
Combinatorial Virtual Library Screening Study of Transforming Growth Factor-β2–Chondroitin Sulfate System


Abstract
:1. Introduction
2. Results
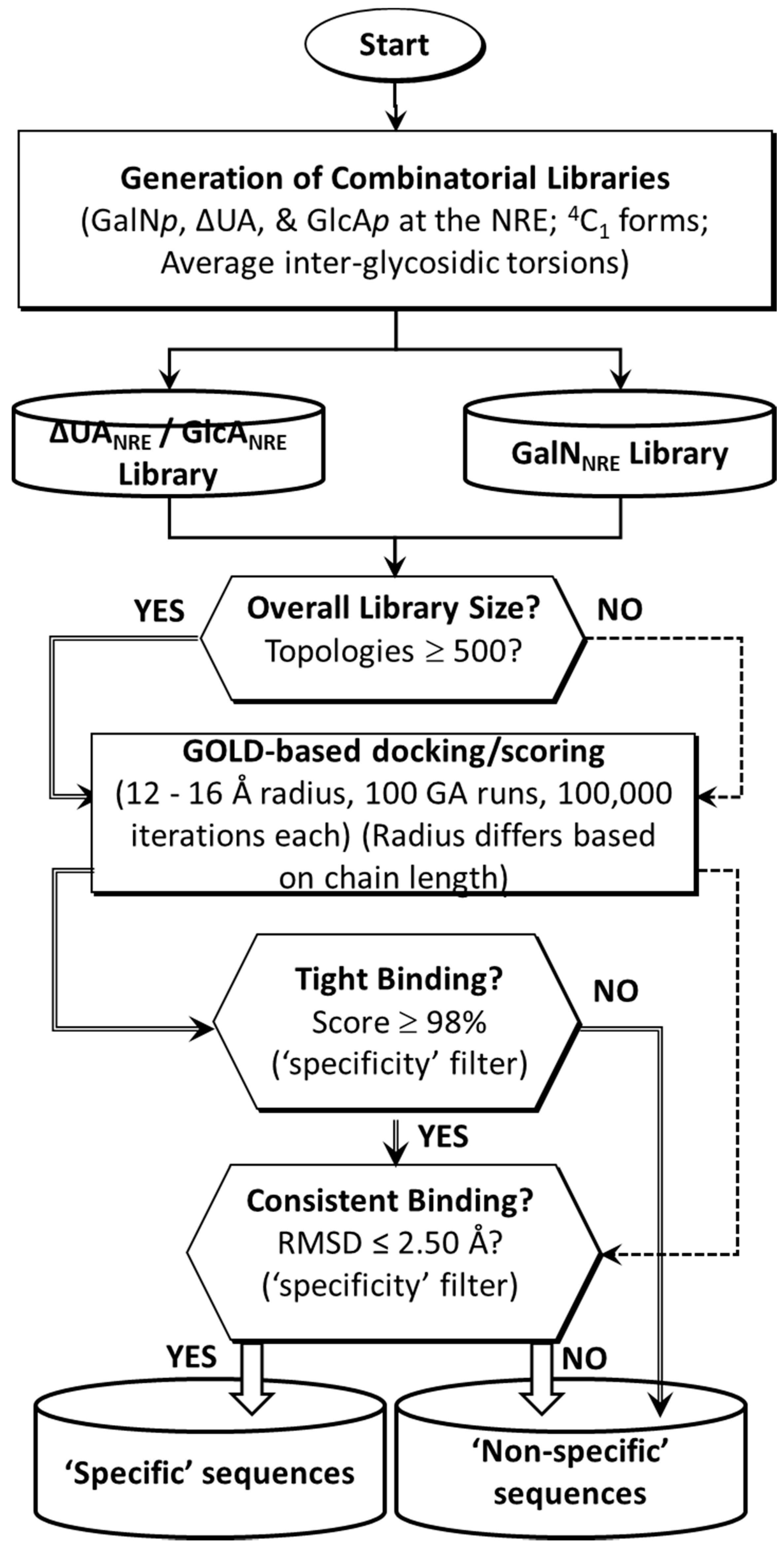
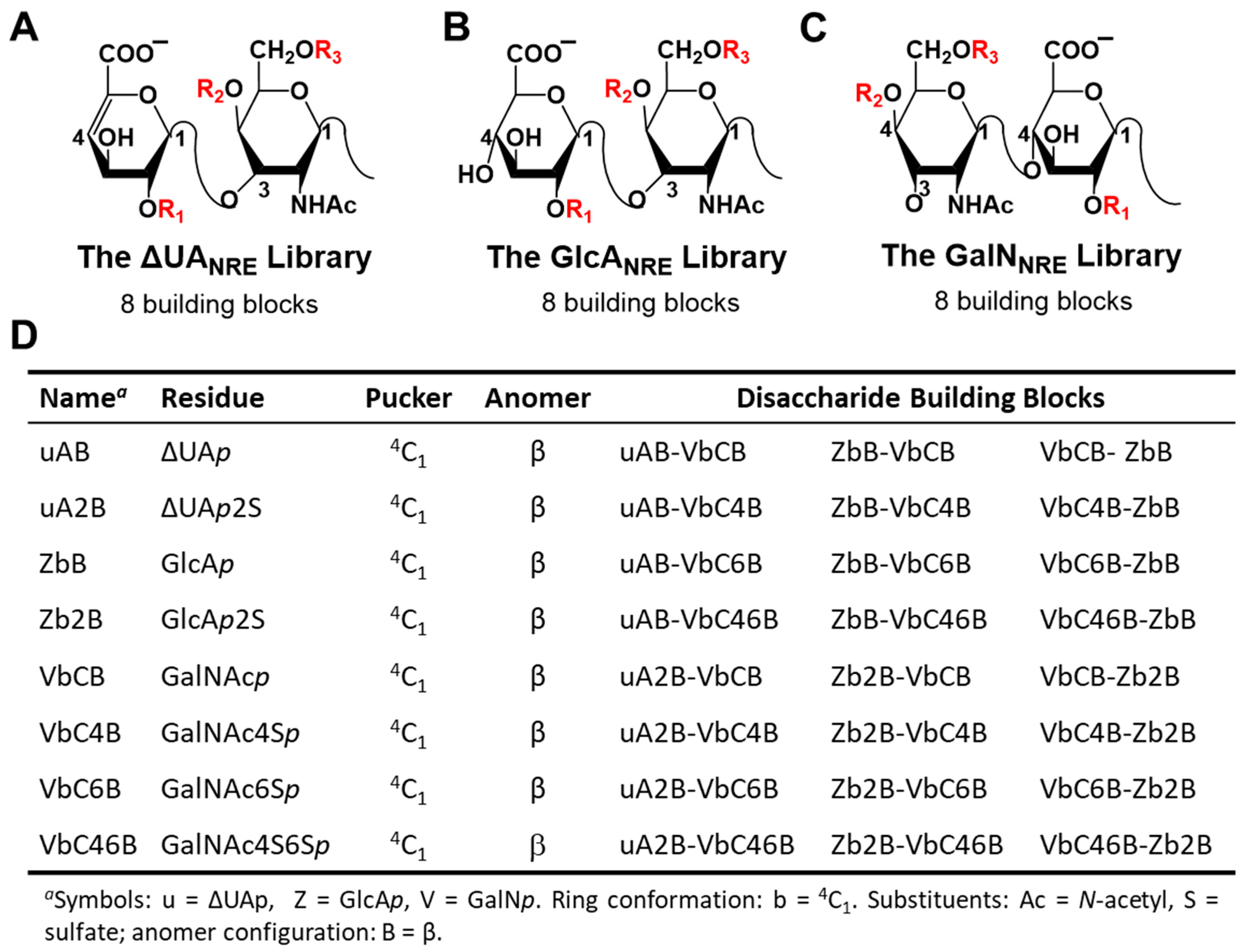
2.1. Application of CVLS Strategy to CS Sequences
2.2. CS Disaccharides Bind to TGF-β2, Albeit with Weak Consistency of Interactions
2.3. Distinct CS Tetrasaccharides Bind TGF-β2 with Moderate Consistency
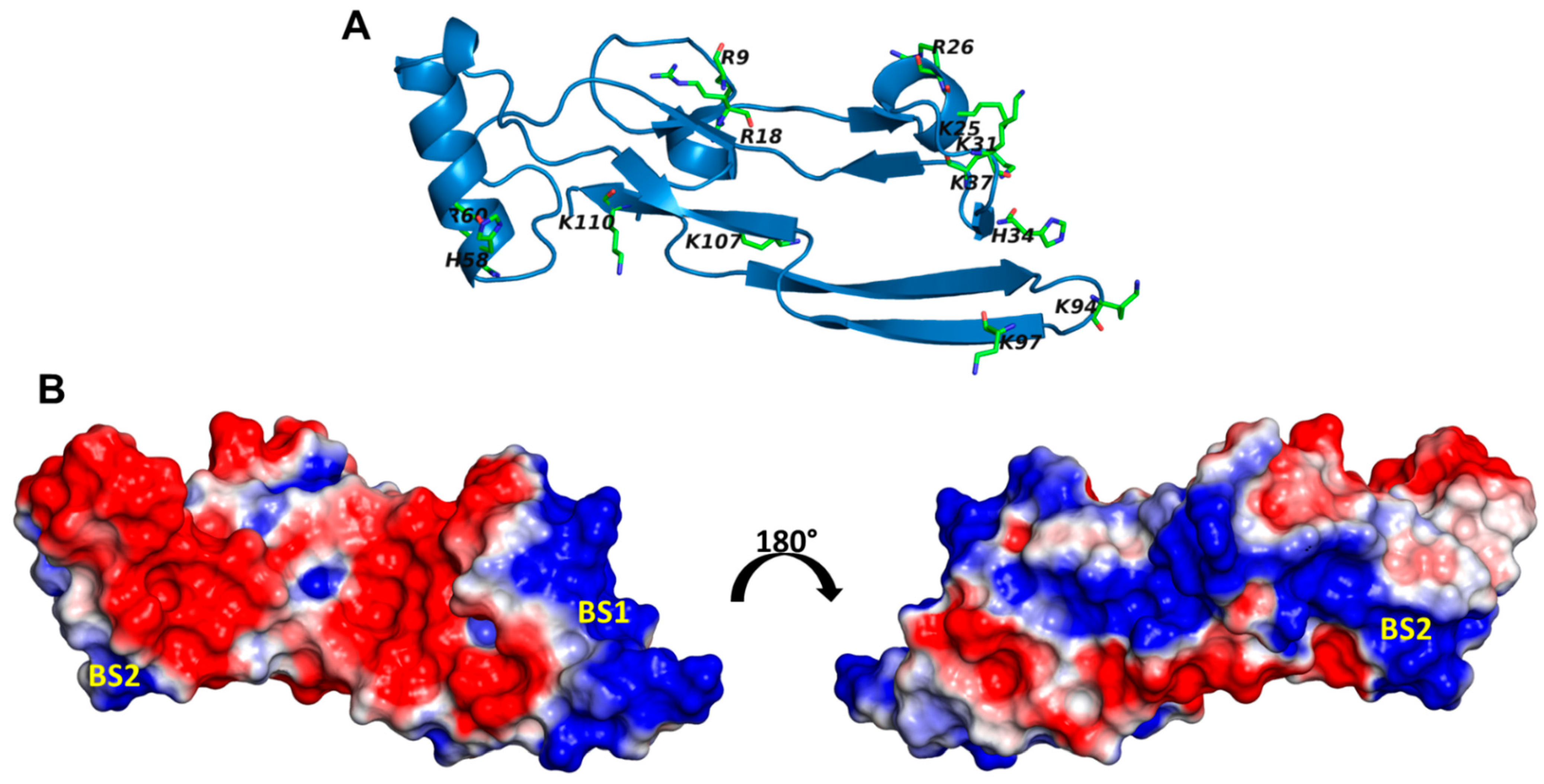
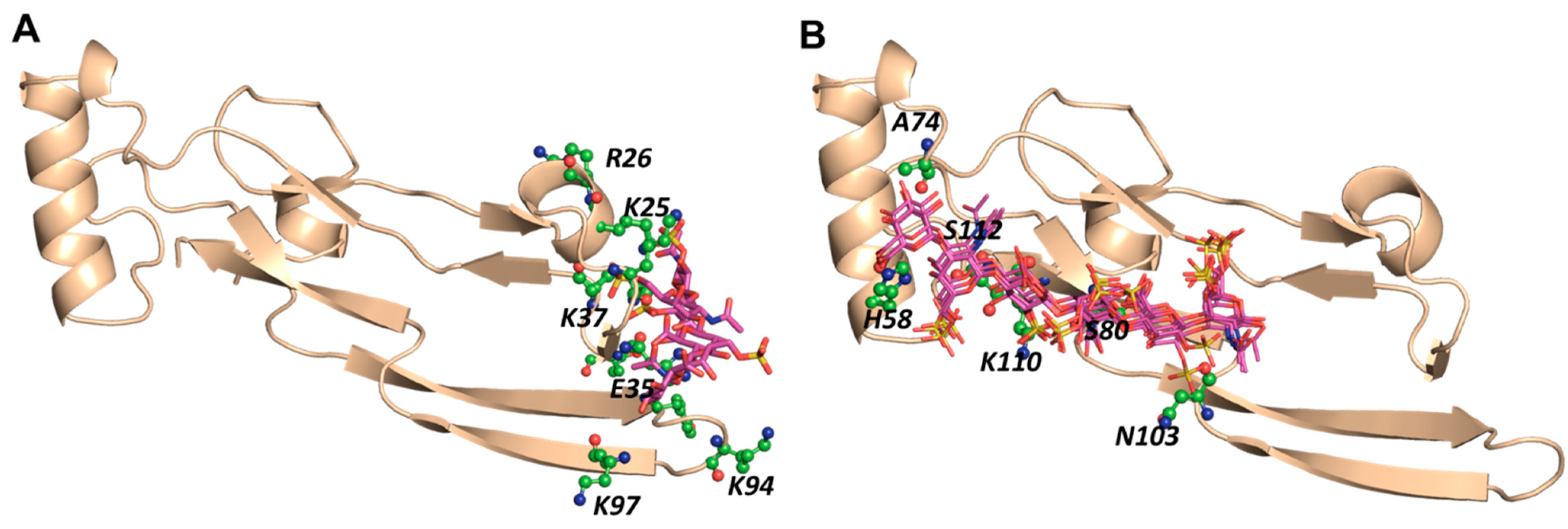
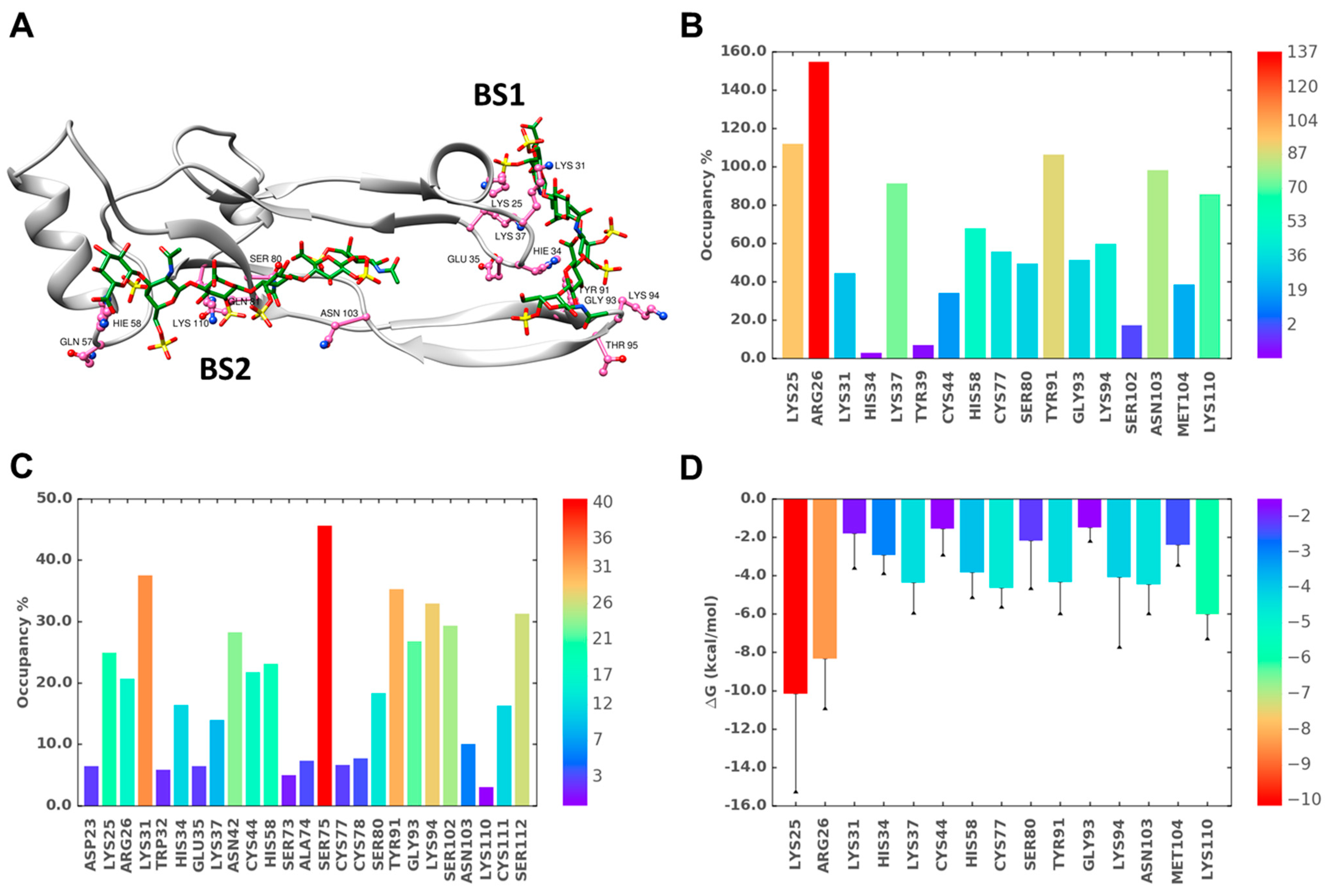
2.4. A Small Group of CS Hexasaccharides Bind in BS1 and BS2 with High Consistency
2.5. MD Simulations Indicate Excellent Stability of the TGF-β2–CS06 Complexes in Both Sites of Binding
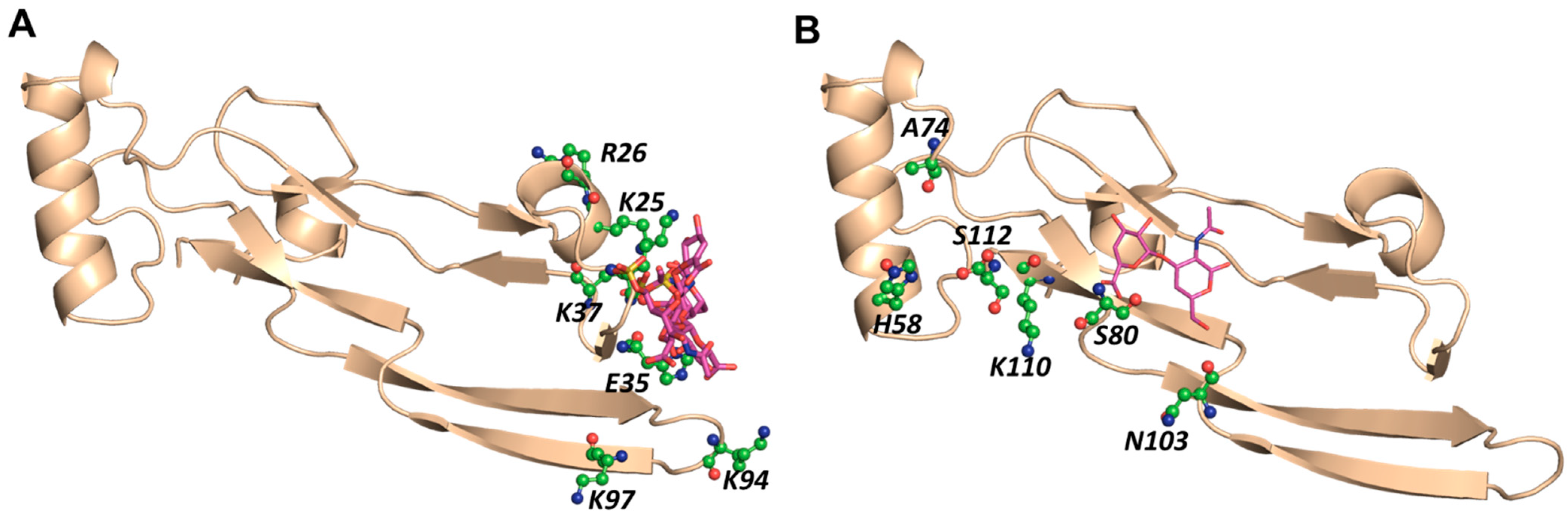
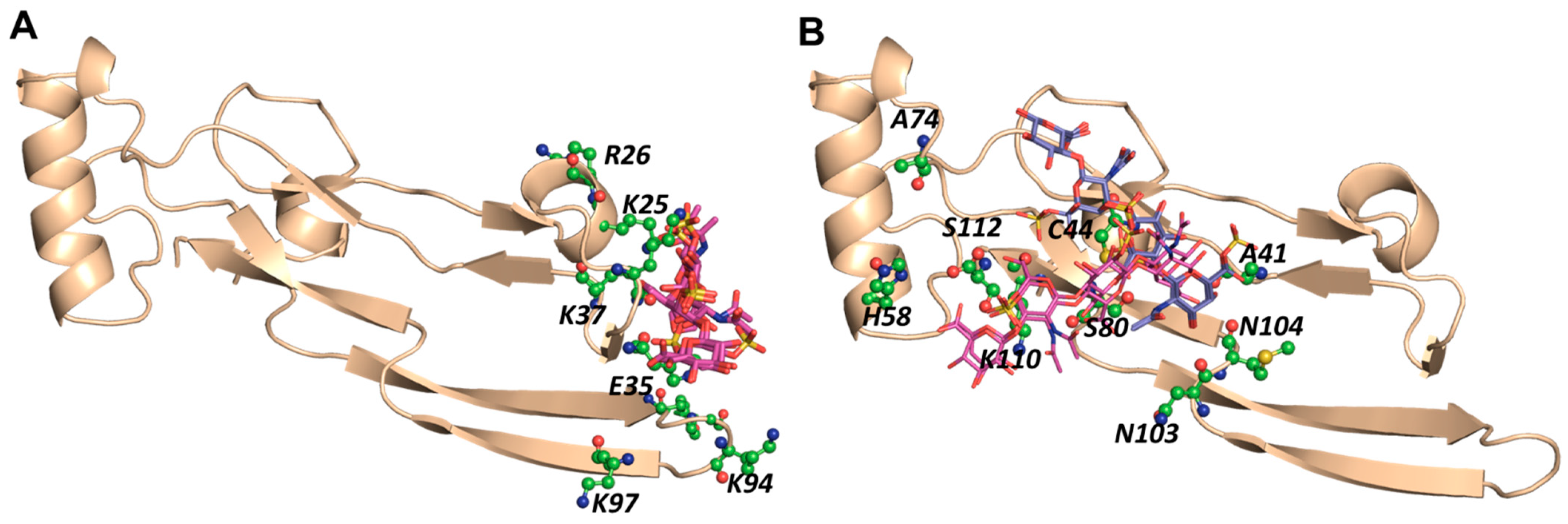
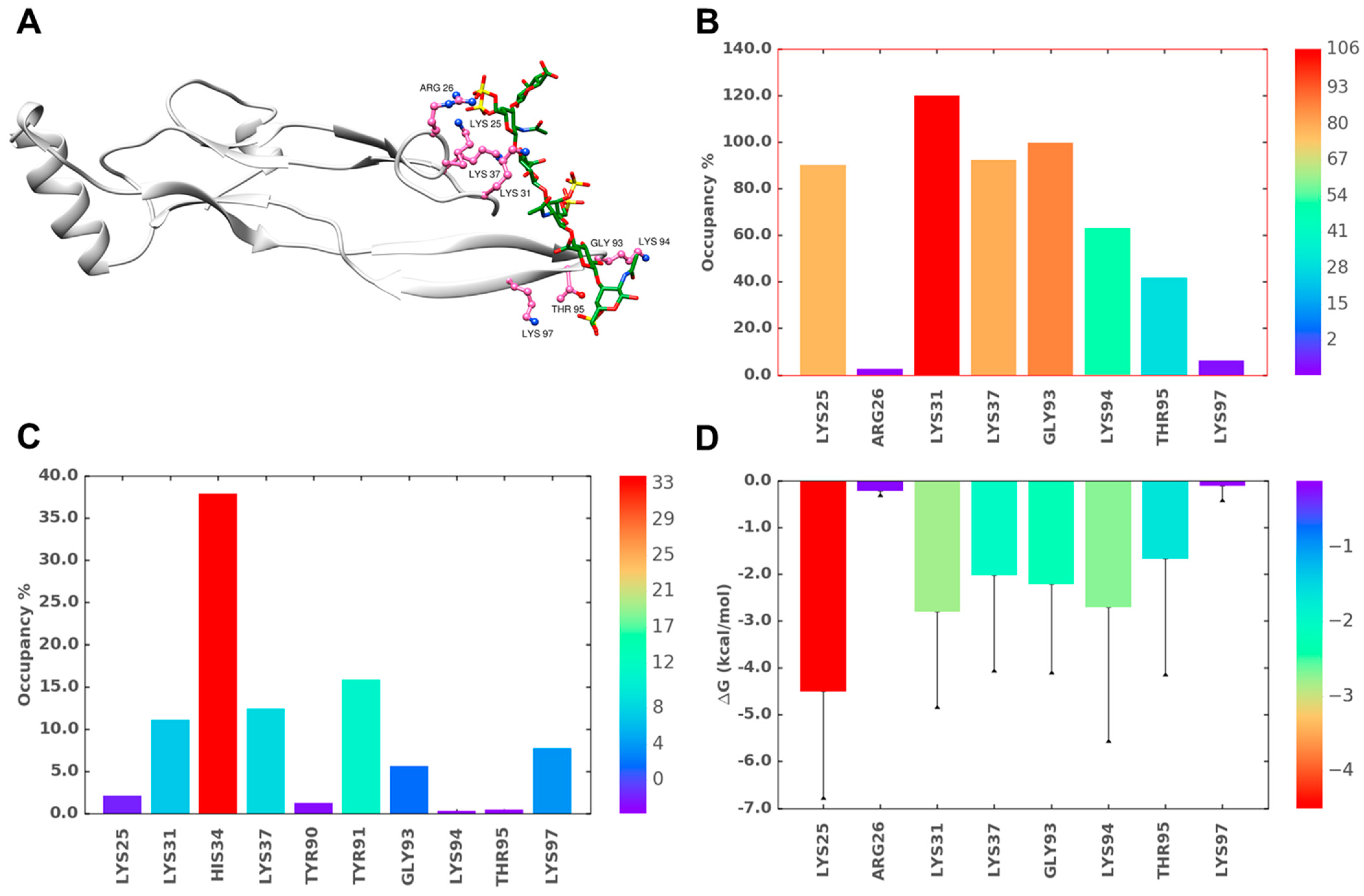
2.6. A Combination of Direct and Water-Mediated Interactions Stabilize CS06 Binding in BS1
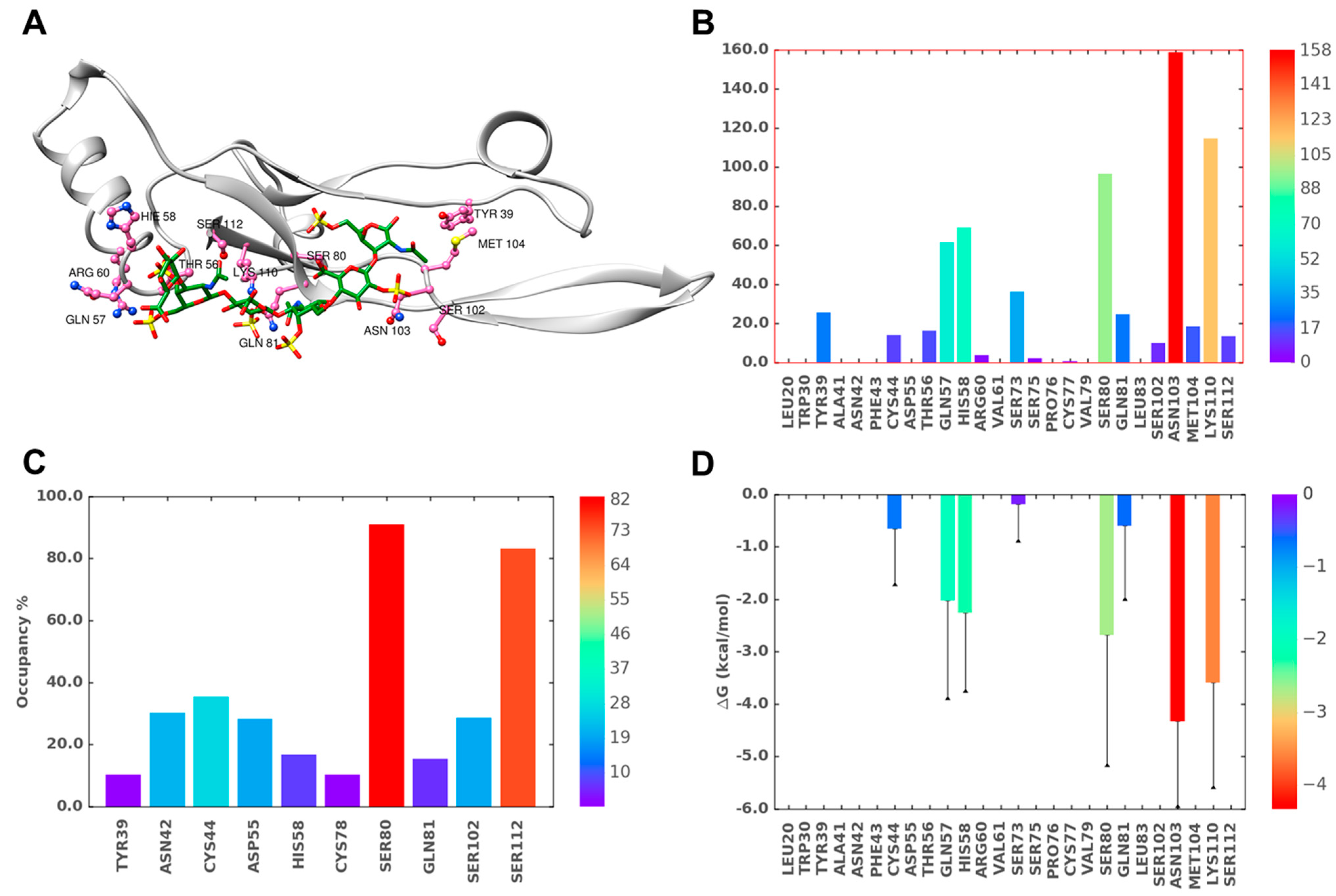
2.7. Role of Direct and Water-Mediated Interactions for BS2
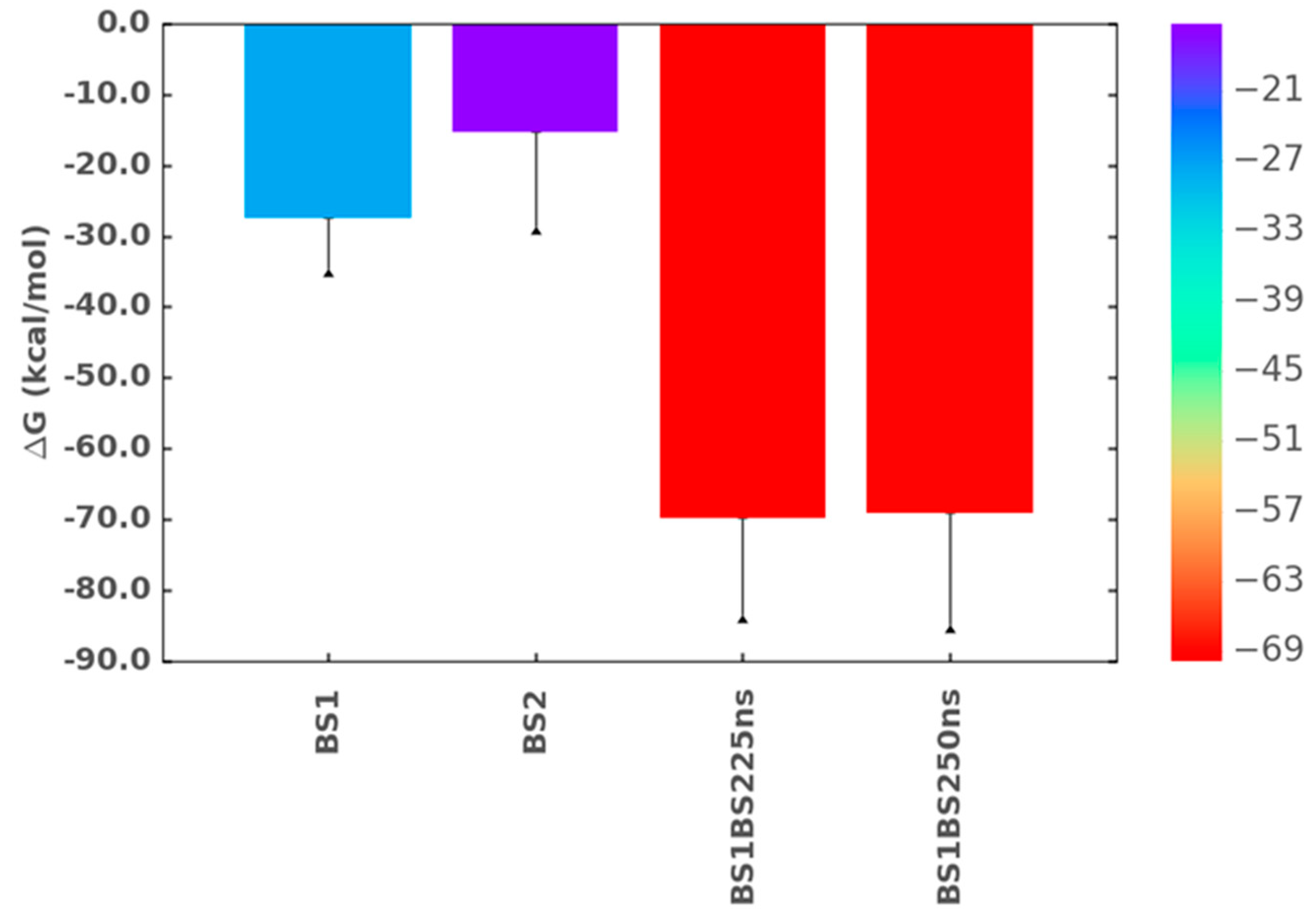
2.8. Simultaneous Engagement of BS1 and BS2 Results in Higher Stability of TGF-β2–CS06 Complex
2.9. Origin of Higher Stability for Dually Occupied TGF-β2 by CS06 Chains
3. Materials and Methods
3.1. Software
3.2. Generation of Library of Chondroitin Sulfate Sequences
3.3. Preparation of TGF-β2 Structure for Docking
3.4. Docking of Library of CS Sequences
3.5. Initial Preparations for Molecular Dynamics (MD)
3.6. MD Simulations
3.7. Analysis of MD Simulations
4. Significance
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Itatani, Y.; Kawada, K.; Sakai, Y. Transforming growth factor-β signaling pathway in colorectal cancer and its tumor microenvironment. Int. J. Mol. Sci. 2019, 20, 5822. [Google Scholar] [CrossRef] [Green Version]
- Birch, J.L.; Coull, B.J.; Spender, L.C.; Watt, C.; Willison, A.; Syed, N.; Chalmers, A.J.; Hossain-Ibrahim, M.K.; Inman, G.J.; Strait, A.A.; et al. Multifaceted transforming growth factor-beta (tgfβ) signalling in glioblastoma the role of transforming growth factor-beta in immune suppression and chronic inflammation of squamous cell carcinomas. Cell. Signal. 2020, 72, 109638–109649. [Google Scholar] [CrossRef] [PubMed]
- Strait, A.A.; Wang, X.J. The role of transforming growth factor-beta in immune suppression and chronic inflammation of squamous cell carcinomas. Mol. Carcinog. 2020, 59, 745–753. [Google Scholar] [CrossRef]
- Galbiati, M.; Crippa, V.; Rusmini, P.; Cristofani, R.; Messi, E.; Piccolella, M.; Tedesco, B.; Ferrari, V.; Casarotto, E.; Chierichetti, M.; et al. Multiple roles of transforming growth factor beta in amyotrophic lateral sclerosis. Int. J. Mol. Sci. 2020, 21, 4291. [Google Scholar] [CrossRef]
- Ismaeel, A.; Kim, J.S.; Kirk, J.S.; Smith, R.S.; Bohannon, W.T.; Koutakis, P. Role of transforming growth factor-β in skeletal muscle fibrosis: A review. Int. J. Mol. Sci. 2019, 20, 2446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Di, C.; Zhang, X.; Wang, J.; Wang, F.; Yan, J.F.; Xu, C.; Zhang, J.; Zhang, Q.; Li, H.; et al. Transforming growth factor β signaling pathway: A promising therapeutic target for cancer. J. Cell. Physiol. 2020, 235, 1903–1914. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Ding, Z.Y.; Li, S.; Liu, S.; Xiao, C.; Li, Z.; Zhang, B.X.; Chen, X.P.; Yang, X. Targeting transforming growth factor-β signaling for enhanced cancer chemotherapy. Theranostics 2021, 11, 1345–1363. [Google Scholar] [CrossRef]
- Patil, A.S.; Sable, R.B.; Kothari, R.M. An update on transforming growth factor-β (TGFβ): Sources, types, functions and clinical applicability for cartilage/bone healing. J. Cell. Physiol. 2011, 226, 3094–3103. [Google Scholar] [CrossRef]
- Daopin, S.; Li, M.; Davies, D.R. Crystal structure of TGFbeta 2 refined at 1.8 a resolution. Proteins 1993, 17, 176–192. [Google Scholar] [CrossRef]
- Schlunegger, M.P.; Grütter, M.G. An unusual feature revealed by the crystal structure at 2.2 a resolution of human transforming growth factor-beta 2. Nature 1992, 358, 430–434. [Google Scholar] [CrossRef]
- Archer, S.J.; Bax, A.; Roberts, A.B.; Sporn, M.B.; Ogawa, Y.; Piez, K.A.; Weatherbee, J.A.; Tsang, M.L.; Lucas, R.; Zheng, B.L.; et al. Transforming growth factor beta 1: Secondary structure as determined by heteronuclear magnetic resonance spectroscopy. Biochemistry 1993, 32, 1164–1171. [Google Scholar] [CrossRef] [PubMed]
- Mittl, P.R.; Priestle, J.P.; Cox, D.A.; McMaster, G.; Cerletti, N.; Grütter, M.G. The crystal structure of TGFbeta 3 and comparison to TGFbeta 2: Implications for receptor binding. Protein Sci. 1996, 5, 1261–1271. [Google Scholar] [CrossRef]
- Daopin, S.; Davies, D.R.; Schlunegger, M.P.; Grütter, M.G. Comparison of two crystal structures of TGFbeta2: The accuracy of refined protein structures. Acta. Crystallogr. D Biol. Crystallogr. 1994, 50, 85–92. [Google Scholar] [CrossRef]
- Hinck, A.P.; Archer, S.J.; Qian, S.W.; Roberts, A.B.; Sporn, M.B.; Weatherbee, J.A.; Tsang, M.L.; Lucas, R.; Zhang, B.L.; Wenker, J.; et al. Transforming growth factor beta 1: Three-dimensional structure in solution and comparison with the X-ray structure of transforming growth factor beta 2. Biochemistry 1996, 35, 8517–8534. [Google Scholar] [CrossRef]
- Daopin, S.; Piez, K.A.; Ogawa, Y.; Davies, D.R. Crystal structure of transforming growth factor-beta 2: An unusual fold for the superfamily. Science 1992, 257, 369–373. [Google Scholar] [CrossRef] [Green Version]
- Rider, C.C. Heparin/heparan sulphate binding in the TGFbeta cytokine superfamily. Biochem. Soc. Trans. 2006, 34, 458–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rider, C.C.; Mulloy, B. Heparin, heparan sulphate and the TGFβ cytokine superfamily. Molecules 2017, 22, 713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hintze, V.; Miron, A.; Moeller, S.; Schnabelrauch, M.; Wiesmann, H.P.; Worch, H.; Scharnweber, D. Sulfated hyaluronan and chondroitin sulfate derivatives interact differently with human transforming growth factor-β1 (TGFβ1). Acta. Biomater. 2012, 8, 2144–2152. [Google Scholar] [CrossRef]
- Koehler, L.; Samsonov, S.; Rother, S.; Vogel, S.; Köhling, S.; Moeller, S.; Schnabelrauch, M.; Rademann, J.; Hempel, U.; Pisabarro, M.T.; et al. Sulfated hyaluronan derivatives modulate TGFβ1: Receptor complex formation: Possible consequences for TGFβ1 signaling. Sci. Rep. 2017, 7, 1210–1220. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Zheng, L.; Cheng, S.; Peng, Y.; Fu, L.; Zhang, X.; Linhardt, R.J. Comparison of the interactions of different growth factors and glycosaminoglycans. Molecules 2019, 24, 3360. [Google Scholar] [CrossRef] [Green Version]
- Namachivayam, K.; Coffing, H.P.; Sankaranarayanan, N.V.; Jin, Y.; MohanKumar, K.; Frost, B.L.; Blanco, C.L.; Patel, A.L.; Meier, P.P.; Garzon, S.A.; et al. Transforming growth factor-β2 is sequestered in preterm human milk by chondroitin sulfate proteoglycans. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 309, G171–G180. [Google Scholar] [CrossRef] [Green Version]
- Lyon, M.; Rushton, G.; Gallagher, J.T. The interaction of the transforming growth factor-betas with heparin/heparan sulfate is isoform-specific. J. Biol. Chem. 1997, 272, 18000–18006. [Google Scholar] [CrossRef] [Green Version]
- Raghuraman, A.; Mosier, P.D.; Desai, U.R. Finding a needle in a haystack: Development of a combinatorial virtual screening approach for identifying high specificity heparin/heparan sulfate sequence(s). J. Med. Chem. 2006, 49, 3553–3562. [Google Scholar] [CrossRef] [Green Version]
- Raghuraman, A.; Mosier, P.D.; Desai, U.R. Understanding dermatan sulfate-heparin cofactor ii interaction through virtual library screening. ACS Med. Chem. Lett. 2010, 1, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Sankaranarayanan, N.V.; Bi, Y.; Kuberan, B.; Desai, U.R. Combinatorial virtual library screening analysis of antithrombin binding oligosaccharide motif generation by heparan sulfate 3-o-sulfotransferase 1. Comput. Struct. Biotechnol. J. 2020, 18, 933–941. [Google Scholar] [CrossRef] [PubMed]
- Sankaranarayanan, N.V.; Desai, U.R. Toward a robust computational screening strategy for identifying glycosaminoglycan sequences that display high specificity for target proteins. Glycobiology 2014, 24, 1323–1333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sankaranarayanan, N.V.; Nagarajan, B.; Desai, U.R. So you think computational approaches to understanding glycosaminoglycan-protein interactions are too dry and too rigid? Think again! Curr. Opin. Struct. Biol. 2018, 50, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Sankaranarayanan, N.V.; Strebel, T.R.; Boothello, R.S.; Sheerin, K.; Raghuraman, A.; Sallas, F.; Mosier, P.D.; Watermeyer, N.D.; Oscarson, S.; Desai, U.R. A hexasaccharide containing rare 2-o-sulfate-glucuronic acid residues selectively activates heparin cofactor II. Angew. Chem. Int. Ed. 2017, 56, 2312–2317. [Google Scholar] [CrossRef] [Green Version]
- Mulloy, B. Progress in the structural biology of chondroitin sulfate. Adv. Pharmacol. 2006, 53, 49–67. [Google Scholar] [PubMed]
- Cardin, A.D.; Weintraub, H.J. Molecular modeling of protein-glycosaminoglycan interactions. Arteriosclerosis 1989, 9, 21–32. [Google Scholar] [CrossRef]
- Kummarapurugu, A.B.; Afosah, D.K.; Sankaranarayanan, N.V.; Navaz Gangji, R.; Zheng, S.; Kennedy, T.; Rubin, B.K.; Voynow, J.A.; Desai, U.R. Molecular principles for heparin oligosaccharide-based inhibition of neutrophil elastase in cystic fibrosis. J. Biol. Chem. 2018, 293, 12480–12490. [Google Scholar] [CrossRef] [Green Version]
- Cael, J.J.; Winter, W.T.; Arnott, S. Calcium chondroitin 4-sulfate: Molecular conformation and organization of polysaccharide chains in a proteoglycan. J. Mol. Biol. 1978, 125, 21–42. [Google Scholar] [CrossRef]
- Sepuru, K.M.; Nagarajan, B.; Desai, U.R.; Rajarathnam, K. Structural basis, stoichiometry, and thermodynamics of binding of the chemokines kc and mip2 to the glycosaminoglycan heparin. J. Biol. Chem. 2018, 293, 17817–17828. [Google Scholar] [CrossRef] [Green Version]
- Winter, W.T.; Arnott, S.; Isaac, D.H.; Atkins, E.D. Chondroitin 4-sulfate: The structure of a sulfated glycosaminoglycan. J. Mol. Biol. 1978, 125, 1–19. [Google Scholar] [CrossRef]
- Sepuru, K.M.; Nagarajan, B.; Desai, U.R.; Rajarathnam, K. Molecular basis of chemokine cxcl5-glycosaminoglycan interactions. J. Biol. Chem. 2016, 291, 20539–20550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monneau, Y.R.; Luo, L.; Sankaranarayanan, N.V.; Nagarajan, B.; Vivès, R.R.; Baleux, F.; Desai, U.R.; Arenzana-Seidedos, F.; Lortat-Jacob, H. Solution structure of cxcl13 and heparan sulfate binding show that gag binding site and cellular signalling rely on distinct domains. Open. Biol. 2017, 7, 170133–170146. [Google Scholar] [CrossRef] [Green Version]
- Roe, D.R.; Cheatham, T.E., III. Ptraj and cpptraj: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Gandhi, N.S.; Mancera, R.L. Free energy calculations of glycosaminoglycan-protein interactions. Glycobiology 2009, 19, 1103–1115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jana, M.; Bandyopadhyay, S. Conformational flexibility of a protein-carbohydrate complex and the structure and ordering of surrounding water. Phys. Chem. Chem. Phys. 2012, 14, 6628–6638. [Google Scholar] [CrossRef]
- Samsonov, S.A.; Teyra, J.; Pisabarro, M.T. Docking glycosaminoglycans to proteins: Analysis of solvent inclusion. J. Comput. Aided Mol. Des. 2011, 25, 477–489. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.Y.; Cheng, C.S.; Lee, T.R.; Chang, W.S.; Lyu, P.C. A clamp-like orientation of basic residues set in a parallelogram is essential for heparin binding. FEBS Lett. 2016, 590, 3089–3097. [Google Scholar] [CrossRef] [PubMed]
- Johnson, Z.; Power, C.A.; Weiss, C.; Rintelen, F.; Ji, H.; Ruckle, T.; Camps, M.; Wells, T.N.; Schwarz, M.K.; Proudfoot, A.E.; et al. Chemokine inhibition—Why, when, where, which and how? Biochem. Soc. Trans. 2004, 32, 366–377. [Google Scholar] [CrossRef] [PubMed]
- Torrent, M.; Nogués, M.V.; Andreu, D.; Boix, E. The “cpc clip motif”: A conserved structural signature for heparin-binding proteins. PLoS ONE 2012, 7, e42692–e42699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkar, A.; Desai, U.R. A simple method for discovering druggable, specific glycosaminoglycan-protein systems. Elucidation of key principles from heparin/heparan sulfate-binding proteins. PLoS ONE 2015, 10, e0141127–e0141144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, N.J.; Sharon, C.; Baranwal, S.; Boothello, R.S.; Desai, U.R.; Patel, B.B. Heparan sulfate hexasaccharide selectively inhibits cancer stem cells self-renewal by activating p38 MAP kinase. Oncotarget 2016, 7, 84608–84622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karamanos, N.K.; Syrokou, A.; Vanky, P.; Nurminen, M.; Hjerpe, A. Determination of 24 variously sulfated galactosaminoglycan- and hyaluronan-derived disaccharides by high-performance liquid chromatography. Anal. Biochem. 1994, 221, 189–199. [Google Scholar] [CrossRef]
- Kirschner, K.N.; Yongye, A.B.; Tschampel, S.M.; González-Outeiriño, J.; Daniels, C.R.; Foley, B.L.; Woods, R.J. Glycam06: A generalizable biomolecular force field. Carbohydrates. J. Comput. Chem. 2008, 29, 622–655. [Google Scholar] [CrossRef] [Green Version]
- Cremer, D.; Pople, J.A. General definition of ring puckering coordinates. J. Am. Chem. Soc. 1975, 97, 1354–1358. [Google Scholar] [CrossRef]
- Miller, B.R., 3rd; McGee, T.D., Jr.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory. Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]
- Forster, M.J.; Mulloy, B. Molecular dynamics study of iduronate ring conformation. Biopolymers 1993, 33, 575–588. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Binding Site | Disaccharide Sequence | RMSD (Å) | No of Sulfates | GOLDScore |
|---|---|---|---|---|
| 1 | uAB–VbC4B | 2.5 | 1 | 53.1 |
| VbC6B–Zb2B | 2.5 | 2 | 56.3 | |
| ZbB–VbCB | 2.5 | 0 | 44.2 | |
| 2 | uAB–VbCB | 2.3 | 1 | 39.2 |
| Binding Site | Tetrasaccharide Sequence | RMSD (Å) | No of Sulfate | GOLDScore |
|---|---|---|---|---|
| 1 | Zb2B–VbC4B–Zb2B–VbC6B | 1.5 | 4 | 61.7 |
| uAB–VbC4B–Zb2B–VbCB | 1.9 | 2 | 50.3 | |
| ZbB–VbC4B–ZbB–VbCB | 2.3 | 1 | 45.1 | |
| 2 | uAB–VbC6B–Zb2B–VbC46B | 2.5 | 4 | 50.6 |
| VbC6B–ZbB–VbC46B–ZbB | 1.8 | 3 | 53.9 | |
| VbC6B–ZbB–VbC4B–ZbB | 1.3 | 2 | 52.5 | |
| VbCB–Zb2B–VbC6B–Zb2B | 2.0 | 3 | 53.8 | |
| VbCB–ZbB–VbC4B–ZbB | 1.0 | 1 | 48.1 | |
| VbCB–ZbB–VbCB–ZbB | 1.6 | 0 | 46.5 | |
| ZbB–VbC4B–ZbB–VbC4B | 1.7 | 2 | 57.8 |
| Binding Site | Hexasaccharide Sequence | RMSD (Å) | No of Sulfates | GOLDScore |
|---|---|---|---|---|
| 1 | ZbB–VbC4B–Zb2B–VbC4B–ZbB–VbC6B | 2.5 | 4 | 87.6 |
| 2 | Zb2B–VbC6B–Zb2B–VbC4B–ZbB–VbC46B | 1.8 | 6 | 85.8 |
| Zb2B–VbC6B–ZbB–VbC46B–Zb2B–VbC6B | 2.0 | 6 | 79.0 | |
| ZbB–VbC46B–Zb2B–VbC46B–Zb2B–VbC4B | 2.2 | 7 | 71.8 | |
| ZbB–VbC46B–Zb2B–VbC4B–Zb2B–VbC46B | 1.6 | 7 | 82.7 | |
| ZbB–VbC46B–Zb2B–VbC4B–Zb2B–VbC6B | 1.6 | 6 | 88.4 | |
| ZbB–VbC46B–Zb2B–VbCB–Zb2B–VbC46B | 2.2 | 6 | 86.7 | |
| ZbB–VbC46B–Zb2B–VbCB–ZbB–VbC6B | 2.1 | 4 | 71.7 | |
| ZbB–VbC46B–ZbB–VbC46B–ZbB–VbC4B | 1.8 | 5 | 70.3 |
| Disaccharide Building Blocks | φ | ψ | Atoms |
|---|---|---|---|
| GlcAp(1→3)GalNAcp | −81 | −129 | O5–C1–O3′–C3′ (φ) C1–O3′–C3′–C2′ (ψ) |
| GalNAcp(1→4)GlcAp | −69 | 131 | O5–C1–O4′–C4′ (φ) C1–O4′–C4′–C3′ (ψ) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sankaranarayanan, N.V.; Nagarajan, B.; Desai, U.R. Combinatorial Virtual Library Screening Study of Transforming Growth Factor-β2–Chondroitin Sulfate System. Int. J. Mol. Sci. 2021, 22, 7542. https://doi.org/10.3390/ijms22147542
Sankaranarayanan NV, Nagarajan B, Desai UR. Combinatorial Virtual Library Screening Study of Transforming Growth Factor-β2–Chondroitin Sulfate System. International Journal of Molecular Sciences. 2021; 22(14):7542. https://doi.org/10.3390/ijms22147542
Chicago/Turabian StyleSankaranarayanan, Nehru Viji, Balaji Nagarajan, and Umesh R. Desai. 2021. "Combinatorial Virtual Library Screening Study of Transforming Growth Factor-β2–Chondroitin Sulfate System" International Journal of Molecular Sciences 22, no. 14: 7542. https://doi.org/10.3390/ijms22147542