
Oral Selective Estrogen Receptor Degraders (SERDs) as a Novel Breast Cancer Therapy: Present and Future from a Clinical Perspective

 , , , , , , and
, , , , , , and
Abstract
:1. Introduction
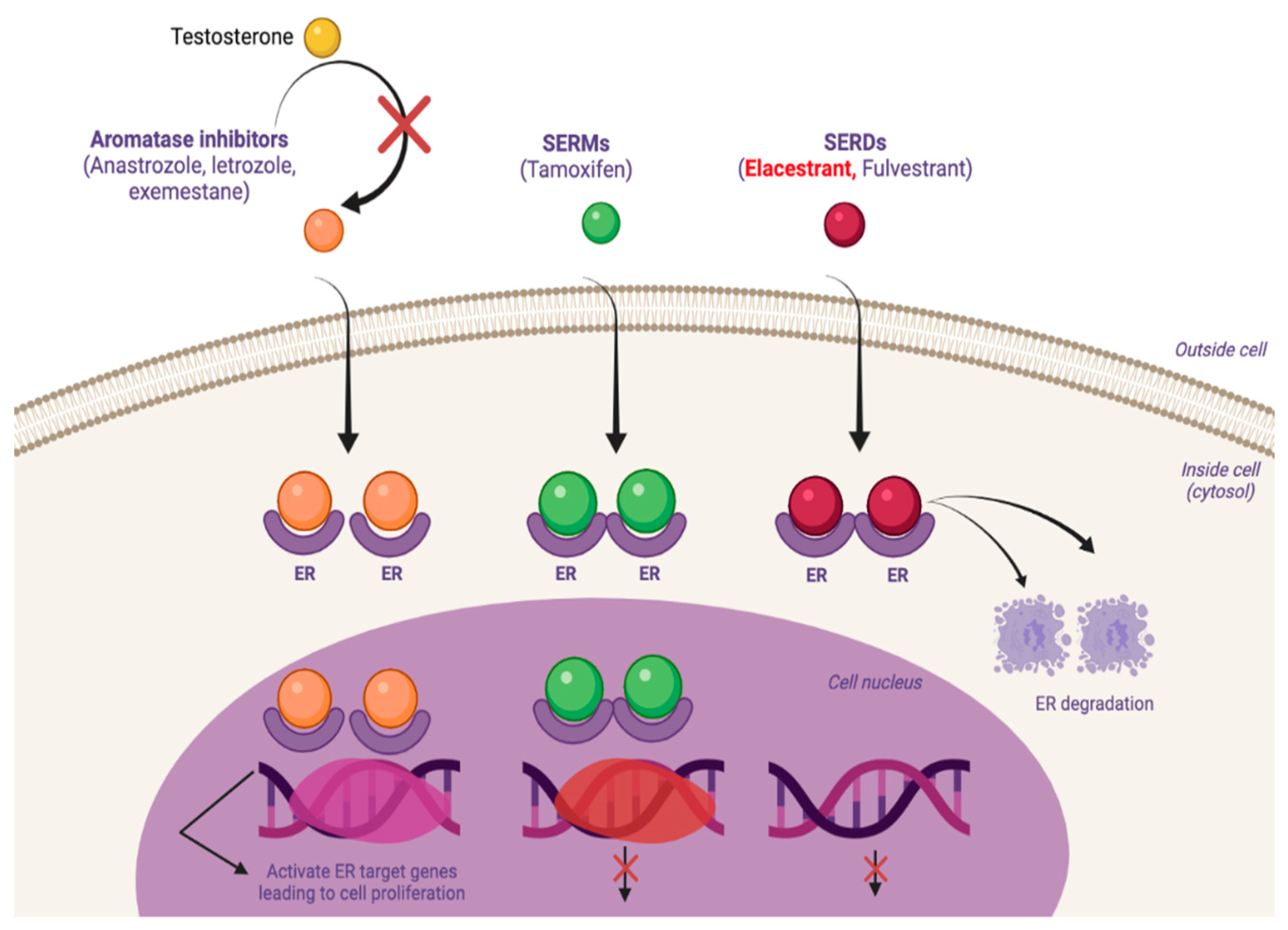
2. Antiestrogen Therapy: Basic Concepts Regarding Old and New Agents
2.1. ERα Structure
2.2. ER Alterations Driving Therapy Resistance: ESR1 Mutations
2.3. Fulvestrant as First SERD
3. New ER-α Targeting Agents in Development
3.1. SERMs
- LASOFOXIFENE
- BAZEDOXIFENE
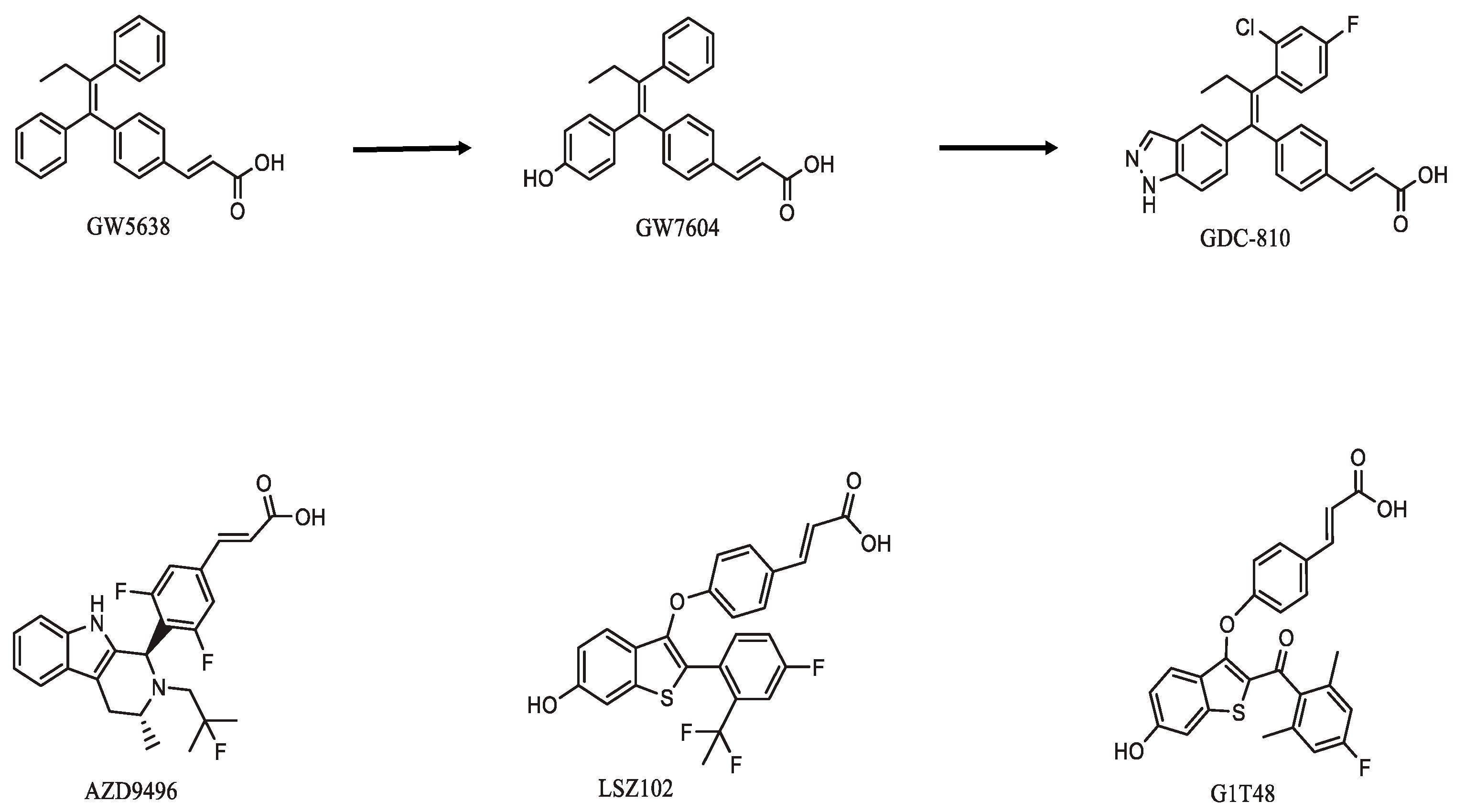
3.2. Nonsteroidal Agents with Acrylic Acid Side Chain as Serds
3.2.1. GDC0810 (Brilanestrant)
3.2.2. AZD9496
3.2.3. LSZ102
3.2.4. G1T48 (Rintodestrant)
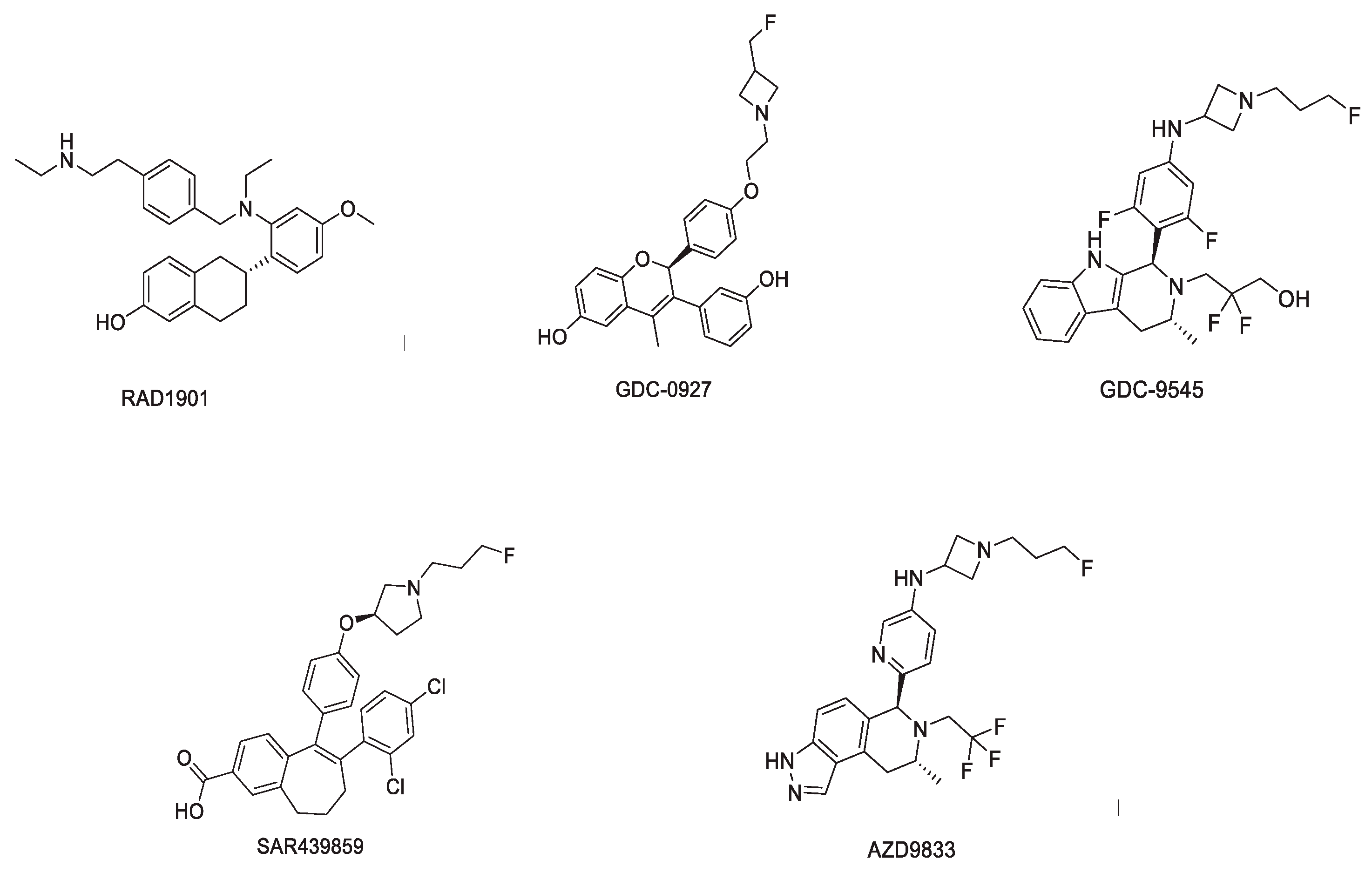
3.3. Nonsteroidal Analogs with a Basic Amino Side Chain as SERDs
3.3.1. RAD1901 (Elacestrant)
3.3.2. GDC-0927
3.3.3. GDC-9545 (Giredestrant)
3.3.4. SAR439859 (Amcenestrant)
3.3.5. AZD9833 (Camizestrant)
3.3.6. Others
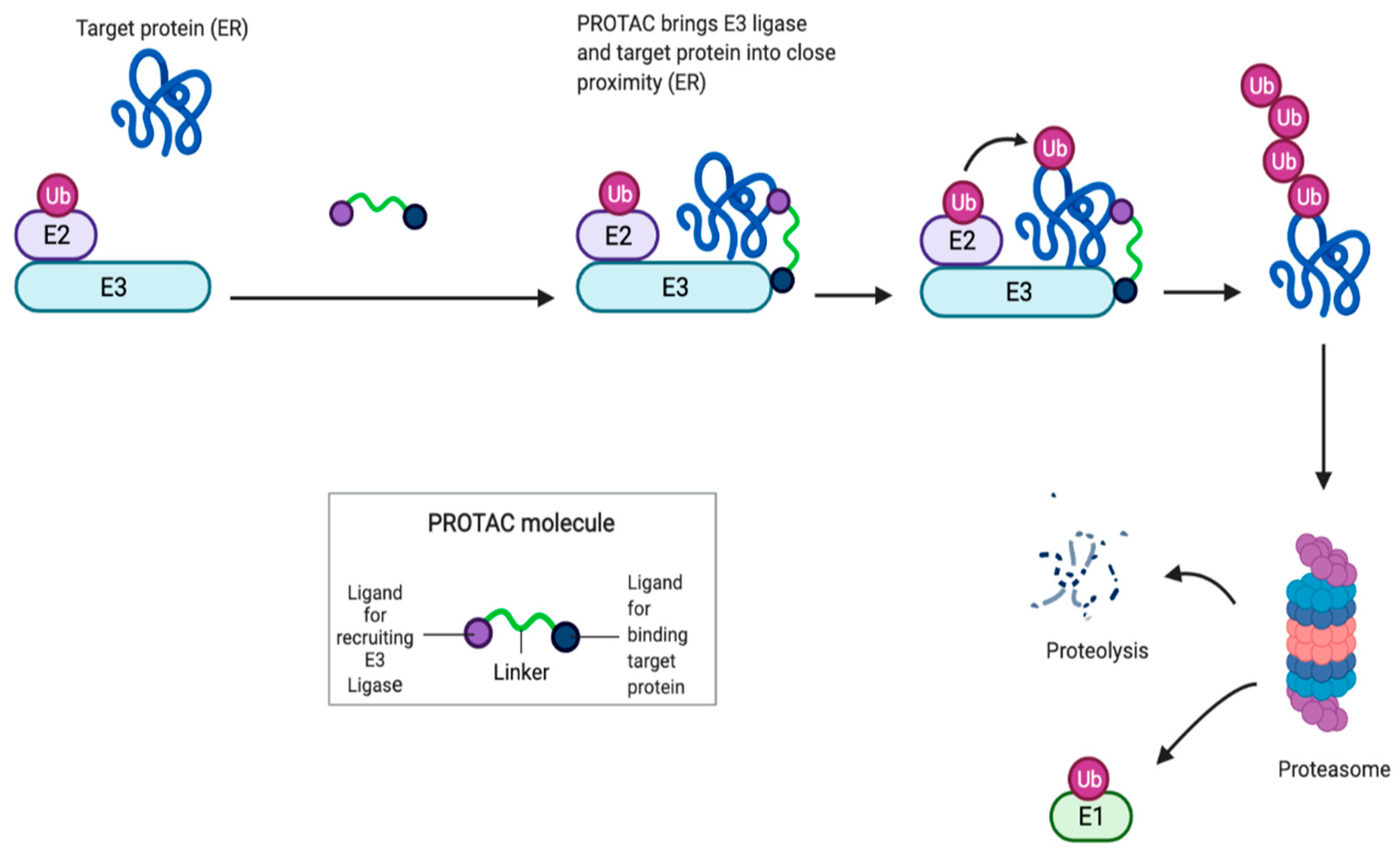
4. Novel Strategies
4.1. Proteolysis Targeting Chimera (PROTAC) as a New Class of Serd
4.2. Selective Estrogen Receptor Covalent Antagonists (Serca)
5. Summary
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AF | Activation function |
| AEs | Adverse events |
| ALT | Alanine aminotransferase |
| AIs | Aromatase inhibitors |
| AST | Aspartate aminotransferase |
| ctDNA | Circulating tumor DNA |
| CBR | Clinical benefit rate |
| CDK4/6i | Cyclin-dependent-kinase 4/6 inhibitors |
| DNA | Deoxynucleic acid |
| DLT | Dose limiting toxicity |
| ET | Endocrine therapy |
| E2 | Estradiol |
| ER | Estrogen receptor |
| ERE | Estrogen responsive element |
| ER+ | Estrogen receptor-positive |
| ERK | Extracellular signal regulated kinase |
| HR | Hormone receptor |
| HR+ | Hormonal receptors-positive |
| HER2 | Human epidermal growth factor receptor 2 |
| LFTs | Liver function tests |
| LTED | Long-term estrogen-deprived |
| MTD | Maximum tolerated dose |
| MBC | Metastatic breast cancer |
| MAPK | Mitogen-activated protein kinase |
| ORR | Objective response rate |
| PR | Partial response |
| PDX | Patient-derived xenograft |
| PK | Pharmacokinetics |
| RP2D | Phase II dose |
| PI3K | Phosphoinositide 3-kinase |
| PR+ | Progesterone receptor-positive |
| PFS | Progression-free survival |
| PD | Progressive disease |
| AKT | Protein kinase B |
| PROTAC | Proteolysis targeting chimera |
| SERCA | Selective estrogen receptor covalent antagonists |
| SERD | Selective estrogen receptor degraders |
| SERM | Selective estrogen receptor modulator |
| SD | Stable disease |
| SOC | Standard of care |
| TRAEs | Treatment-emergent adverse events |
References
- Cardoso, F.; Paluch-Shimon, S.; Senkus, E.; Curigliano, G.; Aapro, M.S.; André, F.; Barrios, C.H.; Bergh, J.; Bhattacharyya, G.S.; Biganzoli, L.; et al. Ohno 5th ESO-ESMO international consensus guidelines for advanced breast cancer (ABC 5). Ann. Oncol. 2020, 31, 1623–1649. [Google Scholar] [CrossRef] [PubMed]
- Santen, R.J.; Brodie, H.; Simpson, E.R.; Siiteri, P.K.; Brodie, A. History of aromatase: Saga of an important biological mediator and therapeutic target. Endocr. Rev. 2009, 30, 343–375. [Google Scholar] [CrossRef]
- Dixon, J.M. Endocrine Resistance in Breast Cancer. N. J. Sci. 2014, 2014, 1–27. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Han, S.J.; Smith, C.L. Cooperative activation of gene expression by agonists and antagonists mediated by estrogen receptor heteroligand dimer complexes. Mol. Pharmacol. 2013, 83, 1066–1077. [Google Scholar] [CrossRef] [Green Version]
- Di Leo, A.; Jerusalem, G.; Petruzelka, L.; Torres, R.; Bondarenko, I.; Khasanov, R.; Verhoeven, D.; Pedrini, J.L.; Smirnova, I.; Lichinitser, M.R.; et al. Results of the CONFIRM Phase III Trial Comparing Fulvestrant 250 mg With Fulvestrant 500 mg in Postmenopausal Women with Estrogen Receptor–Positive Advanced Breast Cancer. J. Clin. Oncol. 2010, 28, 4594–4600. [Google Scholar] [CrossRef]
- Robertson, J.F.; Lindemann, J.; Garnett, S.; Anderson, E.; Nicholson, R.I.; Kuter, I.; Gee, J.M. A Good Drug Made Better: The Fulvestrant Dose-Response Story. Clin. Breast Cancer 2014, 14, 381–389. [Google Scholar] [CrossRef]
- Shiino, S.; Kinoshita, T.; Yoshida, M.; Jimbo, K.; Asaga, S.; Takayama, S.; Tsuda, H. Prognostic Impact of Discordance in Hormone Receptor Status Between Primary and Recurrent Sites in Patients With Recurrent Breast Cancer. Clin. Breast Cancer 2016, 16, e133–e140. [Google Scholar] [CrossRef]
- Miller, T.W.; Balko, J.M.; Fox, E.M.; Ghazoui, Z.; Dunbier, A.; Anderson, H.; Dowsett, M.; Jiang, A.; Smith, R.A.; Maira, S.-M.; et al. ERα-Dependent E2F Transcription Can Mediate Resistance to Estrogen Deprivation in Human Breast Cancer. Cancer Discov. 2011, 1, 338–351. [Google Scholar] [CrossRef] [Green Version]
- Ma, C.X.; Reinert, T.; Chmielewska, I.; Ellis, M.J. Mechanisms of aromatase inhibitor resistance. Nat. Rev. Cancer 2015, 15, 261–275. [Google Scholar] [CrossRef] [PubMed]
- Fribbens, C.; O’Leary, B.; Kilburn, L.; Hrebien, S.; Garcia-Murillas, I.; Beaney, M.; Cristofanilli, M.; Andre, F.; Loi, S.; Loibl, S.; et al. Plasma ESR1 Mutations and the Treatment of Estrogen Receptor–Positive Advanced Breast Cancer. J. Clin. Oncol. 2016, 34, 2961–2968. [Google Scholar] [CrossRef] [PubMed]
- Chandarlapaty, S.; Chen, D.; He, W.; Sung, P.; Samoila, A.; You, D.; Bhatt, T.; Patel, P.; Voi, M.; Gnant, M.; et al. Prevalence of ESR1 mutations in cell-free DNA and outcomes in metastatic breast cancer: A secondary analysis of the BOLERO-2 clinical trial. JAMA Oncol. 2016, 2, 1310–1315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Razavi, P.; Chang, M.T.; Xu, G.; Bandlamudi, C.; Ross, D.S.; Vasan, N.; Cai, Y.; Bielski, C.M.; Donoghue, M.T.A.; Jonsson, P.; et al. The Genomic Landscape of Endocrine-Resistant Advanced Breast Cancers. Cancer Cell 2018, 34, 427–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology: Breast Cancer v3, 2021. Available online: https://www.nccn.org/professionals/physician_gls/pdf/breast.pdf (accessed on 26 April 2021).
- Lee, N.; Park, M.-J.; Song, W.; Jeon, K.; Jeong, S. Currently Applied Molecular Assays for Identifying ESR1 Mutations in Patients with Advanced Breast Cancer. Int. J. Mol. Sci. 2020, 21, 8807. [Google Scholar] [CrossRef] [PubMed]
- Speirs, V. Oestrogen receptor beta in breast cancer: Good, bad or still too early to tell? J. Pathol. 2002, 197, 143–147. [Google Scholar] [CrossRef] [PubMed]
- Omoto, Y.; Iwase, H. Clinical significance of estrogen receptor beta in breast and prostate cancer from biological aspects. Cancer Sci. 2015, 106, 337–343. [Google Scholar] [CrossRef]
- Enmark, E.; Pelto-Huikko, M.; Grandien, K.; Lagercrantz, S.; Lagercrantz, J.; Fried, G.; Nordenskjöld, M.; Gustafsson, J.-A. Human Estrogen Receptor β-Gene Structure, Chromosomal Localization, and Expression Pattern. J. Clin. Endocrinol. Metab. 1997, 82, 4258–4265. [Google Scholar] [CrossRef]
- Menasce, L.P.; White, G.R.M.; Harrison, C.J.; Boyle, J.M. Localization of the estrogen receptor locus (ESR) to chromosome 6q25.1 by FISH and a simple post-FISH banding technique. Genomics 1993, 17, 263–265. [Google Scholar] [CrossRef]
- Lu, Y.; Gutgesell, L.M.; Xiong, R.; Zhao, J.; Li, Y.; Rosales, C.I.; Hollas, M.; Shen, Z.; Gordon-Blake, J.; Dye, K.; et al. Design and Synthesis of Basic. Selective Estrogen Receptor Degraders for Endocrine Therapy Resistant Breast Cancer. J. Med. Chem. 2019, 62, 11301–11323. [Google Scholar] [CrossRef]
- Pawlak, M.; Lefebvre, P.; Staels, B. General molecular biology and architecture of nuclear receptors. Curr. Top. Med. Chem. 2012, 12, 486–504. [Google Scholar] [CrossRef] [Green Version]
- Kumar, V.; Chambon, P. The estrogen receptor binds tightly to its responsive element as a ligand-induced homodimer. Cell 1988, 55, 145–156. [Google Scholar] [CrossRef]
- Hall, J.M.; McDonnell, D.P. Coregulators in nuclear estrogen receptor action: From concept to therapeutic targeting. Mol. Interv. 2005, 5, 343–357. [Google Scholar] [CrossRef]
- Evinger, A.J., III; Levin, E.R. Requirements for estrogen receptor α membrane localization and function. Steroids 2005, 70, 361–363. [Google Scholar] [CrossRef]
- Kahlert, S.; Nuedling, S.; van Eickels, M.; Vetter, H.; Meyer, R.; Grohé, C. Estrogen Receptor α Rapidly Activates the IGF-1 Receptor Pathway. J. Biol. Chem. 2000, 275, 18447–18453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, C.-W.; McNally, C.; Nickbarg, E.; Komm, B.S.; Cheskis, B.J. Estrogen receptor-interacting protein that modulates its nongenomic activity-crosstalk with Src/Erk phosphorylation cascade. Proc. Natl. Acad. Sci. USA 2002, 99, 14783–14788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeselsohn, R.; Buchwalter, G.; DE Angelis, C.; Brown, M.; Schiff, R. ESR1 mutations—A mechanism for acquired endocrine resistance in breast cancer. Nat. Rev. Clin. Oncol. 2015, 12, 573–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basudan, A.; Priedigkeit, N.; Hartmaier, R.J.; Sokol, E.S.; Bahreini, A.; Watters, R.J.; Boisen, M.M.; Bhargava, R.; Weiss, K.R.; Karsten, M.M.; et al. Frequent ESR1 and CDK Pathway Copy-Number Alterations in Metastatic Breast Cancer. Mol. Cancer Res. 2018, 17, 457–468. [Google Scholar] [CrossRef] [Green Version]
- Brown, L.; Hoog, J.; Chin, S.-F.; Tao, Y.; Zayed, A.A.; Chin, K.; Teschendorff, A.E.; Quackenbush, J.F.; Marioni, J.C.; Leung, S.; et al. ESR1 gene amplification in breast cancer: A common phenomenon? Nat. Genet. 2008, 40, 806–807. [Google Scholar] [CrossRef] [PubMed]
- Tomita, S.; Zhang, Z.; Nakano, M.; Ibusuki, M.; Kawazoe, T.; Yamamoto, Y.; Iwase, H. Estrogen receptor α geneESR1amplification may predict endocrine therapy responsiveness in breast cancer patients. Cancer Sci. 2009, 100, 1012–1017. [Google Scholar] [CrossRef]
- Holst, F.; Singer, C.F. ESR1-Amplification-Associated Estrogen Receptor α Activity in Breast Cancer. Trends Endocrinol. Metabolites 2016, 27, 751–752. [Google Scholar]
- Nielsen, K.V.; Ejlertsen, B.; Müller, S.; Møller, S.; Rasmussen, B.B.; Balslev, E.; Lænkholm, A.-V.; Christiansen, P.; Mouridsen, H.T. Amplification of ESR1 may predict resistance to adjuvant tamoxifen in postmenopausal patients with hormone receptor positive breast cancer. Breast Cancer Res. Treat. 2010, 127, 345–355. [Google Scholar] [CrossRef] [Green Version]
- Markiewicz, A.; Welnicka-Jaskiewicz, M.; Skokowski, J.; Jaśkiewicz, J.; Szade, J.; Jassem, J.; Zaczek, A.J. Prognostic Significance of ESR1 Amplification and ESR1 PvuII, CYP2C19*2, UGT2B15*2 Polymorphisms in Breast Cancer Patients. PLoS ONE 2013, 8, e72219. [Google Scholar] [CrossRef] [Green Version]
- Veeraraghavan, J.; Tan, Y.; Cao, X.-X.; Kim, J.A.; Wang, X.; Chamness, G.C.; Maiti, S.N.; Cooper, L.J.N.; Edwards, D.P.; Contreras, A.; et al. Recurrent ESR1–CCDC170 rearrangements in an aggressive subset of oestrogen receptor-positive breast cancers. Nat. Commun. 2014, 5, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiavon, G.; Hrebien, S.; Garcia-Murillas, I.; Cutts, R.J.; Pearson, A.; Tarazona, N.; Fenwick, K.; Kozarewa, I.; Lopez-Knowles, E.; Ribas, R.; et al. Analysis of ESR1 mutation in circulating tumor DNA demonstrates evolution during therapy for metastatic breast cancer. Sci. Transl. Med. 2015, 7, 313ra182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Santo, I.; McCartney, A.; Migliaccio, I.; Di Leo, A.; Malorni, L. The emerging role of ESR1 mutations in luminal breast cancer as a prognostic and predictive biomarker of response to endocrine therapy. Cancers 2019, 11, 1894. [Google Scholar] [CrossRef] [Green Version]
- Toy, W.; Weir, H.; Razavi, P. Activating ESR1 mutations differentially impact the efficacy of ER antagonists. Cancer Discov. 2017, 7, 277–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clatot, F.; Perdrix, A.; Beaussire, L.; Lequesne, J.; Levy, C.; Emile, G.; Bubenheim, M.; Lacaille, S.; Calbrix, C.; Augusto, L.; et al. Risk of early progression according to circulating ESR1 mutation, CA-15.3 and cfDNA increases under first-line anti-aromatase treatment in metastatic breast cancer. Breast Cancer Res. 2020, 22, 56. [Google Scholar] [CrossRef]
- Wakeling, A.E.; Dukes, M.; Bowler, J. A potent specific pure antiestrogen with clinical potential. Cancer Res. 1991, 51, 3867–3873. [Google Scholar]
- Wakeling, A.; Slater, S. Estrogen-receptor binding and biologic activity of tamoxifen and its metabolites. Cancer Treat. Rep. 1980, 64, 741–744. [Google Scholar]
- Fawell, S.E.; White, R.; Hoare, S.; Sydenham, M.; Page, M.; Parker, M.G. Inhibition of estrogen receptor-DNA binding by the “pure” antiestrogen ICI 164,384 appears to be mediated by impaired receptor dimerization. Proc. Natl. Acad. Sci. USA 1990, 87, 6883–6887. [Google Scholar] [CrossRef] [Green Version]
- Dauvois, S.; White, R.; Parker, M.G. The antiestrogen ICI 182780 disrupts estrogen receptor nucleocytoplasmic shuttling. J. Cell Sci. 1993, 106, 1377–1388. [Google Scholar] [CrossRef] [PubMed]
- Osborne, C.; Wakeling, A.; Nicholson, R. Fulvestrant: An oestrogen receptor antagonist with a novel mechanism of action. Br. J. Cancer 2004, 90, S2–S6. [Google Scholar] [CrossRef] [PubMed]
- Long, X.; Nephew, K.P. Fulvestrant (ICI 182,780)-dependent interacting proteins mediate immobilization and degradation of estrogen receptor-α. J. Biol. Chem. 2006, 281, 9607–9615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wijayaratne, A.L.; McDonnell, D.P. The human estrogen receptor-α is a ubiquitinated protein whose stability is affected differentially by agonists, antagonists, and selective estrogen receptor modulators. J. Biol. Chem. 2001, 276, 35684–35692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Khissiin, A.; Leclercq, G. Implication of proteasome in estrogen receptor degradation. FEBS Lett. 1999, 448, 160–166. [Google Scholar] [CrossRef] [Green Version]
- Harrison, M.; Laight, A.; Clarke, D.; Giles, P.; Yates, Y. 564 pharmacokinetics and metabolism of fulvestrant after oral, intravenous and intramuscular administration in healthy volunteers. Eur. J. Cancer Suppl. 2003, 1, S171. [Google Scholar] [CrossRef]
- Wardley, A.M. Fulvestrant: A review of its development, pre-clinical and clinical data. Int. J. Clin. Pract. 2002, 56, 305–309. [Google Scholar]
- Serrano, D.; Lazzeroni, M.; Gandini, S.; Macis, D.; Johansson, H.; Gjerde, J.; Lien, E.; Feroce, I.; Pruneri, G.; Sandri, M.; et al. A randomized phase II presurgical trial of weekly low-dose tamoxifen versus raloxifene versus placebo in premenopausal women with estrogen receptor- positive breast cancer. Breast Cancer Res. 2013, 15, R47. [Google Scholar] [CrossRef] [Green Version]
- Johnston, S.R. Endocrine manipulation in advanced breast cancer: Recent advances with SERM therapies. Clin. Cancer Res. 2001, 7, 4376s–4387s. [Google Scholar]
- Deshmane, V.; Krishnamurthy, S.; Melemed, A.S.; Peterson, P.; Buzdar, A.U. Phase III double-blind trial of arzoxifene compared with tamoxifen for locally advanced or metastatic breast cancer. J. Clin. Oncol. 2007, 25, 4967–4973. [Google Scholar] [CrossRef]
- Vajdos, F.; Hoth, L.R.; Geoghegan, K.F.; Simons, S.P.; Lemotte, P.K.; Danley, D.E.; Ammirati, M.J.; Pandit, J. The 2.0 Å crystal structure of the ERα ligand-binding domain complexed with lasofoxifene. Protein Sci. 2007, 16, 897–905. [Google Scholar] [CrossRef] [Green Version]
- Lacroix, A.Z.; Powles, T.; Osborne, C.K.; Wolter, K.; Thompson, J.R.; Thompson, D.D.; Allred, D.C.; Armstrong, R.; Cummings, S.R.; Eastell, R.; et al. Breast cancer incidence in the randomized pearl trial of lasofoxifene in postmenopausal osteoporotic women. JNCI J. Natl. Cancer Inst. 2010, 102, 1706–1715. [Google Scholar] [CrossRef] [Green Version]
- Cummings, S.R.; Ensrud, K.; Delmas, P.D.; LaCroix, A.Z.; Vukicevic, S.; Reid, D.M.; Eastell, R. Lasofoxifene in postmenopausal women with osteoporosis. Obstet. Gynecol. Surv. 2010, 65, 447–448. [Google Scholar] [CrossRef]
- Lewis-Wambi, J.S.; Kim, H.; Curpan, R.; Grigg, R.; Sarker, M.A.; Jordan, V.C. The selective estrogen receptor modulator bazedoxifene inhibits hormone-independent breast cancer cell growth and down-regulates estrogen receptor and cyclin D1. Mol. Pharmacol. 2011, 80, 610–620. [Google Scholar] [CrossRef] [Green Version]
- Wardell, S.E.; Ellis, M.J.; Alley, H.M.; Eisele, K.; VanArsdale, T.; Dann, S.G.; Arndt, K.T.; Primeau, T.; Griffin, E.; Shao, J. Efficacy of SERD/SERM hybrid-CDK4/6 inhibitor combinations in models of endocrine therapy-resistant breast cancer. Clin. Cancer Res. 2005, 21, 5121–5130. [Google Scholar] [CrossRef] [Green Version]
- Wardell, S.E.; Nelson, E.R.; Chao, C.A.; McDonnell, D.P. Bazedoxifene Exhibits antiestrogenic activity in animal models of tamoxifen-resistant breast cancer: Implications for treatment of advanced disease. Clin. Cancer Res. 2013, 19, 2420–2431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tria, G.S.; Abrams, T.; Baird, J.; Burks, H.E.; Firestone, B.; Gaither, L.A.; Hamann, L.G.; He, G.; Kirby, C.A.; Kim, S.; et al. Discovery of LSZ102, a potent, orally bioavailable selective estrogen receptor degrader (SERD) for the treatment of estrogen receptor positive breast cancer. J. Med. Chem. 2018, 61, 2837–2864. [Google Scholar] [CrossRef]
- Willson, T.M.; Henke, B.R.; Momtahen, T.M.; Charifson, P.S.; Batchelor, K.W.; Lubahn, D.B.; Moore, L.B.; Oliver, B.B.; Sauls, H.R.; Triantafillou, J.A.; et al. 3-[4-(1,2-Diphenylbut-1-enyl)phenyl]acrylic Acid: A non-steroidal estrogen with functional selectivity for bone over uterus in rats. J. Med. Chem. 1994, 37, 1550–1552. [Google Scholar] [CrossRef] [PubMed]
- Lai, A.; Kahraman, M.; Govek, S.; Nagasawa, J.; Bonnefous, C.; Julien, J.; Douglas, K.; Sensintaffar, J.; Lu, N.; Lee, K.J.; et al. Identification of GDC-0810 (ARN-810), an Orally Bioavailable Selective Estrogen Receptor Degrader (SERD) that Demonstrates Robust Activity in Tamoxifen-Resistant Breast Cancer Xenografts. J. Med. Chem. 2015, 58, 4888–4904. [Google Scholar] [CrossRef]
- McDonnell, D.P.; Wardell, S.E.; Norris, J.D. Oral Selective Estrogen Receptor Downregulators (SERDs), a Breakthrough Endocrine Therapy for Breast Cancer. J. Med. Chem. 2015, 58, 4883–4887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weir, H.M.; Bradbury, R.H.; Lawson, M.; Rabow, A.A.; Buttar, D.; Callis, R.J.; Curwen, J.O.; De Almeida, C.; Ballard, P.; Hulse, M.; et al. AZD9496: An Oral Estrogen Receptor Inhibitor That Blocks the Growth of ER-Positive and ESR1-Mutant Breast Tumors in Preclinical Models. Cancer Res. 2016, 76, 3307–3318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamilton, E.P.; Patel, M.R.; Armstrong, A.C.; Baird, R.D.; Jhaveri, K.; Hoch, M.; Klinowska, T.; Lindemann, J.P.O.; Morgan, S.R.; Schiavon, G.; et al. A First-in-Human Study of the New Oral Selective Estrogen Receptor Degrader AZD9496 for ER+/HER2− Advanced Breast Cancer. Clin. Cancer Res. 2018, 24, 3510–3518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robertson, J.F.R.; Evans, A.; Henschen, S.; Kirwan, C.C.; Jahan, A.; Kenny, L.M.; Dixon, J.M.; Schmid, P.; Kothari, A.; Mohamed, O.; et al. A Randomized, Open-label, Presurgical, Window-of-Opportunity Study Comparing the Pharmacodynamic Effects of the Novel Oral SERD AZD9496 with Fulvestrant in Patients with Newly Diagnosed ER (+) HER2(−) Primary Breast Cancer. Clin. Cancer Res. 2020, 26, 4242–4249. [Google Scholar]
- Ahmad, I.; Mathew, S.; Rahman, S. Recent progress in selective estrogen receptor downregulators (SERDs) for the treatment of breast cancer. RSC Med. Chem. 2020, 11, 438–454. [Google Scholar]
- Jhaveri, K.; Juric, D.; Yap, Y.; Cresta, S.; Layman, R.M.; Duhoux, F.P.; Terret, C.; de Vita, S.; Kundamal, N.; He, W.; et al. Interim results of a phase 1/1b study of LSZ102, an oral selective estrogen receptor degrader, in combination with ribociclib or alpelisib in patients with ER+ breast cancer who had progressed after endocrine therapy. Ann. Oncol. 2020, 31, S62. [Google Scholar] [CrossRef]
- Andreano, K.J.; Wardell, S.E.; Baker, J.G.; Desautels, T.K.; Baldi, R.; Chao, C.A. G1T48, an oral selective estrogen receptor degrader, and the CDK4/6 inhibitor lerociclib inhibit tumor growth in animal models of endocrine-resistant breast cancer. Breast Cancer Res. Treat. 2020, 180, 635–646. [Google Scholar] [CrossRef] [Green Version]
- Wardell, S.E.; Baker, J.G.; Baldi, R.M.; Yllanes, A.P.; Krebs, T.K.; Sorrentino, J.A.; Bisi, J.E.; Strum, J.C.; Norris, J.D. Abstract5641: Effects of G1T48, a novel orally bioavailable selective estrogen receptor degrader (SERD), and the CDK4/6 inhibitor, G1T38, on tumor growth in animal models of endocrine resistant breast cancer. Cancer Res. 2017, 77, 5641–15641. [Google Scholar]
- Scott, J.S.; Moss, T.A.; Balazs, A.; Barlaam, B.; Breed, J.; Carbajo, R.J.; Chiarparin, E.; Davey, P.R.J.; Delpuech, O.; Fawell, S.; et al. Discovery of AZD9833, a Potent and Orally Bioavailable Selective Estrogen Receptor Degrader and Antagonist. J. Med. Chem. 2020, 63, 14530–14559. [Google Scholar] [CrossRef] [PubMed]
- Bihani, T.; Patel, H.K.; Arlt, H.; Tao, N.; Jiang, H.; Brown, J.L.; Purandare, D.M.; Hattersley, G.; Garner, F. Elacestrant (RAD1901), a Selective Estrogen Receptor Degrader (SERD), Has Antitumor Activity in Multiple ER+ Breast Cancer Patient-derived Xenograft Models. Clin. Cancer Res. 2017, 23, 4793–4804. [Google Scholar] [CrossRef] [Green Version]
- Garner, F.; Shomali, M.; Paquin, D.; Lyttle, C.R.; Hattersley, G. RAD1901: A novel, orally bioavailable selective estrogen receptor degrader that demonstrates antitumor activity in breast cancer xenograft models. Anti-Cancer Drugs 2015, 26, 948–956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wardell, S.E.; Nelson, E.R.; Chao, C.A.; Alley, H.M.; McDonnell, D.P. Evaluation of the pharmacological activities of RAD1901, a selective estrogen receptor degrader. Endocr. Relat. Cancer 2015, 22, 713–724. [Google Scholar] [CrossRef] [Green Version]
- Patel, H.K.; Tao, N.; Lee, K.-M.; Huerta, M.; Arlt, H.; Mullarkey, T.; Troy, S.; Arteaga, C.L.; Bihani, T. Elacestrant (RAD1901) exhibits anti-tumor activity in multiple ER+ breast cancer models resistant to CDK4/6 inhibitors. Breast Cancer Res. 2019, 21, 1–17. [Google Scholar] [CrossRef]
- Bardia, A.; Kaklamani, V.; Wilks, S.; Weise, A.; Richards, D.; Harb, W.; Osborne, C.; Wesolowski, R.; Karuturi, M.; Conkling, P.; et al. Phase I Study of Elacestrant (RAD1901), a Novel Selective Estrogen Receptor Degrader, in ER-Positive, HER2-Negative Advanced Breast Cancer. J. Clin. Oncol. 2021, 39, 1360–1370. [Google Scholar] [CrossRef]
- Bardia, A.; Aftimos, P.; Bihani, T.; Anderson-Villaluz, A.T.; Jung, J.; Conlan, M.G.; Kaklamani, V.G. EMERALD: Phase III trial of elacestrant (RAD1901) vs. endocrine therapy for previously treated ER+ advanced breast cancer. Future Oncol. 2019, 15, 3209–3218. [Google Scholar] [CrossRef]
- Kahraman, M.; Govek, S.P.; Nagasawa, J.Y.; Lai, A.; Bonnefous, C.; Douglas, K.; Sensintaffar, J.; Liu, N.; Lee, K.; Aparicio, A.; et al. Maximizing ER-α degradation maximizes activity in a tamoxifen-resistant breast cancer model: Identification of GDC-0927. ACS Med. Chem. Lett. 2019, 10, 50–55. [Google Scholar] [CrossRef]
- Labadie, S.S.; Li, J.; Blake, R.A.; Chang, J.H.; Goodacre, S.; Hartman, S.J.; Liang, W.; Kiefer, J.R.; Kleinheinz, T.; Lai, T.; et al. Discovery of a C-8 hydroxychromene as a potent degrader of estrogen receptor alpha with improved rat oral exposure over GDC-0927. Bioorg. Med. Chem. Lett. 2019, 29, 2090–2093. [Google Scholar] [CrossRef] [PubMed]
- Dickler, M.N.; Villanueva, R.; Perez Fidalgo, J.A. A First-in-Human Phase I Study to Evaluate the Oral Selective Estrogen Receptor Degrader (SERD), GDC-0927, in Postmenopausal Women with Estrogen Receptor Positive (ERþ) HER2-Negative Metastatic Breast Cancer (BC). Cancer Res. 2018, 78. [Google Scholar] [CrossRef]
- Spoerke, J.M.; Daemen, A.; Chang, C.-W. Phamacodynamic and circulating tumor DNA evaluation in a phase I study of GDC-0927, a selective estrogen receptor antagonist/degrader (SERD). In Proceedings of the 2018 San Antonio Breast Cancer Symposium, San Antonio, TX, USA, 4–8 December 2018. [Google Scholar]
- Metcalfe, C.; Ingalla, E.; Blake, R.; Chang, J.; Daemen, A.; De Bruyn, T.; Giltnane, J.; Guan, J.; Hafner, M.; Hartman, S.; et al. Abstract P5-04-07: GDC-9545: A novel ER antagonist and clinical candidate that combines desirable mechanistic and pre-clinical DMPK attributes. Cancer Res. 2019, 79. [Google Scholar] [CrossRef]
- Guan, J.; Zhou, W.; Hafner, M.; Blake, R.A.; Chalouni, C.; Chen, I.; De Bruyn, T.; Giltnane, J.M.; Hartman, S.; Heidersbach, A.; et al. Therapeutic Ligands Antagonize Estrogen Receptor Function by Impairing Its Mobility. Cell 2019, 178, 949–963.e18. [Google Scholar] [CrossRef] [PubMed]
- Lim, E.; Jhaveri, K.L.; Perez-Fidalgo, J.A.; Bellet, M.; Boni, V.; Garcia, J.M.P.; Estevez, L.; Bardia, A.; Turner, N.C.; Villanueva, R.; et al. A phase Ib study to evaluate the oral selective estrogen receptor degrader GDC-9545 alone or combined with palbociclib in metastatic ER-positive HER2-negative breast cancer. J. Clin. Oncol. 2020, 38, 1023. [Google Scholar] [CrossRef]
- Shomali, M.; Cheng, J.; Sun, F.; Koundinya, M.; Guo, Z.; Hebert, A.T.; McManus, J.; Levit, M.N.; Hoffmann, D.; Courjaud, A.; et al. SAR439859, a Novel Selective Estrogen Receptor Degrader (SERD), Demonstrates Effective and Broad Antitumor Activity in Wild-Type and Mutant ER-Positive Breast Cancer Models. Mol. Cancer Ther. 2021, 20, 250–262. [Google Scholar] [CrossRef] [PubMed]
- El-Ahmad, Y.; Tabart, M.; Halley, F.; Certal, V.; Thompson, F.; Filoche-Rommé, B.; Gruss-Leleu, F.; Muller, C.; Brollo, M.; Fabien, L.; et al. Discovery of 6-(2,4-Dichlorophenyl)-5-[4-[(3S)-1-(3-fluoropropyl)pyrrolidin-3-yl]oxyphenyl]-8,9-dihydro-7H-benzo[7]annulene-2-carboxylic acid (SAR439859), a Potent and Selective Estrogen Receptor Degrader (SERD) for the Treatment of Estrogen-Receptor-Positive Breast Cancer. J. Med. Chem. 2020, 63, 512–528. [Google Scholar]
- Linden, H.M.; Campone, M.; Bardia, A.; Ulaner, G.A.; Gosselin, A.; Doroumian, S.; Pelekanou, V.; Celanovic, M.; Chandarlapaty, S. A Phase 1/2 Study of SAR439859, an Oral Selective Estrogen Receptor (ER) Degrader (SERD), as Monotherapy and in Combination with Other Anti-Cancer Therapies in Postmenopausal Women with ER-Positive (ER+)/Human Epidermal Growth Factor Receptor 2-Negative (HER2-) Metastatic Breast Cancer (mBC): AMEERA-1. In Proceedings of the 2020 San Antonio Breast Cancer Symposium (Virtual), San Antonio, TX, USA, 8–11 December 2020; Available online: https://www.abstractsonline.com/pp8/#!/9223/presentation/848 (accessed on 15 May 2021).
- Gallardo, A.; Garcia-Valdecasas, B.; Murata, P.; Teran, R.; Lopez, L.; Barnadas, A.; Lerma, E. Inverse relationship between Ki67 and survival in early luminal breast cancer: Confirmation in a multivariate analysis. Breast Cancer Res. Treat. 2017, 167, 31–37. [Google Scholar] [CrossRef]
- Scott, J.S.; Moss, T.; Stokes, S.; Nissink, W.M.; Morrow, C.J.; Lawson, M.; Cureton, N.; Gangl, E.; Gutierrez, P.M.; Mather, R.; et al. Abstract 5674: Discovery of AZD9833, an oral small molecule selective degrader of the estrogen receptor (SERD). Cancer Chem. 2020, 80, 5674. [Google Scholar]
- Baird, R.; Oliveira, M.; Gil, E.M.C.; Patel, M.R.; Heras, B.B.D.L.; Ruiz-Borrego, M.; García-Corbacho, J.; Armstrong, A.; Banerji, U.; Twelves, C.; et al. Abstract PS11-05: Updated data from SERENA-1: A Phase 1 dose escalation and expansion study of the next generation oral SERD AZD9833 as a monotherapy and in combination with palbociclib, in women with ER-positive, HER2-negative advanced breast cancer. Poster Sess. Abstr. 2021, 81. [Google Scholar] [CrossRef]
- Lim, E.; Beeram, M.; Prawira, A.; Patnaik, A.; Wang, X.A.; Young, S.R.; Smyth, L.M.; Hamilton, E.P. Abstract OT-09-03: EMBER: A phase 1a/b trial of LY3484356, a novel, oral selective estrogen-receptor degrader (SERD), in advanced ER+ breast cancer and endometroid endometrial cancer. Cancer Res. 2021, 81 (Suppl. 4). [Google Scholar] [CrossRef]
- Jhaveri, K.L.; Lim, E.; Hamilton, E.P.; Saura, C.; Meniawy, T.; Jeselsohn, R.; Beck, J.T.; Kaufman, P.A.; Sammons, S.; Banda, K.; et al. A first-in-human phase 1a/b trial of LY3484356, an oral selective estrogen receptor (ER) degrader (SERD) in ER+ advanced breast cancer (aBC) and endometrial endometrioid cancer (EEC): Results from the EMBER study. J. Clin. Oncol. 2021, 39, 1050. [Google Scholar] [CrossRef]
- Ahmed, A.S.; Li, J.; Hegde, S.; Huang, P.; Ma, J.; Bunker, K.; Winkler, R.; Donate, F.; Sergeeva, M. Discovery of ZN-c5, a novel potent and oral selective estrogen receptor degrader [abstract]. Cancer Res. 2020, 80 (Suppl. 16), 4373. [Google Scholar] [CrossRef]
- Dougan, D.A.; Micevski, D.; Truscott, K.N. The N-end rule pathway: From recognition by N-recognins, to destruction by AAA +proteases. Biochim. Biophys. Acta Mol. Cell Res. 2012, 1823, 83–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flanagan, J.; Qian, Y.; Gough, S.; Andreoli, M.; Bookbinder, M.; Cadelina, G.; Bradley, J.; Rousseau, E.; Willard, R.; Pizzano, J.; et al. Abstract P5-04-18: ARV-471, an oral estrogen receptor PROTAC degrader for breast cancer. Poster Sess. Abstr. 2019, 79. [Google Scholar] [CrossRef]
- Hu, J.; Hu, B.; Wang, M.; Xu, F.; Miao, B.; Yang, C.Y.; Wang, M.; Liu, Z.; Hayes, D.F.; Chinnaswamy, K.; et al. Discovery of ERD-308 as a Highly Potent Proteolysis Targeting Chimera (PROTAC) Degrader of Estrogen Receptor (ER). J. Med. Chem. 2019, 62, 1420–1442. [Google Scholar] [CrossRef]
- Gao, H.; Sun, X.; Rao, Y. PROTAC technology: Opportunities and challenges. ACS Med. Chem Lett. 2020, 11, 237–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arvinas Releases Interim Clinical Data Further Demonstrating the Powerful Potential of PROTAC Protein Degraders ARV-471 and ARV-110. News Release; Arvinas, Inc.: New Haven, CT, USA, 14 December 2020; Available online: https://ir.arvinas.com/news-releases/news-release-details/arvinas-releases-interim-clinical-data-further-demonstrating/ (accessed on 4 February 2021).
- Puyang, X.; Furman, C.; Banka, D.; Aithal, K.; Agoulnik, S.; Buonamici, S.; Caleb, B.; Das, S.; Eckley, S.; Fekkes, P.; et al. Discovery of Selective Estrogen Receptor Covalent Antagonists for the Treatment of ER alphaWTand ER alphaMUTBreast Cancer. Cancer Discov. 2018, 8, 1176–1193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamilton, E.P.; Wang, J.S.; Pluard, T.; Johnston, S.; Morikawa, A.A.; Dees, C.E.; Jones, R.H.; Haley, B.; Armstrong, A.; Cohen, A.L.; et al. Abstract PD8-06: Phase I/II trial of H3B-6545, a novel selective estrogen receptor covalent antagonist (SERCA), in estrogen receptor positive (ER+), human epidermal growth factor receptor 2 negative (HER2-) advanced breast cancer. In Proceedings of the 2020 San Antonio Breast Cancer Symposium, Virtual Conference, 8–11 December 2020. Abstract 1142. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AGENT | TREATMENT | DISEASE SETTING | PHASE | NAME (INDICATOR) |
|---|---|---|---|---|
| SERMs | ||||
| LASOFOXIFENE | vs. FULVESTRANT +ABEMACICLIB |
Previously treated advanced/metastatic disease with
ESR1
mutations Previously treated advanced/metastatic disease with ESR1 mutations | 2 2 | ELAINE: NCT03781063 ELAINE 2: NCT04432454 |
| BAZEDOXIFENE | +PALBOCICLIB | Previously treated advanced/metastatic | 1/2 | NCT02448771 |
| SERDs | ||||
| LSZ102 | SINGLE AGENT/+ RIBOCICLIB/+ ALPELISIB | Previously treated advanced/metastatic | 1/1b | NCT02734615 |
| G1T48 (RINTODESTRANT) | SINGLE AGENT+/− PALBOCICLIB | Previously treated advanced/metastatic | 1 | NCT03455270 |
| RAD1901 (ELACESTRANT) | vs. SOC (standard of care) | Previously treated advanced/metastatic | 3 | EMERALD: NCT03778931 |
| GDC-9545 (GIREDESTRANT) | GDC-9545 vs. LETROZOLE + PALBOCICLIB GDC-9545 vs. ANASTROZOLE + PALBOCICLIB vs. physician’s choice of endocrine therapy +/− PALBOCICLIB and LHRH agonist Monotherapy | Advanced/metastatic Treatment-naïve early breast cancer (window-of-opportunity -> neoadjuvant) Previously treated advanced/metastatic Advanced/metastatic Treatment-naïve early breast cancer (window-of-opportunity) | 3 2 2 1 1 | persevERA: NCT04546009 coopERA: NCT04436744 acelERA: NCT04576455 NCT03332797 NCT03916744 |
| SAR439859 (AMCENESTRANT) | SAR439859 vs. LETROZOLE + PALBOCICLIB vs. physician’s choice of endocrine therapy vs. LETROZOLE +/− PALBOCICLIB OR ALPELISIB | Advanced/metastatic Previously treated advanced/metastatic Newly diagnosed advanced/metastatic Advanced/metastatic | 3 2 2 1/2 |
AMEERA-5: NCT04478266 AMEERA-3: NCT04059484 AMEERA-4: NCT04191382 AMEERA-1 NCT03284957 |
| AZD9833 (CAMIZESTRANT) | AZD9833 vs. LETROZOLE + PALBOCICLIB MONOTHERAPY vs. FULVESTRANT +/− PALBOCICLIB, EVEROLIMUS OR ABEMACICLIB | Treatment-naïve advanced/metastatic Neoadjuvant treatment Previously treated advanced/metastatic Previously treated advanced/metastatic | 3 2 2 1 | SERENA-4: NCT04711252 SERENA-3: NCT04588298 SERENA-2: NCT04214288 SERENA-1: NCT03616587 |
| LY3484356 | +/− other anticancer therapies MONOTHERAPY | Advanced/metastatic Neoadjuvant treatment | 1 1 | EMBER: NCT04188548 EMBER 2: NCT04647487 |
| Zn-c5 | SINGLE AGENT +/− PALBOCICLIB | Previously treated advanced/metastatic | 1/2 | NCT03560531 |
| D-0502 | SINGLE AGENT +/− PALBOCICLIB | Previously treated advanced/metastatic | 1 | NCT03471663 |
| NOVEL THERAPIES | ||||
| ARV-471 (PROTAC) | +/− PALBOCICLIB | Previously treated advanced/metastatic | 1/2 | NCT04072952 |
| H3B-5942 (SERCA) | MONOTHERAPY + PALBOCICLIB |
Previously treated advanced/metastatic Previously treated advanced/metastatic | 1/2 1 | NCT03250676 NCT04288089 |
| LSZ102 | Phase I/Ib (NCT02734615) (65) Arm A: Monotherapy Arm B: Combination with Arm B: Combination with Alpelisib | Arm A (n: 78): ORR (1.3%), CBR (9.1%), PFS (1.8 m) Arm B (n: 76): ORR (15.8%), CBR (35.5%), PFS (6.2 m) Arm C (n: 39): ORR (5.4%), CBR (18.9%), PFS 3.5m | Arm A, B, C: Grade 3/4: Nausea (3.1%) and Diarrhea (6.7%) Arm B (Grade 3 AEs): Neutropenia (13.2%) and Increased AAT (3.9%) Arm C (Grade 3 AEs): Hyperglycemia (10%), Skin Rashes (15.4%) |
|---|---|---|---|
| RAD1901 (ELACESTRANT) | Phase I (NCT02338349) (73) | N: 50 (dose-escalation) ORR 19.4% N: 47 (dose expansion) CBR 42.6% | No DLTs Grade 1/2: nausea (33.3%), increased triglycerides (25%), decreased blood phosphorus (25%) |
| GDC-9545 (GIREDESTRANT) | Phase Ib/II (NCT03332797) (88) Dose expansion: Cohort A: monotherapy Cohort B: combination with palbociclib | N: 88 Cohort A, n: 39 ORR (13%), PFS (7.8 m) Cohort B, n:43 ORR (33%), PFS (9.3 m) | Cohort A: Grade 1/2 fatigue, arthralgia. Grade 3: Fatigue (1), diarrhea (1), transaminase increased (1) Cohort B: Grade 1/2: neutropenia, fatigue, bradycardia, diarrhea, constipation, dizziness, nauseas, anemia, asthenia, pruritus and visual impairment. Grade 3: neutropenia (50%) |
| SAR439859 (AMCENESTRANT) | Phase I: AMEERA-1 (NCT03284957). (84) Monotherapy dose-escalation (Part A) | Part A: n: 59 ORR 8.5%, CBR (33.5%) | Part A: hot flushes (16.1%), constipation (9.7%), arthralgia (9.7%), decreased appetite (8.1%), vomiting (8.1%), diarrhea (8.1%), nausea (8.1%), and fatigue (6.5%) |
| AZD9833 (CAMIZESTRANT) | Phase I: SERENA-1 (NCT03616587) (87) Part A and B: monotherapy Part C and D: Combination with palbociclib | Part A and B, n: 98 ORR (10%), CDR (35.3%), PFS (5.4m) Part B and C, n: 48 ORR (6.3%), CBR (50%) | 5 dose-limiting toxicities (3 for monotherapy and 2 for combination therapy) Monotherapy (≥Grade 2 instances of AZD9833-related adverse events): fatigue (9%), bradicardia (3.1%), nausea (3%), visual disturbances (1.1%) Combination: grade 1–2: anemia, fatigue, lymphopenia, nausea, neutropenia, thrombocytopenia, and reduced white blood cell count |
| LY3484356 | Phase I/Ib (89) EMBER (NCT04188548) | N: 28 | Grade 1–2: nausea (32%), fatigue (25%), and diarrhea (18%) |
| H3B-5942 (SERCA) | Phase 1/2 (NCT03250676) (97) | N: 130 (phase I n: 47/phase II n: 83) PR (12%). SD (45%) and CBR (33%), PFS (3.7m) | Grade 2 or higher adverse: anemia (20%), fatigue (16%), nausea (14%), diarrhea (11%) and AST increase (11%). |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hernando, C.; Ortega-Morillo, B.; Tapia, M.; Moragón, S.; Martínez, M.T.; Eroles, P.; Garrido-Cano, I.; Adam-Artigues, A.; Lluch, A.; Bermejo, B.; et al. Oral Selective Estrogen Receptor Degraders (SERDs) as a Novel Breast Cancer Therapy: Present and Future from a Clinical Perspective. Int. J. Mol. Sci. 2021, 22, 7812. https://doi.org/10.3390/ijms22157812
Hernando C, Ortega-Morillo B, Tapia M, Moragón S, Martínez MT, Eroles P, Garrido-Cano I, Adam-Artigues A, Lluch A, Bermejo B, et al. Oral Selective Estrogen Receptor Degraders (SERDs) as a Novel Breast Cancer Therapy: Present and Future from a Clinical Perspective. International Journal of Molecular Sciences. 2021; 22(15):7812. https://doi.org/10.3390/ijms22157812
Chicago/Turabian StyleHernando, Cristina, Belén Ortega-Morillo, Marta Tapia, Santiago Moragón, María Teresa Martínez, Pilar Eroles, Iris Garrido-Cano, Anna Adam-Artigues, Ana Lluch, Begoña Bermejo, and et al. 2021. "Oral Selective Estrogen Receptor Degraders (SERDs) as a Novel Breast Cancer Therapy: Present and Future from a Clinical Perspective" International Journal of Molecular Sciences 22, no. 15: 7812. https://doi.org/10.3390/ijms22157812