016, 67, 2996–3010. [Google Scholar] [CrossRef]
- Zhu, S.; Law, A.H.Y.; Deng, R.; Poon, E.N.Y.; Lo, C.W.; Kwong, A.K.Y.; Liang, R.; Chan, K.Y.K.; Wong, W.L.; Tan-Un, K.C.; et al. Generation of genomic-integration-free human induced pluripotent stem cells and the derived cardiomyocytes of x-linked dilated cardiomyopathy from dmd gene mutation. Stem Cell Res. 2020, 49, 102040. [Google Scholar] [CrossRef] [PubMed]
- De Palma, C.; Morisi, F.; Cheli, S.; Pambianco, S.; Cappello, V.; Vezzoli, M.; Rovere-Querini, P.; Moggio, M.; Ripolone, M.; Francolini, M.; et al. Autophagy as a new therapeutic target in duchenne muscular dystrophy. Cell Death Dis. 2012, 3, e418. [Google Scholar] [CrossRef] [Green Version]
- Guan, X.; Mack, D.L.; Moreno, C.M.; Strande, J.L.; Mathieu, J.; Shi, Y.; Markert, C.D.; Wang, Z.; Liu, G.; Lawlor, M.W.; et al. Dystrophin-deficient cardiomyocytes derived from human urine: New biologic reagents for drug discovery. Stem Cell Res. 2014, 12, 467–480. [Google Scholar] [CrossRef] [Green Version]
- Takami, Y.; Takeshima, Y.; Awano, H.; Okizuka, Y.; Yagi, M.; Matsuo, M. High incidence of electrocardiogram abnormalities in young patients with duchenne muscular dystrophy. Pediatr. Neurol. 2008, 39, 399–403. [Google Scholar] [CrossRef] [PubMed]
- Maroto, R.; Raso, A.; Wood, T.G.; Kurosky, A.; Martinac, B.; Hamill, O.P. Trpc1 forms the stretch-activated cation channel in vertebrate cells. Nat. Cell Biol. 2005, 7, 179–185. [Google Scholar] [CrossRef]
- Meikle, P.J.; Hopwood, J.J.; Clague, A.E.; Carey, W.F. Prevalence of lysosomal storage disorders. JAMA 1999, 281, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Poorthuis, B.J.; Wevers, R.A.; Kleijer, W.J.; Groener, J.E.; de Jong, J.G.; van Weely, S.; Niezen-Koning, K.E.; van Diggelen, O.P. The frequency of lysosomal storage diseases in the netherlands. Hum. Genet. 1999, 105, 151–156. [Google Scholar] [CrossRef]
- Spada, M.; Pagliardini, S.; Yasuda, M.; Tukel, T.; Thiagarajan, G.; Sakuraba, H.; Ponzone, A.; Desnick, R.J. High incidence of later-onset fabry disease revealed by newborn screening. Am. J. Hum. Genet. 2006, 79, 31–40. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.Y.; Lelis, A.; Mirocha, J.; Wilcox, W.R. Heterozygous fabry women are not just carriers, but have a significant burden of disease and impaired quality of life. Genet. Med. 2007, 9, 34–45. [Google Scholar] [CrossRef] [Green Version]
- Zarate, Y.A.; Hopkin, R.J. Fabry’s disease. Lancet 2008, 372, 1427–1435. [Google Scholar] [CrossRef]
- Gieselmann, V. Cellular pathophysiology of lysosomal storage diseases. In Fabry Disease: Perspectives from 5 Years of Fos; Mehta, A., Beck, M., Sunder-Plassmann, G., Eds.; Oxford PharmaGenesis: Oxford, UK, 2006. [Google Scholar]
- Morrissey, R.P.; Philip, K.J.; Schwarz, E.R. Cardiac abnormalities in anderson-fabry disease and fabry’s cardiomyopathy. Cardiovasc. J. Afr. 2011, 22, 38–44. [Google Scholar] [PubMed]
- Linhart, A. The heat in fabry disease. In Fabry Disease: Perspectives from 5 Years of Fos; Mehta, A.B., Beck, M., Sunder-Plassmann, G., Eds.; Oxford PharmaGenesis: Oxford, UK, 2006. [Google Scholar]
- Schiffmann, R.; Kopp, J.B.; Austin, H.A., 3rd; Sabnis, S.; Moore, D.F.; Weibel, T.; Balow, J.E.; Brady, R.O. Enzyme replacement therapy in fabry disease: A randomized controlled trial. JAMA 2001, 285, 2743–2749. [Google Scholar] [CrossRef] [PubMed]
- Borger, D.K.; McMahon, B.; Roshan Lal, T.; Serra-Vinardell, J.; Aflaki, E.; Sidransky, E. Induced pluripotent stem cell models of lysosomal storage disorders. Dis. Model. Mech. 2017, 10, 691–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawagoe, S.; Higuchi, T.; Otaka, M.; Shimada, Y.; Kobayashi, H.; Ida, H.; Ohashi, T.; Okano, H.J.; Nakanishi, M.; Eto, Y. Morphological features of ips cells generated from fabry disease skin fibroblasts using sendai virus vector (sevdp). Mol. Genet. Metab. 2013, 109, 386–389. [Google Scholar] [CrossRef] [PubMed]
- Renieri, A.; Meloni, I.; Longo, I.; Ariani, F.; Mari, F.; Pescucci, C.; Cambi, F. Rett syndrome: The complex nature of a monogenic disease. J. Mol. Med. 2003, 81, 346–354. [Google Scholar] [CrossRef]
- CENTOGENE. Application of iPSC Technology in Orphan Drug Development. Available online: https://www.centogene.com/science/whitepapers/application-of-ipsc-technology-in-orphan-drug-development.html (accessed on 20 July 2021).
- Kubota, T. Advantage in human induced pluripotent stem cells research of x-linked genetic diseases for drug screening. Int. J. Stem Cell Res. Ther. 2015, 2, 12. [Google Scholar] [CrossRef]
- Huang, H.P.; Chuang, C.Y.; Kuo, H.C. Induced pluripotent stem cell technology for disease modeling and drug screening with emphasis on lysosomal storage diseases. Stem Cell Res. Ther. 2012, 3, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chien, Y.; Chou, S.J.; Chang, Y.L.; Leu, H.B.; Yang, Y.P.; Tsai, P.H.; Lai, Y.H.; Chen, K.H.; Chang, W.C.; Sung, S.H.; et al. Inhibition of arachidonate 12/15-lipoxygenase improves alpha-galactosidase efficacy in ipsc-derived cardiomyocytes from fabry patients. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef] [Green Version]
- Acampa, M.; Guideri, F. Cardiac disease and rett syndrome. Arch. Dis. Child. 2006, 91, 440–443. [Google Scholar] [CrossRef] [Green Version]
- Muffat, J.; Li, Y.; Yuan, B.; Mitalipova, M.; Omer, A.; Corcoran, S.; Bakiasi, G.; Tsai, L.H.; Aubourg, P.; Ransohoff, R.M.; et al. Efficient derivation of microglia-like cells from human pluripotent stem cells. Nat. Med. 2016, 22, 1358–1367. [Google Scholar] [CrossRef] [Green Version]
- Bienvenu, T.; Carrie, A.; de Roux, N.; Vinet, M.C.; Jonveaux, P.; Couvert, P.; Villard, L.; Arzimanoglou, A.; Beldjord, C.; Fontes, M.; et al. Mecp2 mutations account for most cases of typical forms of rett syndrome. Hum. Mol. Genet. 2000, 9, 1377–1384. [Google Scholar] [CrossRef]
- Zachariah, R.M.; Rastegar, M. Linking epigenetics to human disease and rett syndrome: The emerging novel and challenging concepts in mecp2 research. Neural Plast. 2012, 2012, 415825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gold, W.A.; Krishnarajy, R.; Ellaway, C.; Christodoulou, J. Rett syndrome: A genetic update and clinical review focusing on comorbidities. ACS Chem. Neurosci. 2018, 9, 167–176. [Google Scholar] [CrossRef]
- Sekul, E.A.; Moak, J.P.; Schultz, R.J.; Glaze, D.G.; Dunn, J.K.; Percy, A.K. Electrocardiographic findings in rett syndrome: An explanation for sudden death? J. Pediatr. 1994, 125, 80–82. [Google Scholar] [CrossRef]
- Zhu, Y.C.; Xiong, Z.Q. Molecular and synaptic bases of cdkl5 disorder. Dev. Neurobiol. 2019, 79, 8–19. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; McMahon, C.J.; Smith, L.R.; Bersola, J.; Adesina, A.M.; Breinholt, J.P.; Kearney, D.L.; Dreyer, W.J.; Denfield, S.W.; Price, J.F.; et al. Danon disease as an underrecognized cause of hypertrophic cardiomyopathy in children. Circulation 2005, 112, 1612–1617. [Google Scholar] [CrossRef] [Green Version]
- Myerowitz, R.; Puertollano, R.; Raben, N. Impaired autophagy: The collateral damage of lysosomal storage disorders. EBioMedicine 2021, 63, 103166. [Google Scholar] [CrossRef]
- Harris, K.M.; Spirito, P.; Maron, M.S.; Zenovich, A.G.; Formisano, F.; Lesser, J.R.; Mackey-Bojack, S.; Manning, W.J.; Udelson, J.E.; Maron, B.J. Prevalence, clinical profile, and significance of left ventricular remodeling in the end-stage phase of hypertrophic cardiomyopathy. Circulation 2006, 114, 216–225. [Google Scholar] [CrossRef]
- Sugie, K.; Yamamoto, A.; Murayama, K.; Oh, S.J.; Takahashi, M.; Mora, M.; Riggs, J.E.; Colomer, J.; Iturriaga, C.; Meloni, A.; et al. Clinicopathological features of genetically confirmed danon disease. Neurology 2002, 58, 1773–1778. [Google Scholar] [CrossRef] [Green Version]
- D’Souza, R.S.; Levandowski, C.; Slavov, D.; Graw, S.L.; Allen, L.A.; Adler, E.; Mestroni, L.; Taylor, M.R. Danon disease: Clinical features, evaluation, and management. Circ. Heart Fail. 2014, 7, 843–849. [Google Scholar] [CrossRef] [Green Version]
- Hashem, S.I.; Murphy, A.N.; Divakaruni, A.S.; Klos, M.L.; Nelson, B.C.; Gault, E.C.; Rowland, T.J.; Perry, C.N.; Gu, Y.; Dalton, N.D.; et al. Impaired mitophagy facilitates mitochondrial damage in danon disease. J. Mol. Cell Cardiol. 2017, 108, 86–94. [Google Scholar] [CrossRef]
- Chi, C.; Leonard, A.; Knight, W.E.; Beussman, K.M.; Zhao, Y.; Cao, Y.; Londono, P.; Aune, E.; Trembley, M.A.; Small, E.M.; et al. Lamp-2b regulates human cardiomyocyte function by mediating autophagosome–lysosome fusion. Proc. Natl. Acad. Sci. USA 2019, 116, 556–565. [Google Scholar] [CrossRef] [Green Version]
- Del Favero, G.; Bonifacio, A.; Rowland, T.J.; Gao, S.; Song, K.; Sergo, V.; Adler, E.D.; Mestroni, L.; Sbaizero, O.; Taylor, M.R.G. Danon disease-associated lamp-2 deficiency drives metabolic signature indicative of mitochondrial aging and fibrosis in cardiac tissue and hipsc-derived cardiomyocytes. J. Clin. Med. 2020, 9. [Google Scholar] [CrossRef]
- Goldberg, A.L. Protein degradation and protection against misfolded or damaged proteins. Nature 2003, 426, 895–899. [Google Scholar] [CrossRef]
- Lee, Y.K.; Ng, K.M.; Lai, W.H.; Chan, Y.C.; Lau, Y.M.; Lian, Q.; Tse, H.F.; Siu, C.W. Calcium homeostasis in human induced pluripotent stem cell-derived cardiomyocytes. Stem Cell Rev. 2011, 7, 976–986. [Google Scholar] [CrossRef] [Green Version]
- Koivumaki, J.T.; Naumenko, N.; Tuomainen, T.; Takalo, J.; Oksanen, M.; Puttonen, K.A.; Lehtonen, S.; Kuusisto, J.; Laakso, M.; Koistinaho, J.; et al. Structural immaturity of human ipsc-derived cardiomyocytes: In silico investigation of effects on function and disease modeling. Front. Physiol. 2018, 9, 80. [Google Scholar] [CrossRef]
- Itzhaki, I.; Rapoport, S.; Huber, I.; Mizrahi, I.; Zwi-Dantsis, L.; Arbel, G.; Schiller, J.; Gepstein, L. Calcium handling in human induced pluripotent stem cell derived cardiomyocytes. PLoS ONE 2011, 6, e18037. [Google Scholar] [CrossRef]
- Law, M.L.; Cohen, H.; Martin, A.A.; Angulski, A.B.B.; Metzger, J.M. Dysregulation of calcium handling in duchenne muscular dystrophy-associated dilated cardiomyopathy: Mechanisms and experimental therapeutic strategies. J. Clin. Med. 2020, 9. [Google Scholar] [CrossRef] [Green Version]
- Lewandowski, J.; Rozwadowska, N.; Kolanowski, T.J.; Malcher, A.; Zimna, A.; Rugowska, A.; Fiedorowicz, K.; Labedz, W.; Kubaszewski, L.; Chojnacka, K.; et al. The impact of in vitro cell culture duration on the maturation of human cardiomyocytes derived from induced pluripotent stem cells of myogenic origin. Cell Transpl. 2018, 27, 1047–1067. [Google Scholar] [CrossRef] [Green Version]
- Abilez, O.J.; Tzatzalos, E.; Yang, H.; Zhao, M.T.; Jung, G.; Zollner, A.M.; Tiburcy, M.; Riegler, J.; Matsa, E.; Shukla, P.; et al. Passive stretch induces structural and functional maturation of engineered heart muscle as predicted by computational modeling. Stem Cells 2018, 36, 265–277. [Google Scholar] [CrossRef] [Green Version]
- Ronaldson-Bouchard, K.; Ma, S.P.; Yeager, K.; Chen, T.; Song, L.; Sirabella, D.; Morikawa, K.; Teles, D.; Yazawa, M.; Vunjak-Novakovic, G. Advanced maturation of human cardiac tissue grown from pluripotent stem cells. Nature 2018, 556, 239–243. [Google Scholar] [CrossRef]
- Yang, X.; Rodriguez, M.; Pabon, L.; Fischer, K.A.; Reinecke, H.; Regnier, M.; Sniadecki, N.J.; Ruohola-Baker, H.; Murry, C.E. Tri-iodo-l-thyronine promotes the maturation of human cardiomyocytes-derived from induced pluripotent stem cells. J. Mol. Cell Cardiol. 2014, 72, 296–304. [Google Scholar] [CrossRef] [Green Version]
- Kuppusamy, K.T.; Jones, D.C.; Sperber, H.; Madan, A.; Fischer, K.A.; Rodriguez, M.L.; Pabon, L.; Zhu, W.Z.; Tulloch, N.L.; Yang, X.; et al. Let-7 family of microrna is required for maturation and adult-like metabolism in stem cell-derived cardiomyocytes. Proc. Natl. Acad. Sci. USA 2015, 112, E2785–E2794. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | Gene | Type of Cardiomyopathy | Extra-Cardiac Manifestations |
|---|---|---|---|
| Duchenne muscular dystrophy | DMD | Dilated [10] | Muscle weakness [11] Growth delay [12] |
| Fabry disease | GLA | Hypertrophic [13] | Neuropathic pain Renal impairment Angiokeratoma [14] |
| Familial cardiac filaminopathy | FLNA | Dilated [15] | Periventricular heterotopia [16] |
| Hypertrophic [17] | |||
| Both dilated and hypertrophic [18] | |||
| Danon disease | LAMP2 | Hypertrophic [19] | Skeletal myopathy Retinopathy [20] Cognitive impairment [21] |
| Dilated [22] Left-ventricular non-compaction [23] | |||
| Rett syndrome | MECP2 | Arrhythmogenic [24] | Extrapyramidal motor dysfunction [25] Epilepsy [26] Bone fracture [27] |
| X-linked myotubular myopathy | MTM1 | Dilated [28] | Respiratory failure [29] Muscle weakness |
| Disease | Pathophysiological Changes | References |
|---|---|---|
| Duchenne muscular dystrophy | Express truncated non-functional dystrophin protein, disrupted myofibrils, calcium overloads, disrupted membrane fragility, increased DAD and OPPs | [7,117,118,119,120] |
| Fabry disease | Gb3 accumulation, deficient enzyme, high ANP expression Decreased contractility, cellular hypertrophy, disturbed ion channel electrical currents, peripheral displacement of myofibrils | [121,122,123] |
| Danon disease | Deficiency of LAMP2, accumulation of autophagy materials, increased mitochondrial oxidative stress, increased apoptosis, altered metabolism, impaired contractile function, reduced calcium transients | [32,124,125] |
| Rett syndrome | Decreased soma size, fewer glutamatergic synapses, reduced spine density, altered calcium signalling and electrophysiological defects, decreased axon outgrowth, dendritic morphogenesis Changes in exosome protein cargo and signalling bioactivity | [124,125] |
| Disease | Drug/Treatment Tested | References |
|---|---|---|
| Duchenne muscluar dystrophy | Proteasome inhibitors, polaxamer188, human artificial chromosomes | [7,81,89] |
| Fabry disease | glucosylceramide synthase inhibitor | [126] |
| Danon disease | N-acetylcystein, rotenone, DNA-demethylating drugs | [32,127] |
| Rett syndrome | IGF-1, gentamycin, exosomes hstone deacetylase inhibitors, medhya rasayana | [128,129,130,131] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, J.; Chou, O.H.-I.; Tse, Y.-L.; Ng, K.-M.; Tse, H.-F. Application of Patient-Specific iPSCs for Modelling and Treatment of X-Linked Cardiomyopathies. Int. J. Mol. Sci. 2021, 22, 8132. https://doi.org/10.3390/ijms22158132
Zhang J, Chou OH-I, Tse Y-L, Ng K-M, Tse H-F. Application of Patient-Specific iPSCs for Modelling and Treatment of X-Linked Cardiomyopathies. International Journal of Molecular Sciences. 2021; 22(15):8132. https://doi.org/10.3390/ijms22158132
Chicago/Turabian StyleZhang, Jennifer, Oscar Hou-In Chou, Yiu-Lam Tse, Kwong-Man Ng, and Hung-Fat Tse. 2021. "Application of Patient-Specific iPSCs for Modelling and Treatment of X-Linked Cardiomyopathies" International Journal of Molecular Sciences 22, no. 15: 8132. https://doi.org/10.3390/ijms22158132