1. Introduction
Idiopathic pulmonary fibrosis (IPF) is an aging-associated, progressive, and irreversible disease of unknown etiology. Currently, therapeutic options remain limited, and there is no efficient treatment to improve expectancy and quality of life [
1,
2]. Furthermore, the pathogenic mechanisms have not been fully elucidated. However, strong evidence suggests that IPF begins with micro-lesions and activation of the alveolar epithelium, which secretes various mediators that induce migration, proliferation, and activation of fibroblasts that produce excessive extracellular matrix amounts with progressive destruction of the lung parenchyma [
3,
4]. An intriguing aspect of the pathogenesis of IPF is the resistance shown by fibroblasts and myofibroblasts to apoptosis, while on the contrary, alveolar epithelial cells are very susceptible to this process [
3,
4,
5]. The mechanisms of this paradoxical behavior are uncertain, but in the case of epithelial cells, their susceptibility has been attributed, at least partially, to mitochondrial dysfunction [
5].
Fibroblasts and myofibroblasts are the critical effector cells in tissue fibrosis due to their participation in elaborating the extracellular matrix and contraction mechanisms and remodeling of damaged tissue [
6]. During physiological regeneration, fibroblasts are eliminated by apoptosis, and evasion or resistance to this process has been associated with progressive fibrosis. In this context, it has been reported that fibroblasts and myofibroblasts from patients with IPF are resistant to apoptosis, although the molecular mechanisms involved are uncertain [
7,
8,
9,
10]. Due to the central role that mitochondria play in the implementation, amplification, and regulation of cellular apoptosis, this organelle would be involved in the apoptosis resistance of IPF fibroblasts, but this has not yet been studied in depth.
Mitochondria are central organelles that participate in vital metabolic processes, synthesize most ATP, and regulate several signaling cascades, including apoptosis [
11]. The transduction of apoptotic signaling of cell death requires permeabilization of the mitochondrial membrane and the subsequent release of pro-apoptotic factors from the mitochondrial inter-membrane space (IMS) (i.e., cytochrome c, apoptosis-binding protein with low pI (Smac/Diablo), Endonuclease G, Serine protease Htra2 (Htra2/Omi), and apoptosis-inducing factor (AIF)) [
12]. The release of cytochrome c from mitochondria is commonly considered as the “point-of-no-return” in the sequence of events leading to apoptosis and involves changes in the mitochondrial membrane permeability transition (mtMPT) [
13]. mtMPT results in the drop of the mitochondrial membrane potential (Δψm), osmotic swelling of the mitochondrial matrix, rupture of the outer mitochondrial membrane, and release of cytochrome c [
14].
The mitochondrial permeability transition pore (mPTP) is a non-specific voltage-dependent channel that forms in the inner mitochondrial membrane. The molecular nature of mPTP is controversial due to the lack of knowledge about the identity of the proteins that form it. There are different hypotheses about the components that constitute the mPTP. One of them suggests the participation of the proteins adenine nucleotide translocator (ANT), voltage-dependent anion-selective channel (VDAC), and cyclophilin D (CypD), suggesting that ANT/VDAC form the basic unit of the mPTP, and that the recruitment of additional proteins, such as CypD, Bax, hexokinase, or translocator protein (TSPO), modulate the activation of the mPTP. Another hypothesis recently developed has implicated different subunits of the F
1F
0–ATP synthase as the inner membrane pore-forming unit of the mPTP [
15,
16].
Urbani et al. [
17] proposed that active F
1F
0-ATP synthase is responsible for the formation of mPTP; they used highly purified F
1F
0-ATP synthase and showed that calcium treatment mimics the response with specific agonists and inhibitors of mPTP. Recently, Pinke and Sazanov [
18] also stated that the entire mammalian ATP synthase is part of the mPTP, based on an atomic model of ATP opening developed through cryogenic electron microscopy (cryo-EM) data. Despite these findings, the formation of the mPTP from F
1F
0-ATP synthase has been questioned in recent studies, where some authors showed that mPTP persists in the absence of several subunits of F
1F
0-ATP synthase. Since deleting such subunits prevents the assembly of functional F
1F
0-ATP synthase, the authors concluded that ATP synthase does not participate directly in the mPTP formation [
19,
20].
mPTP opening is triggered by matrix Ca
2+, but its activity can be modulated by several other factors such as oxidative stress, adenine nucleotide depletion, high inorganic phosphate (Pi) concentrations, mitochondrial membrane depolarization, or uncoupling [
21]. The prolonged mPTP opening leads to an abrupt increase in the permeability of the inner mitochondrial membrane to solutes with molecular mass up to 1.5 kDa, provoking mitochondrial depolarization, followed by respiratory inhibition and the generation of reactive oxygen species (ROS), and massive release of matrix Ca
2+.
This event induces the swelling of mitochondria, leading to breaks in the outer mitochondrial membrane that induce the release of pro-apoptotic factors [
22]. For this reason, the mPTP has been proposed as a pivotal effector in the process of cell death [
23,
24].
Based on this accumulating evidence, we hypothesized that the resistance of IPF fibroblasts to apoptosis might be associated with a dysregulation in the mitochondrial function, mainly by resistance to the mPTP opening. Therefore, we aimed to analyze the mitochondrial function and the mPTP opening on the apoptosis resistance of human IPF fibroblasts. In the present study, we demonstrate that in IPF fibroblasts, mitochondria are dysmorphic and dysfunctional and that the resistance to apoptosis is related to altered mPTP opening. These findings may provide novel insights into the mechanisms involved in the resistance to apoptosis by IPF fibroblasts.
3. Discussion
A growing body of evidence demonstrates that IPF fibroblasts are resistant to apoptosis [
7,
8,
9,
10]. The mechanisms are unclear, but evidence indicates the participation of the extrinsic pathway of apoptosis, such as the inhibition of Fas-mediated apoptotic cell death [
8]. However, no studies have investigated the role of the critical intrinsic pathway of apoptosis mediated by mitochondria. In this context, our study demonstrates that IPF fibroblasts show resistance to mPTP opening, low cytochrome c levels, mitochondrial dysfunction, and a fragmented mitochondrial network. Recently, mitochondrial dysfunction has been reported in IPF fibroblasts associated with the induction and maintenance of the senescent phenotype [
27], but its specific role in the apoptosis of these cells is uncertain. Mitochondria-induced apoptosis arises from various internal cell stresses, resulting in the release of cytochrome c toward the cytosol. The above is a crucial process in the intrinsic apoptosis pathway and is related to the mPTP opening. Once in the cytosol, cytochrome c activates the apoptosome formation, which is a cytosolic multiprotein complex composed of cytochrome c, Apaf-1, and ATP. This complex induces the caspase 9 activation, triggering the effector caspases activation that provokes cell death [
28]. Therefore, in the fibrotic process, the dysregulation of cytochrome c release can result in the accumulation of apoptosis-resistant fibroblasts, excessive extracellular matrix deposition, and disruption of the lung architecture.
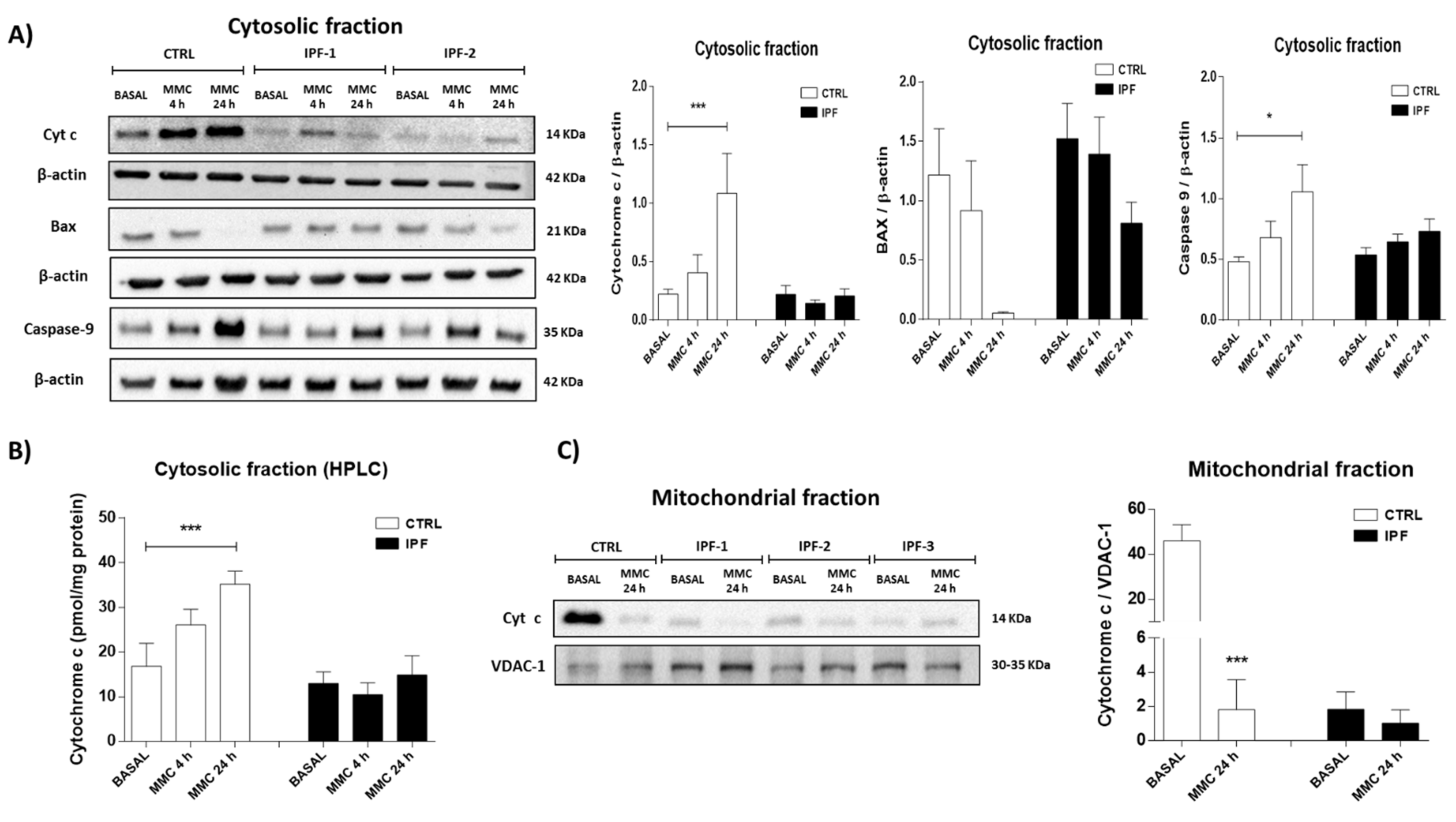
In this work, we demonstrated that IPF fibroblasts presented a marked decrease in basal cytochrome c levels and its release after stimulation with mitomycin C. Cytochrome c is a crucial signaling molecule during apoptosis, but it also plays an essential role in oxidative phosphorylation by transferring electrons from complex III to complex IV of the electron transport chain [
29]. A substantial portion of cytochrome c is cardiolipin-associated, embedded in mitochondrial cristae, while the remaining cytochrome c is free in the intramembrane space. The release of cytochrome c from mitochondria is carried out during the early apoptosis stages and requires a two-step process. First, the solubilization of cytochrome c involves a breaking of the electrostatic and hydrophobic forces that it usually maintains with cardiolipin. Second, the mitochondrial permeability transition is sufficient to release cytochrome c into the cytosol [
30]. Thus, the decrease in basal cytochrome c levels observed in fibroblasts from patients with IPF may be related to decreased electron transfer, oxygen consumption, and ATP synthesis. In this context, various studies have reported that the lack of cytochrome c disrupts the assembly and stability of respiratory complexes I and IV in fibroblasts [
31].
Interestingly, embryonic fibroblasts derived from mice deficient in the somatic isoform of cytochrome c are resistant to apoptosis by agents known to trigger the intrinsic apoptotic pathway. This effect was associated with respiratory chain defects [
32]. The mitochondrial apoptosis pathway is regulated mainly by changes in the permeability of the mitochondrial membrane or by alterations in its function. Mitochondrial outer membrane permeabilization is the ultimate step of apoptotic signal transduction pathways, which converge on mitochondria. mPTP is one of the representative systems proposed to be responsible for mitochondrial outer membrane permeabilization [
33]. Although the concept of the mPTP is still evolving, mounting evidence indicates that the mPTP is directly responsible for cytochrome c release. For this reason, it is considered a strategic regulator of cell death. The mPTP is a non-specific conductance channel in the inner mitochondrial membrane that allows the flux of metabolites with a molecular weight of up to 1.5 kDa.
Induction of the mPTP leads to mitochondrial depolarization, inhibition of ATP synthesis, Ca2+ uptake, respiratory inhibition, generation of ROS, mitochondrial swelling, and potentially the rupture of the outer mitochondrial membrane leading to release of mitochondrial apoptogenic proteins such as cytochrome c, Smac/DIABLO, endonuclease G, and AIF. Therefore, it is not surprising that studies of the mPTP attract substantial attention. However, despite significant effort, the exact molecular composition of the mPTP is still a matter of debate.
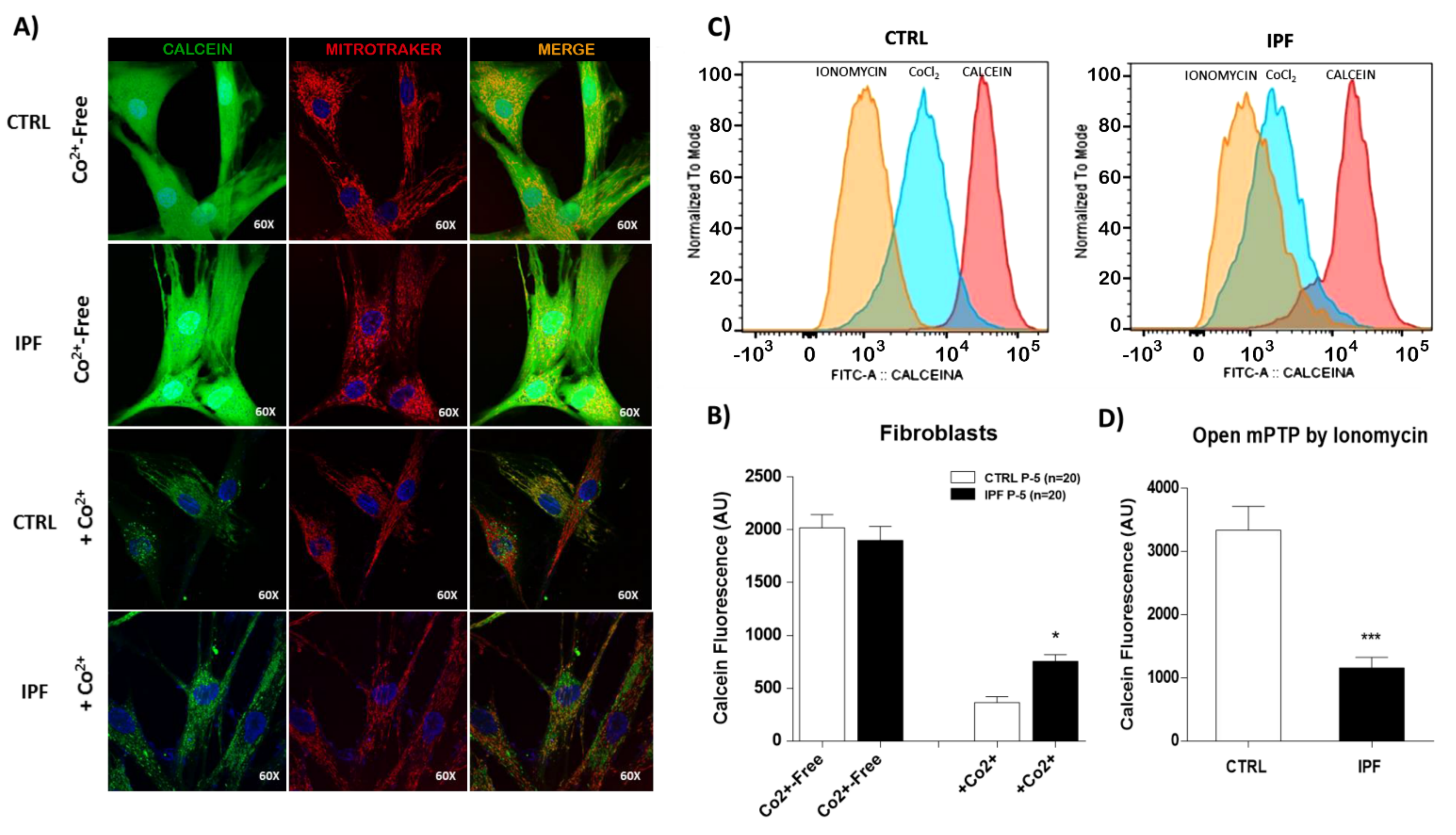
The mPTP opening can be induced by high concentrations of mitochondrial Ca
2+, oxidative stress, and Pi and can be inhibited by cyclosporin A, adenine nucleotides, Mg
2+, acidic pH, and reducing agents in the cells [
16]. The mPTP has been implicated in several diseases, but to our knowledge, no studies had been performed on IPF fibroblasts. We demonstrated for the first time that IPF fibroblasts are resistant to ionomycin-induced mPTP opening; this is an intriguing finding, because high basal ROS production by IPF fibroblasts has been reported [
34], and an mPTP opening should be expected as a consequence of high mitochondrial ROS production. However, a similar effect has been reported in cancer cells. For example, Norman et al. demonstrated that mPTP inhibition by cyclosporin A promotes skin cancer in transplanted patients by allowing keratinocyte survival under conditions of genotoxic stress, highlighting the critical role of mPTP inhibition in tumor development [
35].
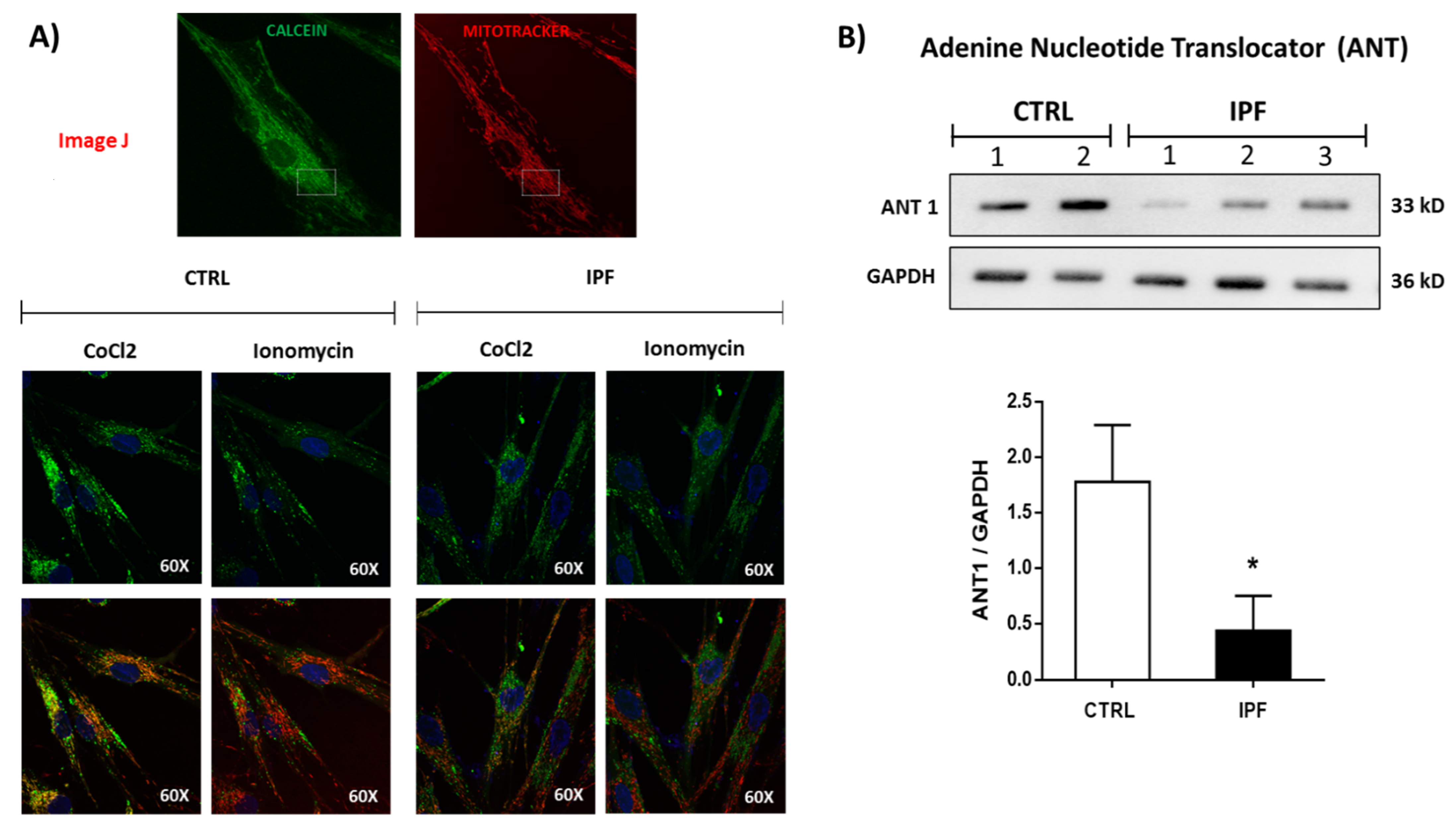
On the other hand, different factors can influence this resistance to ionomicyn-induced mPTP opening, such as the expression of protein components. Recently, it has been proposed that in addition to the ANT, VDAC, cyclophilin D, the ATP synthase is part of the mPTP [
17,
18].
However, this idea has been questioned in recent studies, where some authors showed that mPTP persists in the absence of several subunits of ATP synthase [
19,
20]. We analyzed the expression levels of ATP synthase by WB and did not find significant differences between control and IPF fibroblasts, suggesting that the content of ATP synthase cannot influence the function of mPTP. Interestingly, when we analyzed the expression of ANT, we found that this protein shows decreased levels in IPF fibroblasts, which could suggest its participation in the mPTP inhibition observed in IPF fibroblasts.
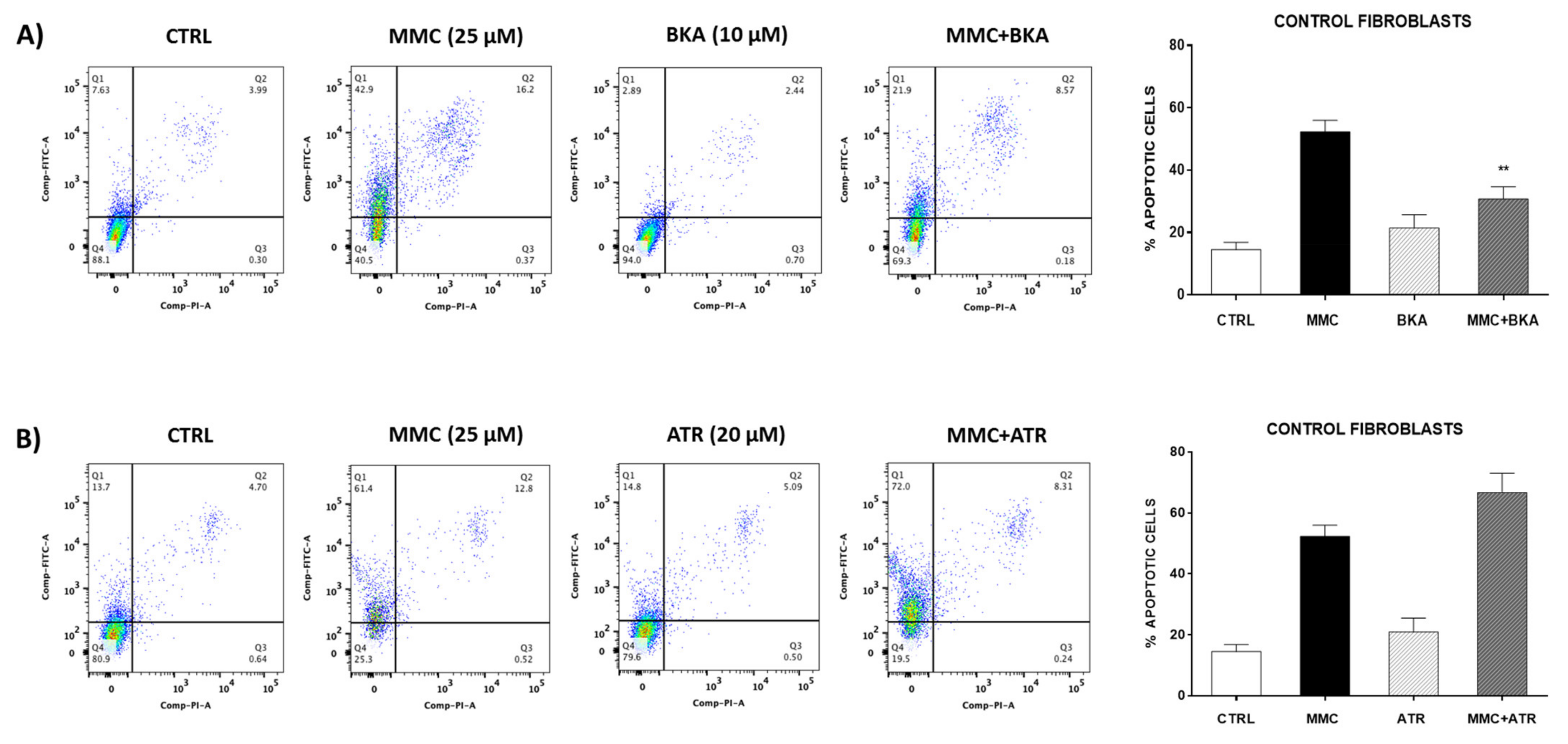
Diverse studies show that drugs that stabilize the conformation of ANT in the cytosol enhanced mPTP opening, whereas others that stabilize ANT in the matrix inhibited mPTP opening by decreasing the sensitivity to [Ca
2+]. When the ANT is stabilized in the cytosolic conformation by carboxyattrayloside, it provides a structural basis for mPTP formation by increasing sensibility to [Ca
2+]. While on the other hand, bongkrekic acid, an inhibitor of ANT that stabilizes the matrix conformation, inhibits mPTP opening by decreasing the sensitivity to [Ca
2+] [
36]. This effect was observed in our results when using attrayloside or bongkrekic acid in the control lung fibroblasts. We observed an increase in apoptosis when stimulated fibroblasts with attrayloside and inhibition with bongkrekic acid.
Other factors that influence the opening of the mPTP may be related to cytochrome c interaction with lipids of the inner mitochondrial membrane or the capacity to accumulate calcium in the mitochondria, as calcium is a well-established mPTP activator. The reduction of mitochondrial calcium release induces mPTP inhibition.
Inhibition of the mPTP augments Ca
2+ accumulation in mitochondria, stabilizes mitochondrial membrane potential, and defers Ca
2+ dysregulation [
36].
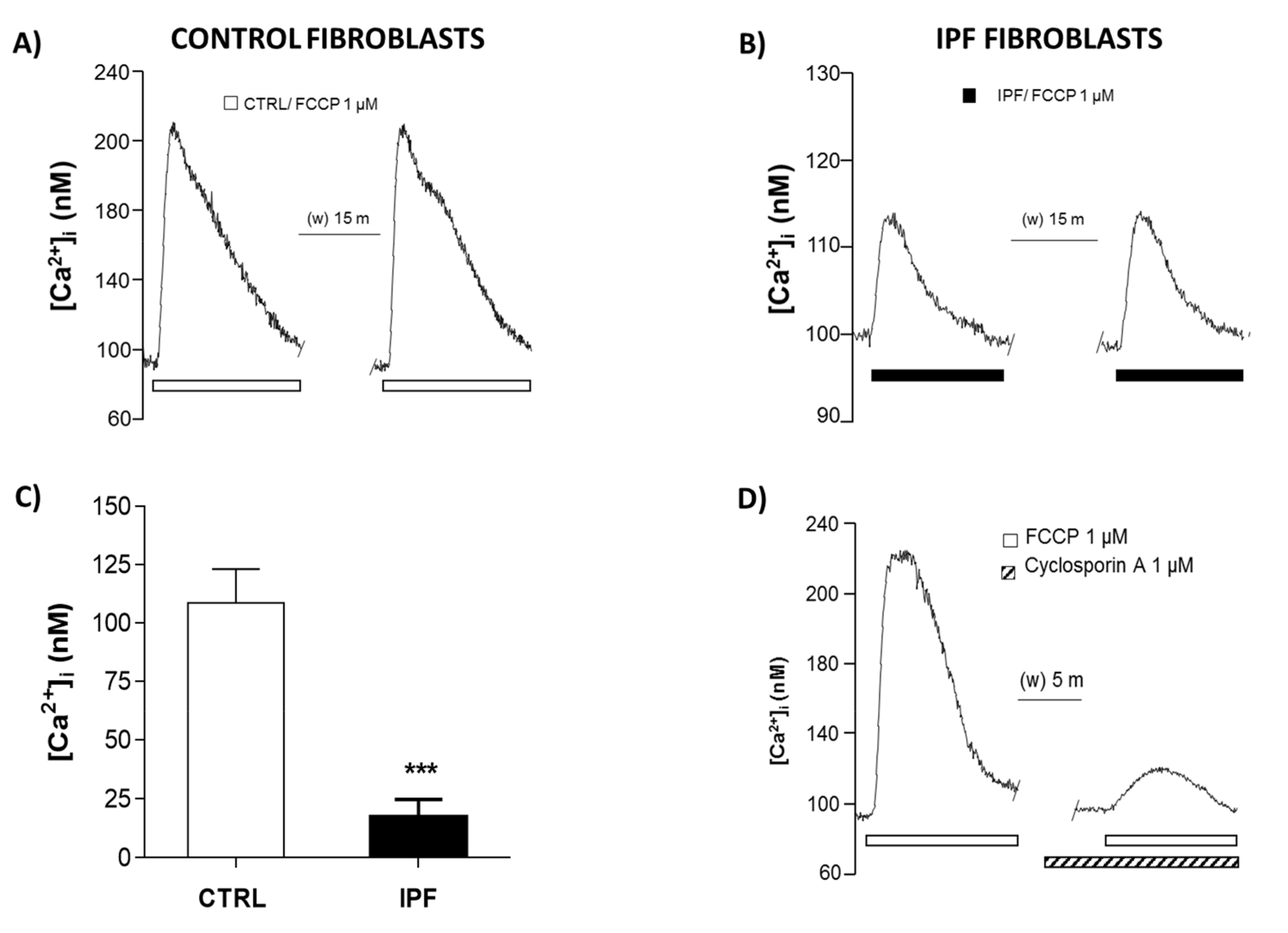
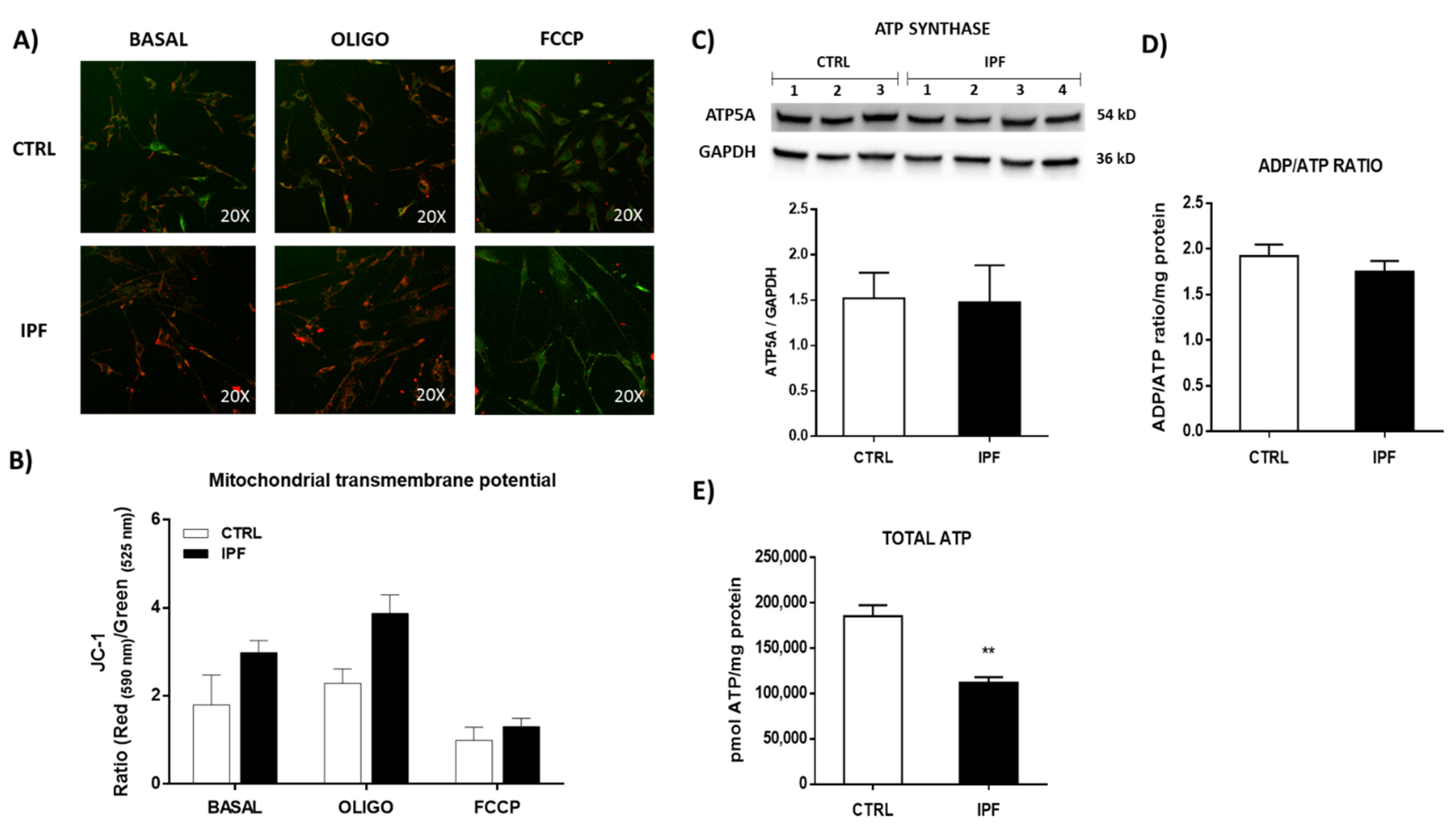
Interestingly, our results on mitochondrial calcium quantification show a marked decrease in mitochondria calcium release in patients with IPF, confirming that mPTP activity decreases in these cells. In addition, we did not observe mitochondrial potential membrane changes, which support that mPTP is inhibited in IPF fibroblasts.
Opening of the mPTP might also stimulate autophagy to eliminate abnormal mitochondria. Thus, the mitochondrial outer membrane recruits the autophagy proteins ATG5 and LC3, not only for mitophagy but also for the anchorage and share of the lipid moieties required for the elongation of the initial phagophore [
37]. Interestingly, we have previously found that IPF fibroblasts show a decrease in autophagy caused by activation of the mechanistic target of rapamycin complex 1 (mTORC1) pathway, contributing to the resistance to cell death [
38].
As mentioned, we also showed that stimulus with BKA inhibited the mitomycin-induced cell death in control fibroblasts, while the mPTP inductor ATR renders the cell more susceptible to cell death. BKA prevents acidification and is a ligand for the ANT that can inhibit apoptosis. Our results concur with Lui et al., who demonstrated that inhibition of mPTP by cyclosporine A (CsA) and BKA affected p53 translocation in mitochondria, leading to protection against the loss of mitochondrial membrane potential and complex I activity and eventually suppression of apoptosis [
39].
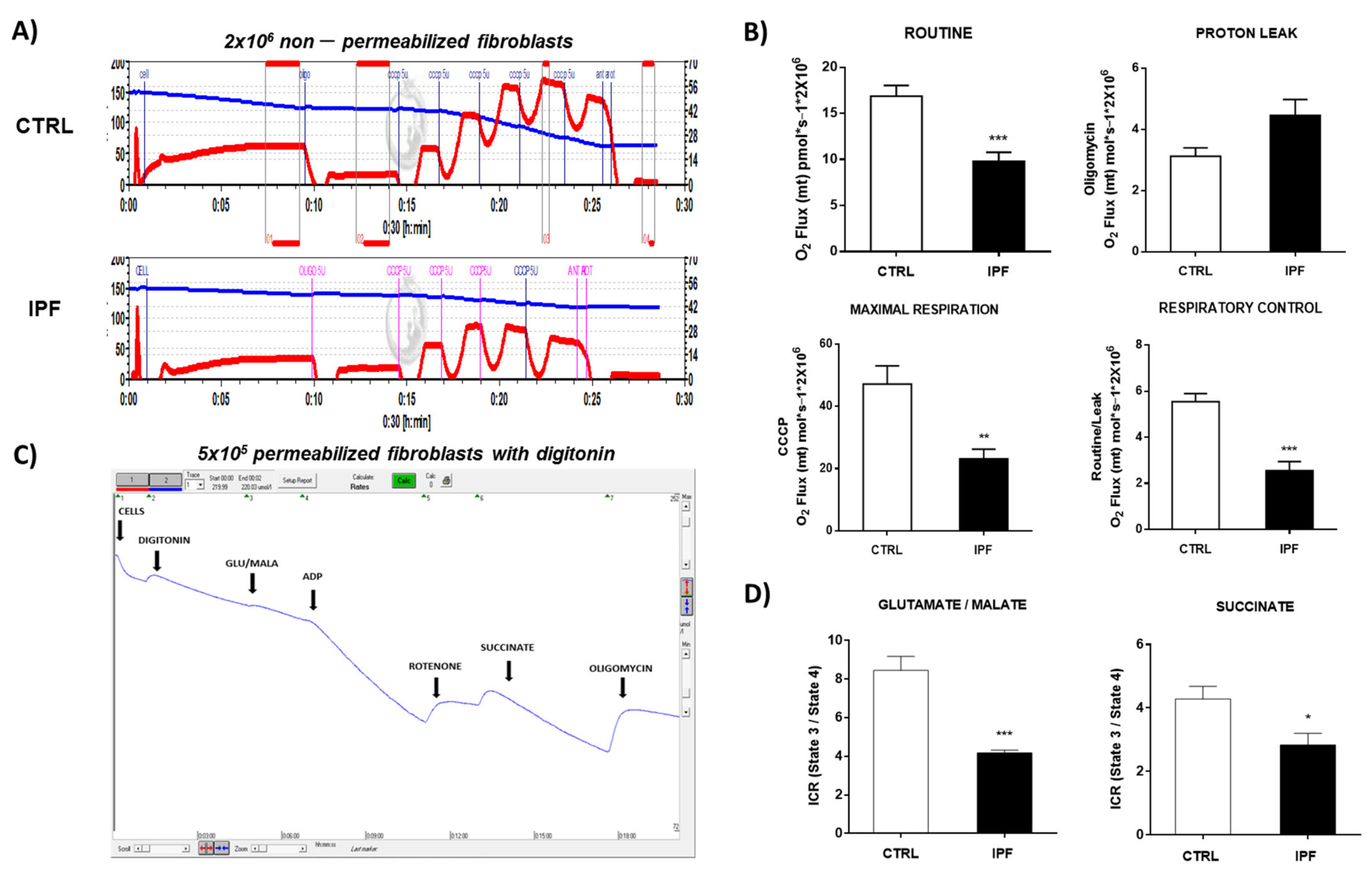
We also found that IPF fibroblasts show mitochondrial dysfunction, as evidenced by decreased oxygen consumption and decreased ATP production, although we did not observe significant changes in the ratio ADP/ATP and mitochondrial membrane potential. The fact that the ADP/ATP ratio does not change suggests that the cell adapts to maintain a minimal mitochondrial activity to supply enough ATP and keep the cell alive with basal transmembrane potential, avoiding cell death through the intrinsic pathway of apoptosis.
These findings agree with those previously reported by Álvarez et al. [
40], who found low ATP levels in senescent IPF fibroblasts, which were associated with decreased OCR, diminished glycolytic capacity, and abnormalities in oxidative phosphorylation [
28,
41].
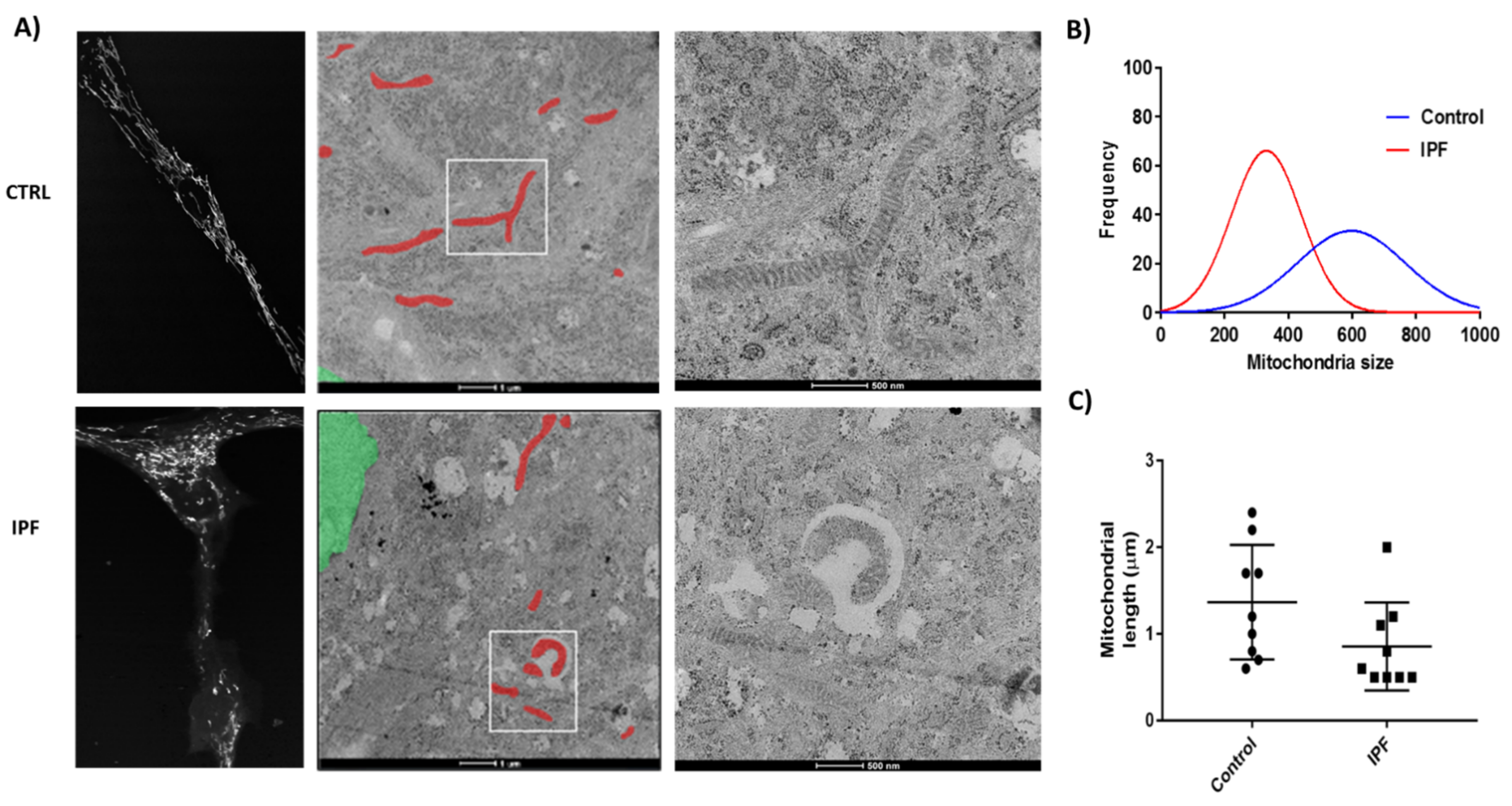
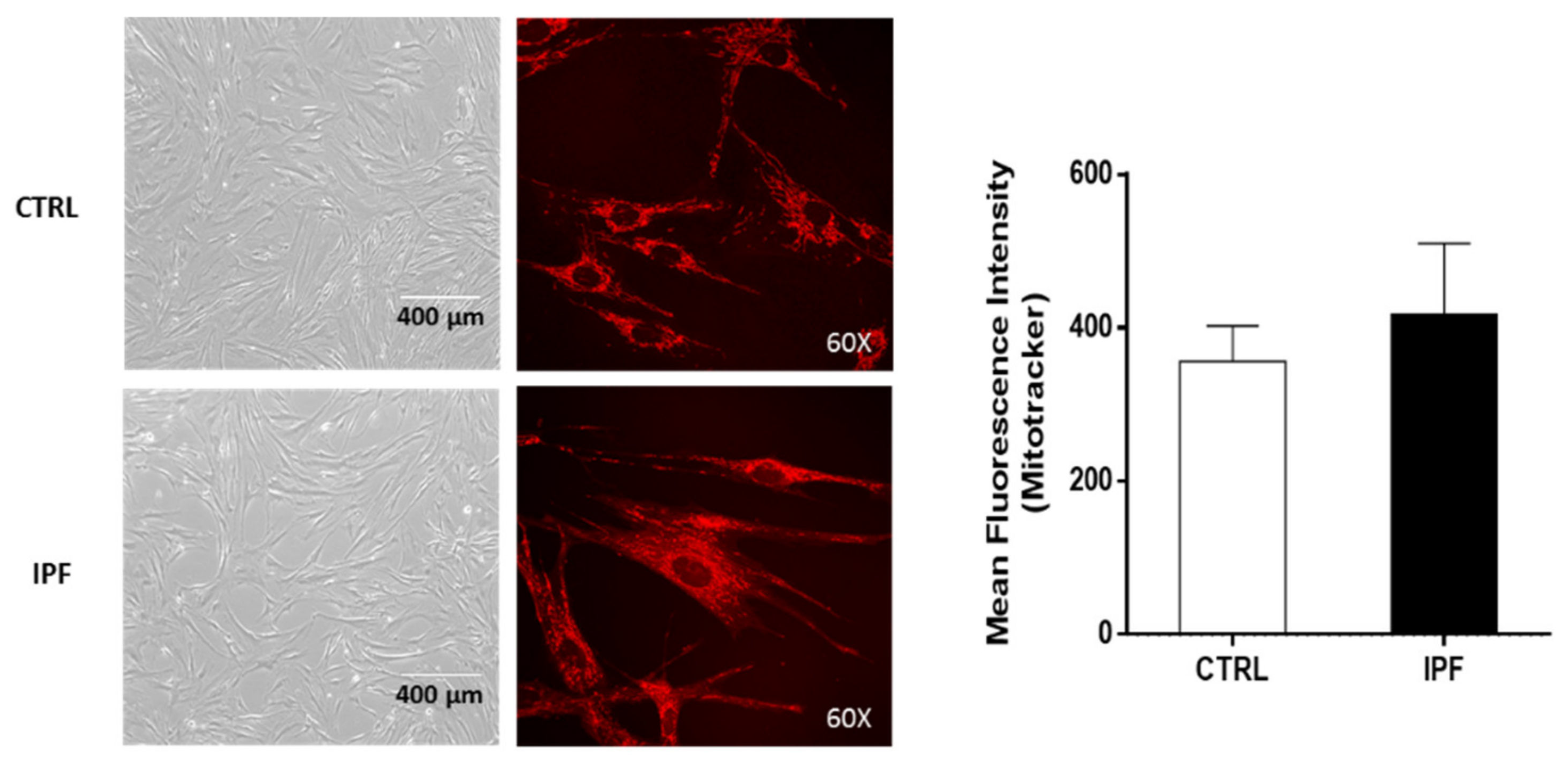
Considerable evidence indicates that the mitochondrial network organization is associated directly with the bioenergetic function and ROS generation. In this sense, our results showed that IPF fibroblasts exhibited a fragmented mitochondrial network and scarce, thinned, and disordered mitochondria with a few electrodense cristae. Diverse studies have shown that cristae architecture is determined by cristae-shaping proteins and depends on the dimeric state of the ATP-synthase [
42]. The formation of mitochondrial cristae increases the mitochondrial surface, locating a higher number of enzyme complexes in the mitochondria, improving oxidative phosphorylation (OXPHOS). Alterations in respiration and changes in ATP levels might be associated with mitochondrial morphology modulators. Benard et al. [
43] demonstrated that the silencing of DRP1 in HeLa cells resulted in alterations of the mitochondrial network morphology. This effect was associated with reducing sensitivity to apoptosis inducers, potent inhibition of energy production, abnormal connectivity, and multiple budding areas, which suggested a perturbation of mitochondrial dynamics.
Furthermore, we observed a reduction in coupled respiration and ATP production. Studies with the effect of OXPHOS inhibition on mitochondrial network organization in primary human fibroblasts suggest that it may contribute to these abnormalities. Thus, the formation of vesicles due to the inhibition of complex I by rotenone can be regarded as a direct consequence of impaired OXPHOS function [
43]. In general, a highly efficient OXPHOS correlates with a highly interconnected and ramified network and enlarged cristae compartments, whereas low OXPHOS activity and high glycolysis correlate with bulkier, more spherical tubules, and isolated mitochondria, displaying reduced intra-cristae space.
We found alterations in the respiratory chain, but we cannot describe it exactly. Therefore, we propose that it is necessary to study the formation of mitochondrial complexes and respirasomes because there is evidence supporting the fact that cytochrome c can influence the function of the chain. Furthermore, morphology observed in IPF fibroblasts increases the evidence that there are issues with the respiratory chain, since the mitochondrial ridges exhibit lesser electro-density than in control fibroblasts. The relationship between mitochondrial morphology and function has gained significant attention in recent years, and it has been verified that there is a direct relationship between the cristae structure and the respiratory chain.
Mitochondria are very complex organelles that control many diverse functions that, as a whole, define the fate of the cell, and mitochondrial dysfunction is usually understood as an event leading to cell death. However, we found that, on the contrary, mitochondrial dysfunction is associated with resistance to apoptosis in IPF fibroblasts, as it occurs in cancer cells. Therefore, the study of the in-depth mechanisms through which mitochondria dysfunction leads to proliferation or death in different pathologies will be helpful to identify new therapeutic targets against IPF, cancer, or other pathologies.
Tissue fibrosis is likely an evolutionarily conserved adaptive process, and persistent fibrosis almost always accompanies incomplete or unsuccessful regeneration. However, in this case, the underlying mechanisms that contribute to the persistence of myofibroblasts and continuous extracellular matrix accumulation in fibrotic tissues remain poorly understood. A growing body of evidence indicates that one of them is the evasion of apoptosis, which usually occurs during physiological wound healing. However, the molecular pathways that are involved in the apoptosis resistance of myofibroblast in fibrotic tissues are still unclear and may even involve different mechanisms. Some evidence indicates the participation of the extrinsic pathway of apoptosis, but no studies had investigated the role of the intrinsic pathway mediated by mitochondria.
In this context, we thought that changes in the mPTP opening, given its relevance in regulating cell death, could be involved. Our results revealed that mPTP is inhibited in IPF fibroblasts, and that calcium, a well-established activator of mPTP, is decreased and pro-apoptotic proteins such as cytochrome c are released as well. We consider that this finding is relevant to understand some of the biopathological mechanisms that participate in the apoptosis resistance of fibroblasts during progressive fibrosis as in IPF. A better understanding of the molecular composition of the mPTP and apoptosis mechanisms will provide clues for effective and selective therapeutic strategies for the treatment of this devastating disease.
4. Materials and Methods
4.1. Reagents
We purchased the Cell Proliferation Reagent WST-1 (11644807001) from Roche Diagnostics, Mannheim, Germany. The FITC Annexin V Apoptosis Detection Kit (556547) was from BD Biosciences, San Diego, CA, USA. The Mitochondria Isolation Kit for Cultured Cells (89874), Pierce™ BCA Protein Assay Kit (23225), HRP-anti-goat IgG (611620), and Anti-GAPDH (PA1-987) were from Thermo Fisher Scientific, Rockford, IL, USA. The Image-IT™ LIVE Mitochondrial Transition Pore Assay Kit (I35103), MitoProbe™ Transition Pore Assay Kit (M34153), ATP Determination Kit (A22066), and tetraethyl benzimidazolyl carbocyanine iodide dye JC-1 (T3168) were from Molecular Probes, Eugene, OR, USA. The ADP/ATP ratio assay kit (MAK135), anti-β-actin (A5441), oligomycin A, rotenone, antimycin A, L-glutamic acid, L( )-malic acid disodium salt, sodium succinate dibasic hexahydrate, carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazine (FCCP), carbonyl cyanide-3-chlorophenyl hydrazone (CCCP), ethylene glycol-bis (2-aminoethylether)-N,N,N′,N′-tetra acetic acid (EGTA), potassium salt, 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid potassium salt (HEPES), bovine serum albumin (BSA), free fatty acid, magnesium chloride (MgCl2), potassium phosphate monobasic (KH2PO4), cytochrome c, bongkrekic acid (BKA), and atractyloside (ATR) were purchased from Sigma-Aldrich from St. Louis, MO, USA. Mitomycin C (sc-3514); anti-VDAC-1 (sc-390996), and anti-caspase-3 (sc-7272) were from Santa Cruz Biotechnology, Heidelberg, Germany. Anti-cytochrome c (612504) and HRP-anti-rabbit IgG (406401) were from BioLegend, San Diego, CA, USA. The anti-caspase-9 (ab32539) was from Abcam Cambridge, MA, USA. HRP-Anti-Mouse IgG (115-035-003) was acquired from Jackson Immuno Research, West Grove, PA, USA.
4.2. Bioethics and Experimental Design
The Bioethics Committee of the Instituto Nacional de Enfermedades Respiratorias approved this research, with ethic approval code B05-21, (20 September 2018). Fibroblasts from IPF patients were obtained from lung biopsies performed for diagnostic purposes; all participants provided informed consent, and their personal and identification data were protected. As controls, healthy lung fibroblasts were obtained by lobectomy from patients without morphological data of interstitial disease, and the commercial human normal fibroblasts line NHLF (Normal Human Lung Fibroblasts CC-2512 Lonza Clonetics) was used. Healthy and IPF lung fibroblasts matched by age were studied under basal conditions and after mitomycin C-induced apoptosis.
4.3. Cell Culture
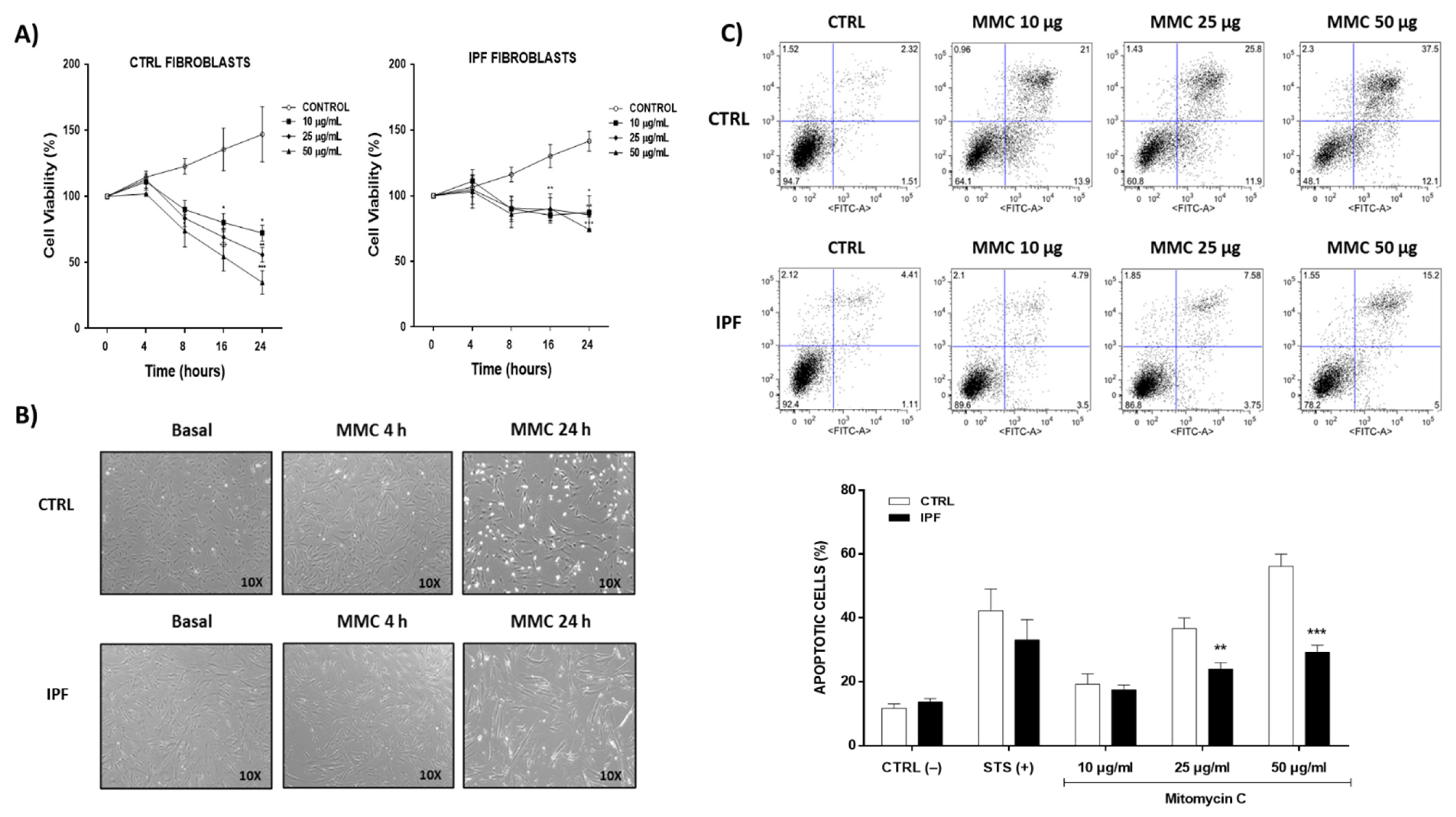
The fibroblasts were cultured in Ham medium (F-12), supplemented with penicillin (100 U/mL), streptomycin (100 mg/mL), and 10% fetal bovine serum (SFB), and incubated in a humid atmosphere at 37 °C with 5% CO2, until 80% confluence. Fibroblasts were treated with different doses of mitomycin C (10, 25, or 50 μg/mL) in F-12 medium with 1% SFB at different times (4, 8, 16, and 24 h). Control fibroblasts were incubated in F-12 medium with 1% SFB for the same period. After the treatment, fibroblasts were washed three times with phosphate-buffered saline (PBS) and kept in the F-12 medium for subsequent experiments.
4.4. Cell Viability
Cell viability was assessed using the Cell Proliferation Reagent WST-1 assay following the manufacturer’s instructions. Cells were seeded in 96-well plates at a final density of 5 × 103 cells per well in 100 μL of culture medium. After treatment with mitomycin-C, 10 μL of WST-1 solution was added to each well, and the cells were incubated for 3 h at 37 °C in a 5% CO2 atmosphere. The optical density was measured using a multimodal microplate reader (Synergy HTX BioTek, Winooski, VT, USA) at a wavelength of 450 nm.
4.5. Cell Apoptosis
Fibroblasts were stimulated with mitomycin-C for 24 h and stained with Annexin V-IP for flow cytometric analysis. Staurosporine (1 μM) was used as a positive control. Cells were acquired using a FACSAria flow cytometer (BD Biosciences). Data were analyzed using FlowJo 7.8 software (FlowJo, Ashland, OR, USA, Becton Dickinson and Company).
4.6. Preparation of the Cytosolic and Mitochondrial Fractions
Control and IPF fibroblasts were cultured in a cell culture flask of 175 cm2 until 80% confluence (approximately two or three flasks by experimental condition). Fibroblasts were stimulated with mitomycin C (25 μg/mL) for 4 and 24 h. After treatment with mitomycin C, the cells were washed and trypsinized, and their viability was analyzed by a Trypan Blue exclusion test. For each experimental condition, 1 × 107 viable cells were used.
The isolation of mitochondrial and cytosolic fractions of fibroblast cultures was performed with the Mitochondria Isolation Kit for Cultured Cells, following the manufacturer’s instructions. For this assay, we used a Dounce homogenizer to disrupt cells, and subsequently, we separated the fractions by differential centrifugation. The mitochondrial and cytosolic fractions were used for analysis by Western blotting. The mitochondrial pellet was lysed with 50 μL of 2% detergent 3-(3-cholamidopropyl) dimethylammonio)-1-propane sulfonate (CHAPS) in Tris buffer saline (TBS; 25 mM Tris, 0.15 M NaCl; pH: 7.2). The supernatant contained the soluble mitochondrial protein that was quantified by the Pierce BCA Protein Assay Kit.
4.7. Western Blotting
Cytosolic and mitochondrial fractions and cell lysates were analyzed by Western blot. Cellular lysates from human normal and IPF fibroblasts were prepared using radioimmunoprecipitation assay buffer (RIPA) with phenylmethylsulfonyl fluoride (PMSF) and protease inhibitors. The protein concentration was quantified using the Bradford assay. Briefly, 16 μg of protein were separated by Sodium Dodecyl Sulfate-Polyacrylamide SDS-PAGE (110 V for 1 h at room temperature) and transferred onto a nitrocellulose membrane using a wet chamber blotting system. The nitrocellulose membranes were blocked with 5% skim milk for 1 h and afterwards incubated overnight at 4 °C with the primary antibodies: anti-cytochrome c (1:500); anti-caspase 9 (1:500); anti-VDAC-1 (1:100); anti-Bax (1:100); anti-ATP5A (1:500); anti-ANT1 (1:1000); anti-GAPDH (1:2000); anti-β-actin (1:10,000). The secondary antibodies were incubated for 1 h at room temperature with the following dilutions: anti-mouse (1:10,000), anti-rabbit (1:2500), and anti-goat (1:1000).
4.8. Cytochrome C Release
The cytosolic fraction was filtered through a polyvinylidene fluoride membrane PVDF (0.45 μm), and the cytochrome c content was determined by high-performance liquid chromatography (HPLC) using a 300 Å Delta Pack C4 column with 5 μm particles (3.9 × 150 mm; Waters). A gradient of 30% acetonitrile in water with trifluoroacetic acid (0.1%
vol/
vol) to 70% acetonitrile in water with trifluoroacetic acid (0.1%
vol/
vol) over 15 min with a flow rate of 1 mL/min was used as was described elsewhere [
44]. Cytochrome c was detected at 393 nm, and its concentration was quantified using a standard curve of cytochrome c from bovine heart.
4.9. mPTP Opening
The mPTP opening was assessed by cobalt quenching of calcein-AM fluorescence. After treatment, cells were loaded with calcein-AM (1 μM, Molecular Probes, Life Technologies) at 37 °C in the dark, CoCl2 (1 mM) was added, and cells were incubated for another 15 min. Then, the fluorescence of 30,000 cells for each experiment was measured with a flow cytometer (FACS Aria II, BD, San Jose, CA, USA), and the data were processed with the FlowJo software. For imaging experiments, 1 × 104 fibroblasts were cultured on glass-bottom culture dishes. First, cells were loaded with calcein-AM (1 μM) and MitoTracker Red (150 nM), with or without CoCl2 (1 mM), in Hanks’ Balanced Salt Solution (HBSS) 1X for 15 min at 37 °C. Then, cells were washed three times with HBSS 1X. Live images of the cells were captured with the Olympus FluoView FV1000 Confocal Microscope and analyzed using the ImageJ software version 1.53i (Bethesda, MD, USA).
4.10. Intracellular Calcium Concentration Assay ([Ca2+]i)
To evaluate [Ca
2+]
i changes induced by trifluoromethoxy carbonyl cyanide phenylhydrazone (FCCP) in control and IPF fibroblasts, cells were plated in round coverslips coated with poly-L-Lysine and cultured for 2 days. Then, cells were loaded with Fura 2-AM (2.5 μM) in a low concentration of Ca
2+ (0.1 mM) and at room temperature. Then, fibroblasts were incubated for 1 h at 37 °C under a 5% CO
2 atmosphere. Afterward, cells were transferred to a heated perfusion chamber (37 °C) mounted on an inverted Nikon Diaphot 200 microscope (Nikon, Tokyo, Japan). Cells were recorded under continuous perfusion and carbogen bubbling (to maintain pH at 7–4) at a rate of 2–2.5 mL/min with Krebs–Ringer buffer (NaCl 118 mM, NaHCO
3 25 mM, KCl 4.7 mM, KH
2PO
4 1.2 mM, MgSO
3 1.2 mM, glucose 11 mM, and CaCl
2 2.5 mM) at 37 °C. After the recording of basal fluorescence, cells were exposed to FCCP 1 μM. Next, fibroblasts loaded with Fura 2-AM were alternately submitted to Xe lamp at 340 nm and 380 nm excitation light, and the emission fluorescence was measured at 510 nm using a microphotometer (model D-104), from Photon Technology International (PTI, Princeton, NJ, USA). Fluorescence was measured at intervals of 0.5 s for 10 min, and the intracellular Ca
2+ concentration ([Ca
2+]
i) was calculated according to the Grynkiewicz [
45] formula as follows:
where Kd is the dissociation constant of Fura-2AM, b is the ratio of fluorescent signals (R) at 380 nm for Ca
2+-free and Ca
2+-saturated dye, R
min is R in the absence of external Ca
2+, and R
max is R in saturating [Ca
2+]
i [
46]. These parameters were determined in vitro. The mean 340–380 nm fluorescence ratios for R
max (6.06) and R
min (0.39) were obtained by exposing the cells to Ca
2+ (10 mM) in the presence of ionomycin (10 μM), and Ca
2+ free Krebs with EGTA (10 mM), respectively. The fluorescence ratio at 380 nm light excitation in Ca
2+ free medium and Ca
2+ saturated cells was 4.23. The Kd of Fura 2-AM was assumed to be 386 nM [
46]. To evaluate the Ca
2+ response to FCCP, cells were perfused with 1 μM FCCP to stimulate the mitochondrial permeability transition pore (mPTP). To pharmacologically determine if mPTP were involved in this response, a blocker cyclosporin A (CspA) 10 μM plus ATP was used.
4.11. Cell Respirometry
4.11.1. Respirometry in Non-Permeabilized Cells
The oxygen consumption experiments in intact cells were performed as previously described using a high-resolution respirometry O2 k meter (Oroboros Instruments, Innsbruck, Austria) [
47]. Briefly, measures were made at 37 °C using 2 mL of culture medium. The respiratory parameters were defined as (a) basal respiration, corresponding to the oxygen consumption in the presence only of cells, (b) the leak of the respiration, corresponding to cellular oxygen consumption in the presence of 5 μM oligomycin, (c) respiratory control corresponding to the ratio basal/leak, (d) the maximum uncoupled respiration was achieved by titrations of 0.5 μL of 1 μM CCCP. All parameters were normalized by the number of cells and corrected by the non-mitochondrial residual respiration, which was obtained by adding 5 μM rotenone plus 5 μM antimycin A.
4.11.2. Respirometry in Permeabilized Cells with Digitonin
The oxygen consumption in permeabilized fibroblasts was performed as was previously described by Kuznetsov et al. [
48]. Control and IPF fibroblasts were seeded in standard conditions, and the trypan blue exclusion test was used to determine cell density. For each experimental condition, 5 × 10
5 fibroblasts were added, and the system was equilibrated again for 5 min. The reaction was carried out in respiration medium (EGTA 0.5 mM; MgCl
2 3 mM; KH
2PO
4 10 mM; HEPES 20 mM; BSA 1 g; mannitol 110 mM for liter) pH 7.1, adjusted with 5 N KOH. Next, the fibroblasts were permeabilized with digitonin (20 μg/mL) and incubated for 5 min at 37 °C. After the addition of digitonin, the respiration rate should markedly decline for 3–5 min. Then, glutamate/malate (10 mM/5 mM) was used as a substrate for complex I activity, ADP was added to induce ATP production, and the respiration was inhibited by 0.5 μM rotenone, which is a specific inhibitor of complex I. Afterward, succinate (10 mM) was used as a substrate to induce complex II supported respiration, ADP was added to induce ATP production, and respiration was inhibited with oligomycin (5 μM) (state 4). Finally, 2 mM ADP was added for maximal (state 3) mitochondrial respiration. The respiratory control ratio (RCR) was calculated by dividing state 3/state 4 respiratory values.
4.12. Mitochondrial Membrane Potential (Δψm)
Mitochondrial Δψ was measured using JC-1, which is a fluorescent dye that exhibits potential-dependent accumulation in mitochondria. Polarized mitochondria are marked by punctate orange-red fluorescent staining. On depolarization, the orange-red punctate staining is replaced by diffuse green monomer fluorescence. Thus, mitochondrial depolarization is indicated by a decrease in the red/green fluorescence intensity ratio. Briefly, IPF and control fibroblasts were stained with JC-1 in Ham F-12 culture medium for 30 min at 37 °C in the dark. Then, cells were washed twice with PBS and stimulated with the uncoupling agent FCCP (100 μM) or oligomycin (5 μM). Fibroblasts were observed with a confocal microscope using the 20x objective, and both green (520 nm) and red (572 nm) fluorescence were measured to detect the emission shift. This shift was calculated as the red/green fluorescence intensity ratio. Images were analyzed using ImageJ software.
4.13. ATP Production
ATP production was determined quantitatively in IPF and control fibroblasts using the ATP Determination Kit, which uses a bioluminescent assay. For this assay, 10,000/well cells were plated on a white 96-well microplate with a clear bottom, and the assay was performed following the manufacturer’s instructions. The bioluminescence was recorded using a multimodal microplate reader (Synergy HTX BioTek, Winooski, VT, USA) in the luminometer mode.
4.14. ADP/ATP Ratio
The ADP/ATP ratio in IPF and control fibroblasts was measured using the ADP/ATP ratio assay kit. Briefly, IPF and control fibroblasts were plated at a density of 10,000 cells/cm2 in a white 96-well microplate with a clear bottom. Following overnight incubation, the ADP/ATP ratio was determined following the manufacturer’s guidelines. Next, the bioluminescence was measured using a multimodal microplate reader (Synergy HTX BioTek, Winooski, VT, USA).
4.15. Transmission Electron Microscopy (TEM)
TEM was done with the support of the Center for Advanced Microscopy (Northwestern University). In brief, lung fibroblasts were cultured on Thermanox coverslips placed in a 24-well plate. After 48 h of incubation, samples were fixed in 0.1 M sodium cacodylate (pH 7.2) containing 2% paraformaldehyde and 2.5% glutaraldehyde and post-fixed with 2% osmium tetroxide and stained with 3% uranyl acetate. Samples were dehydrated in ascending ethanol grades, transitioned with a 1:1 mixture of ethanol and resin, and embedded in the resin mixture of the EMbed-812 kit. Using a Leica Ultracut UC6 ultramicrotome, ultra-thin sections (70 nm) were collected on 200 mesh copper grids and post-stained with 3% uranyl acetate and Reynolds lead citrate. Ultra-thin sections were used to obtain images from each sample with the Tecnai G2 Spirit transmission electron microscope (FEI Company, Hillsboro, OR, USA) at 120 kV. The length of mitochondria was used to determine mitochondrial elongation mean per image; at least 70 mitochondria per condition were measured. Images were analyzed using the ImageJ software.
4.16. Statistical Analysis
All the data were expressed as the mean ± standard error mean (SEM). Two-way or one-way analysis of variance (ANOVA) followed by Bonferroni test assessed the significance of the differences. To analyze the significant differences between the control and IPF group, we used an unpaired t-test. p-values of less than 0.05 were considered to be statistically significant. Statistical analysis was performed using the GraphPad Prism 5.01 software (San Diego, CA, USA).


 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}









