
Comparison of Gut Bacterial Communities of Grapholita molesta (Lepidoptera: Tortricidae) Reared on Different Host Plants


Abstract
:1. Introduction
2. Results
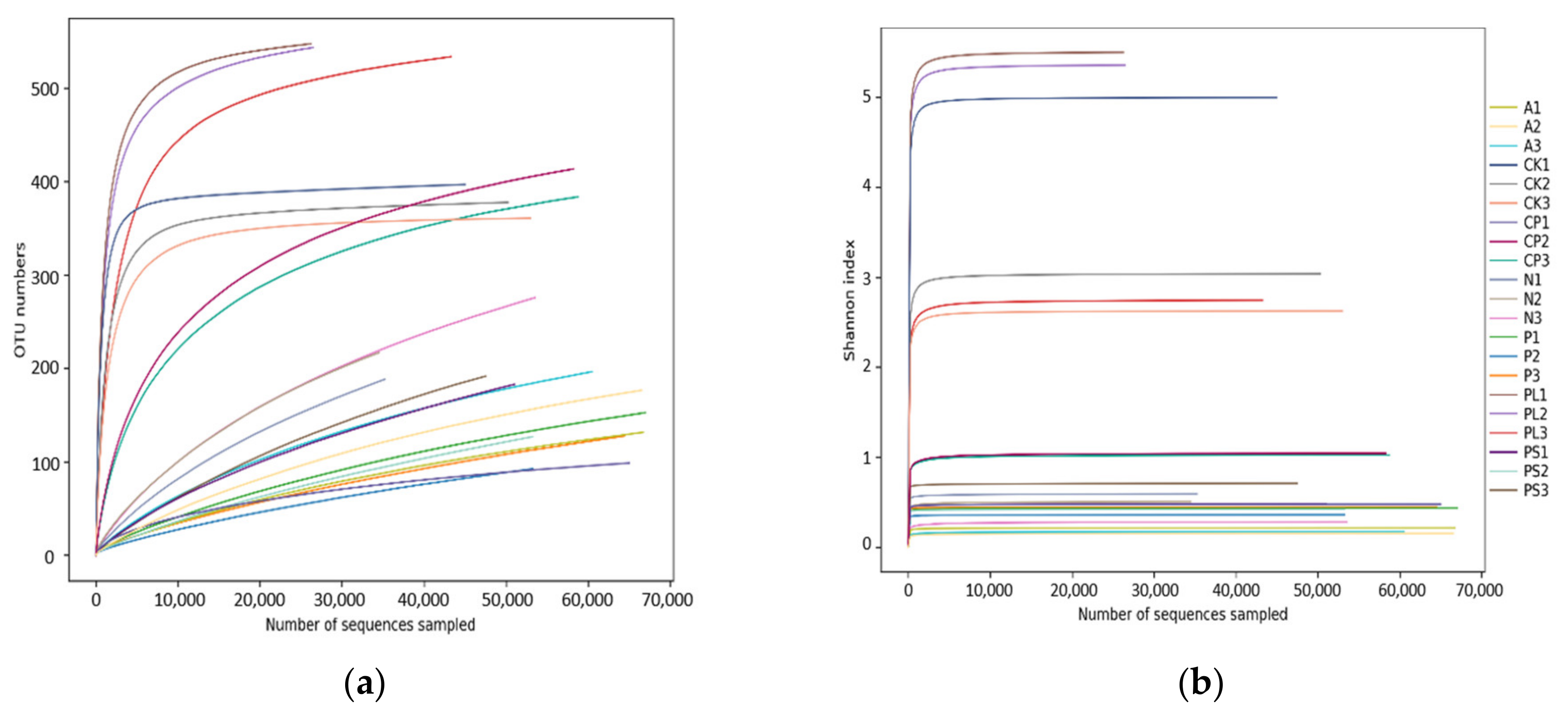
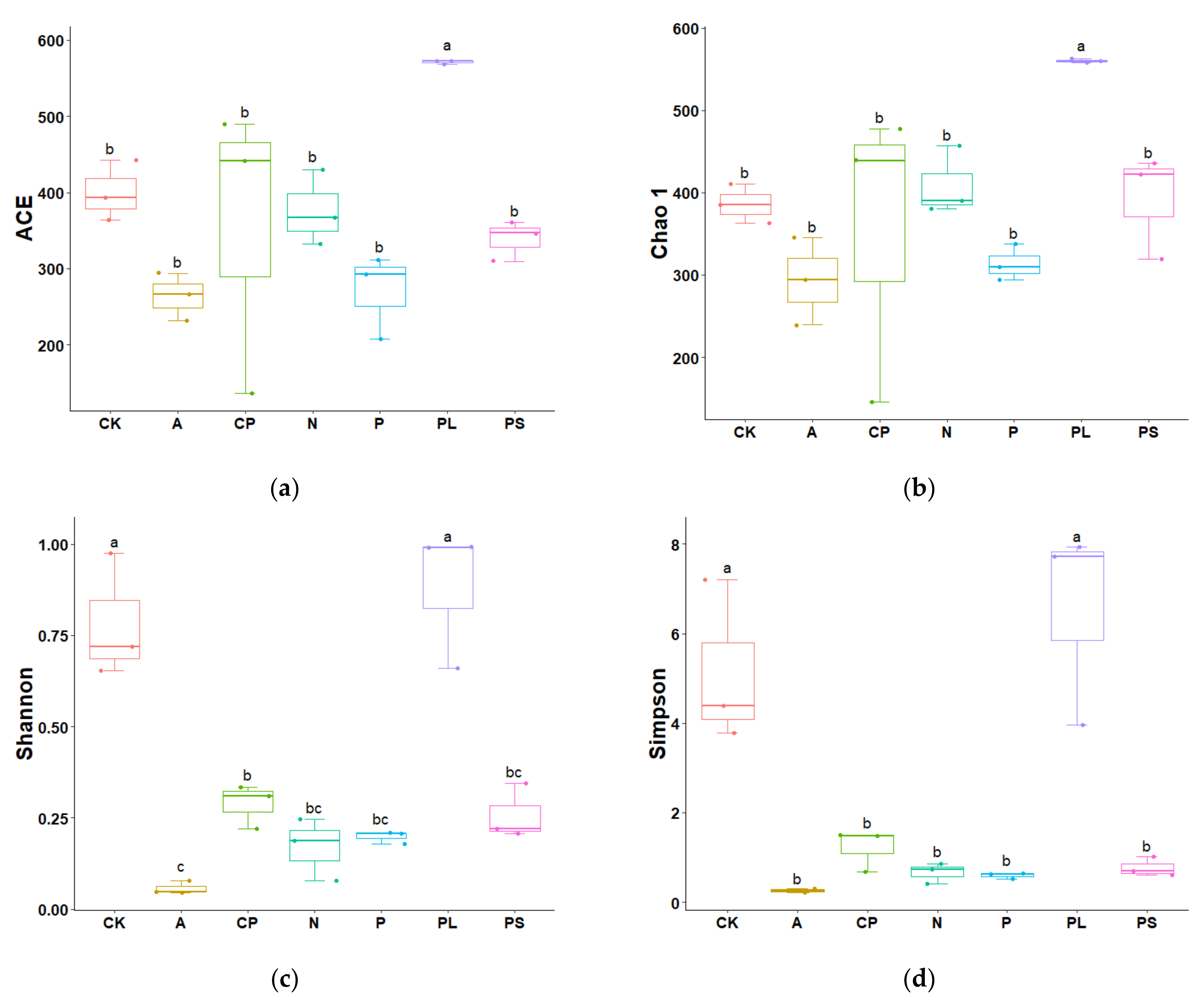
2.1. Analysis of 16S rDNA Sequencing Results
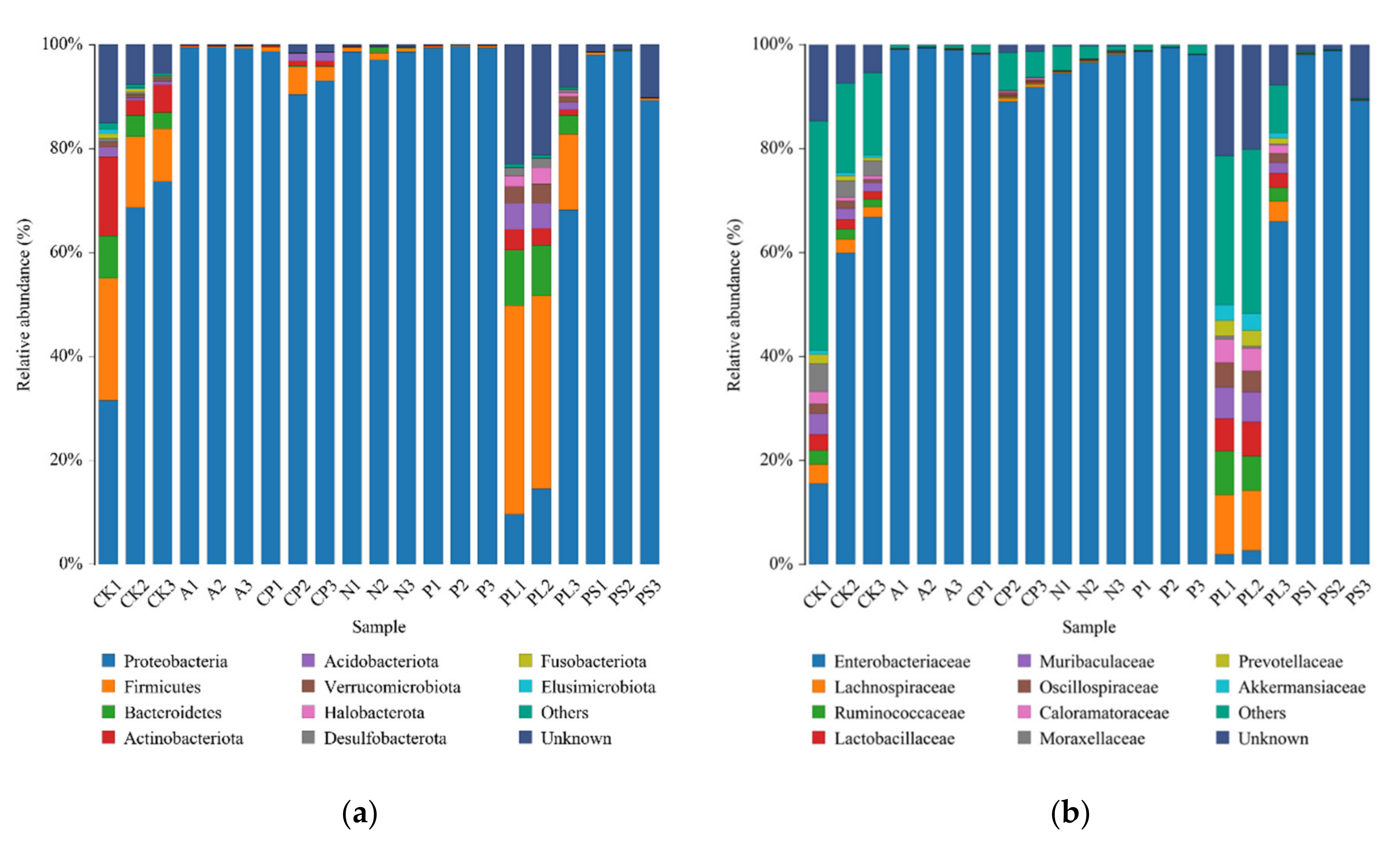
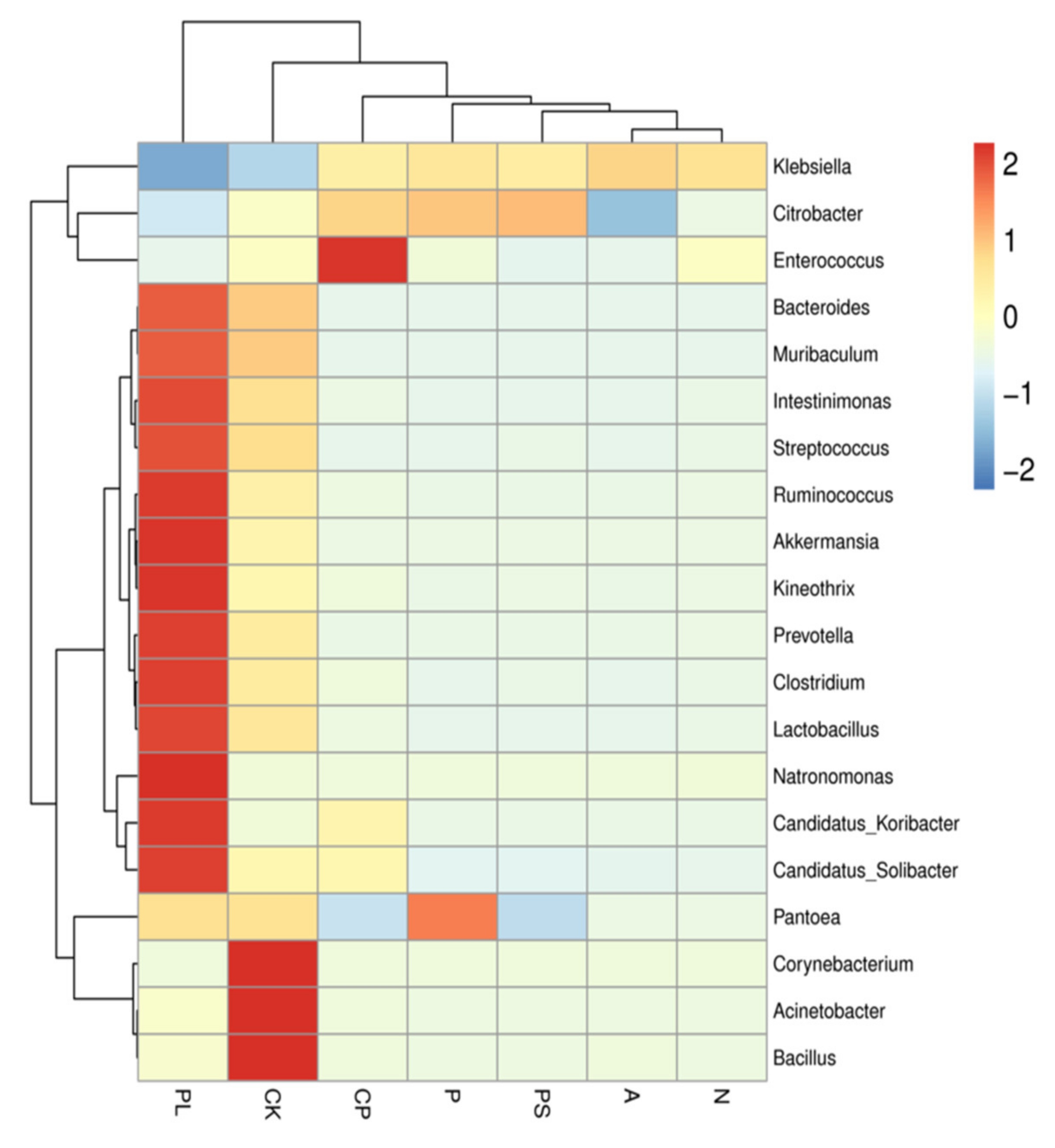
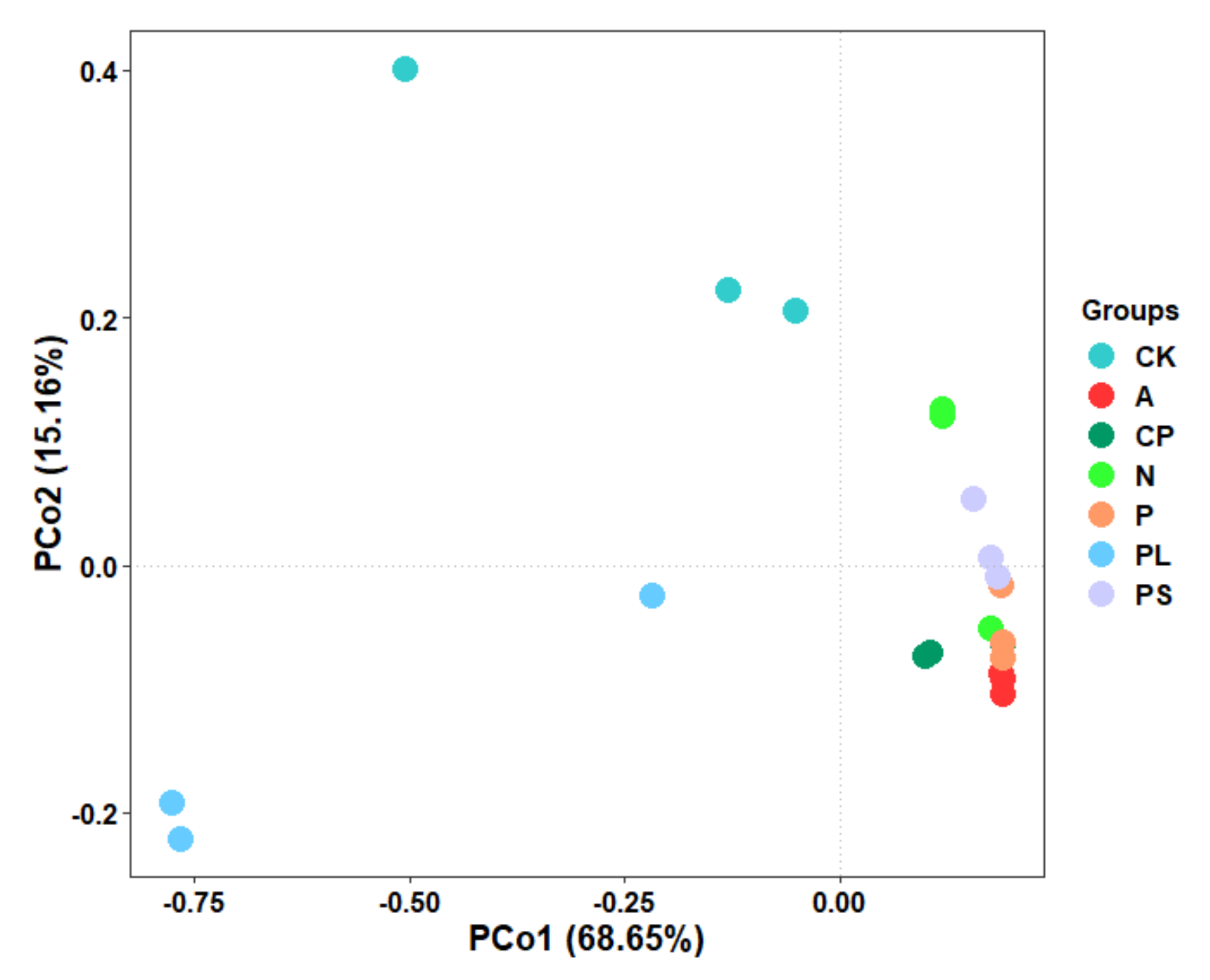
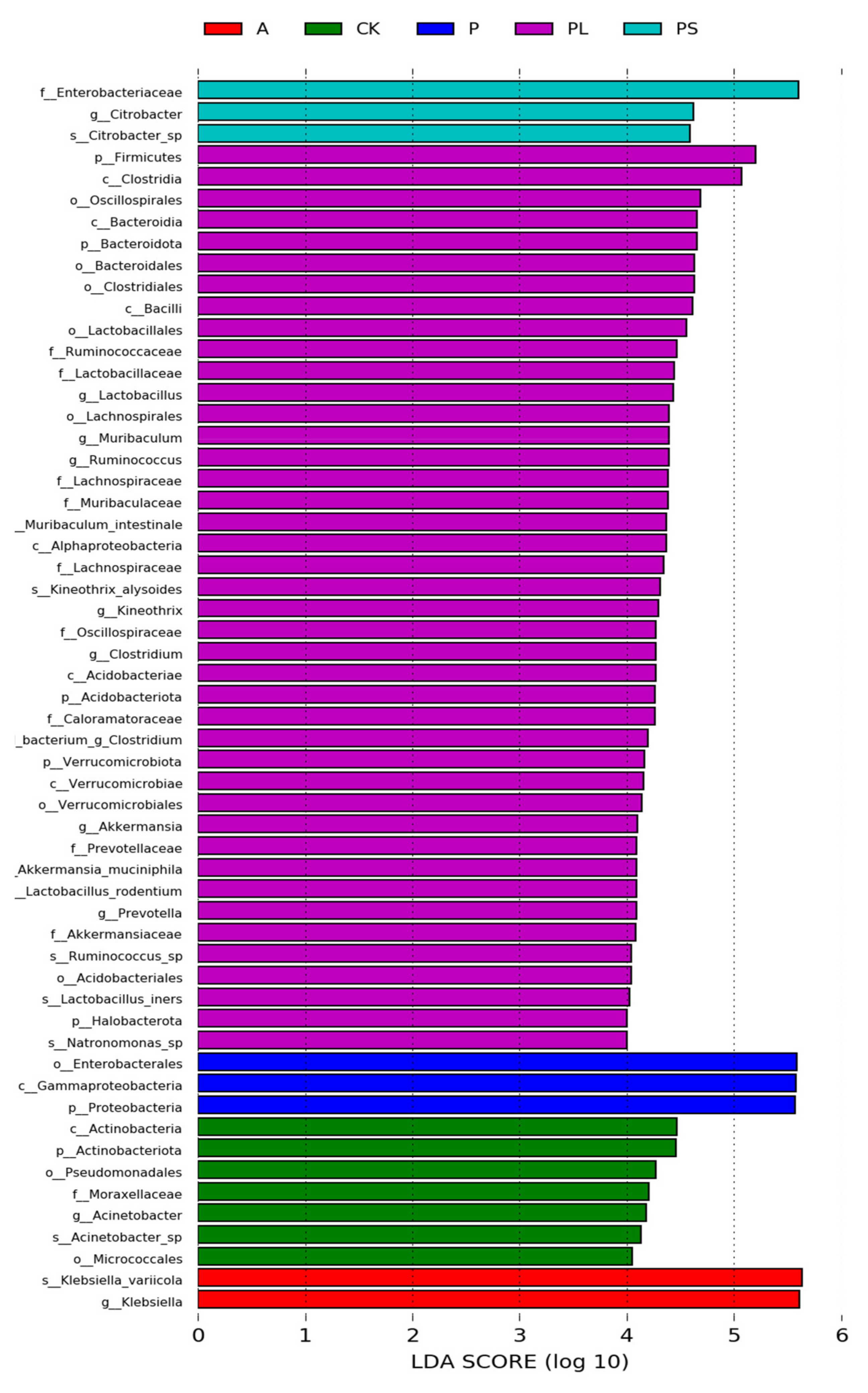
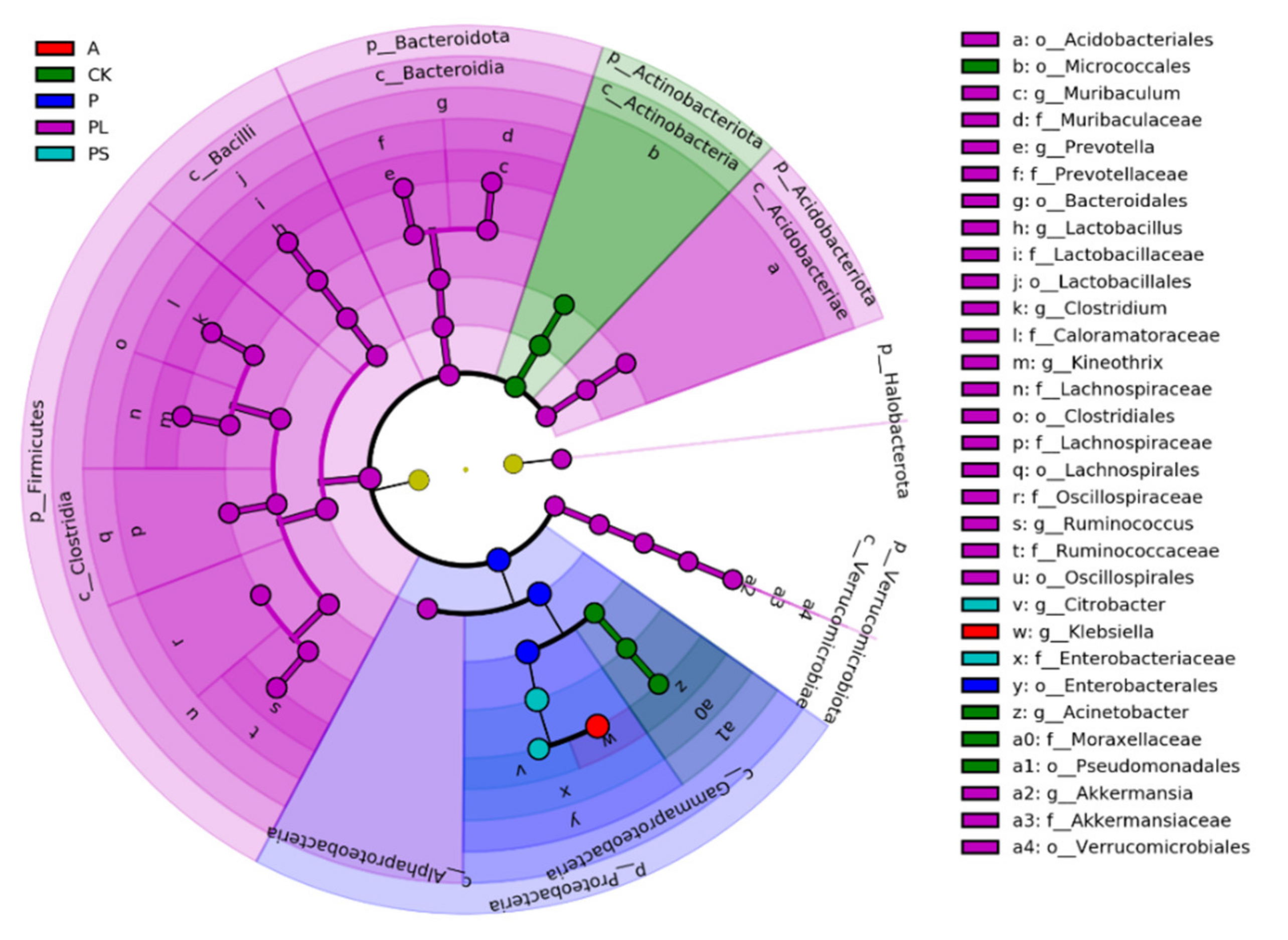
2.2. Comparison of the Gut Microbiota between Different Diets
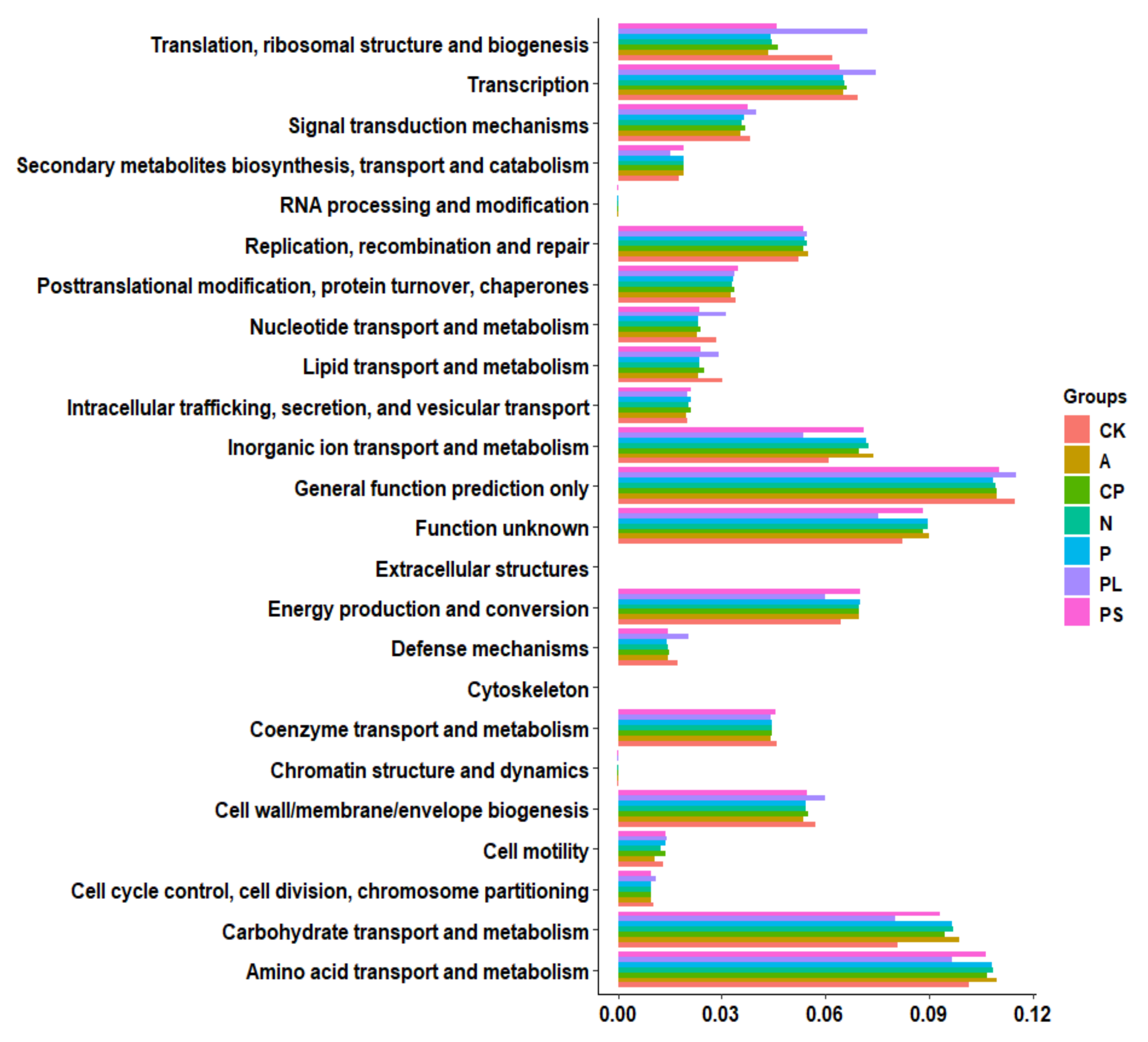
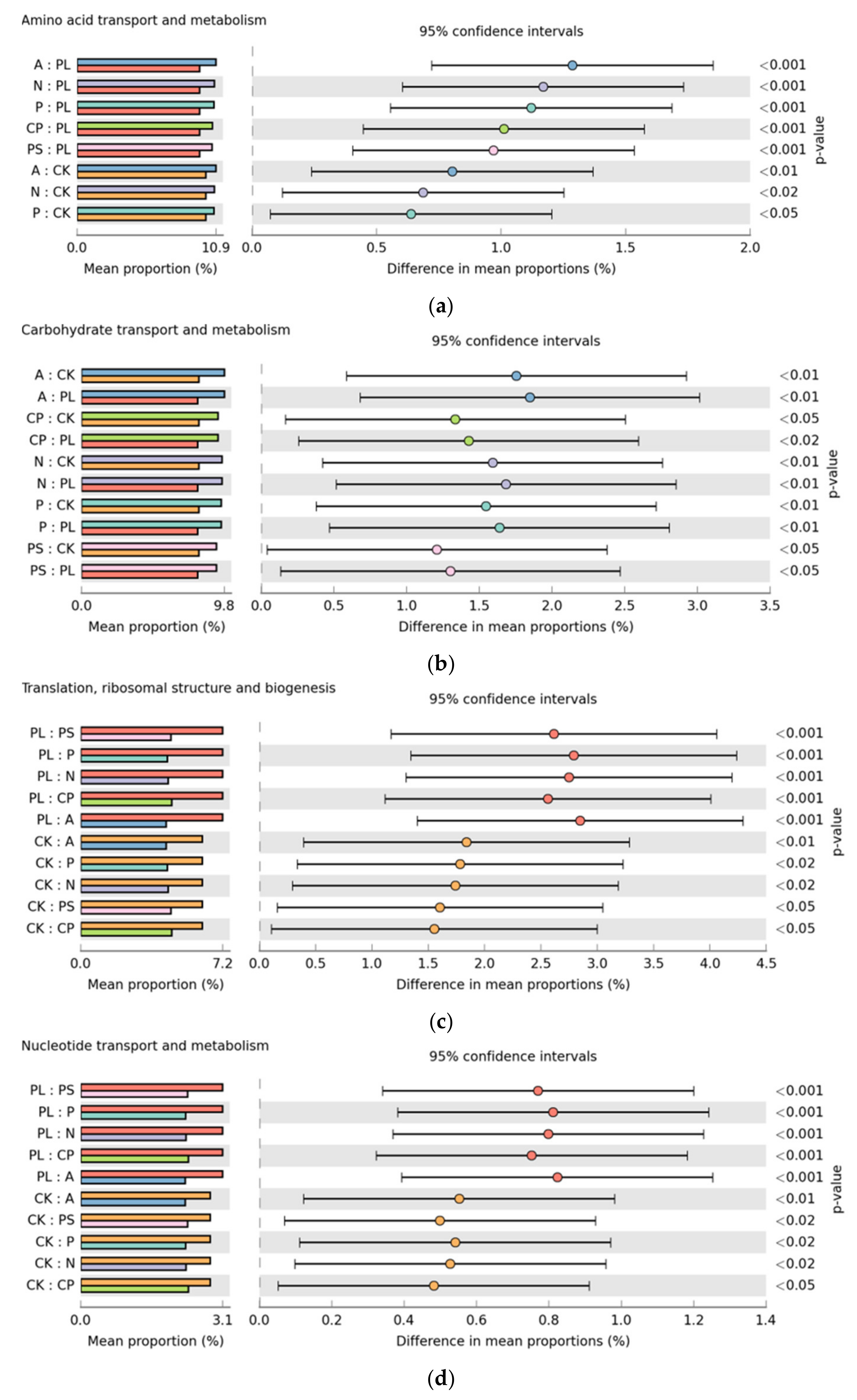
2.3. Functional Prediction of Gut Microbiota
3. Discussion
4. Materials and Methods
4.1. Insect Rearing and Sample Processing
4.2. DNA Extraction
4.3. PCR Amplification and High-Throughput Sequencing
4.4. Statistical and Bioinformatics Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shin, S.C.; Kim, S.H.; You, H.; Kim, B.; Kim, A.C.; Lee, K.A.; Yoon, J.H.; Ryu, J.H.; Lee, W.J. Drosophila microbiome modulates host developmental and metabolic homeostasis via insulin signaling. Science 2011, 334, 670–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cazemier, A.E.; Hackstein, J.H.P.; Op Den Camp, H.J.M.; Rosenberg, J.; Van Der Drift, C. Bacteria in the intestinal tract of different species of arthropods. Microb. Ecol. 1997, 33, 189–197. [Google Scholar] [CrossRef]
- Ceja-Navarro, J.A.; Vega, F.E.; Karaoz, U.; Hao, Z.; Jenkins, S.; Lim, H.C.; Kosina, P.; Infante, F.; Northen, T.R.; Brodie, E.L. Gut microbiota mediate caffeine detoxification in the primary insect pest of coffee. Nat. Commun. 2015, 6, 7618. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Shu, J.; Xue, H.; Zhang, W.; Zhang, Y.; Liu, Y.; Fang, L.; Wang, Y.; Wang, H. The gut microbiota in Camellia Weevils are influenced by plant secondary metabolites and contribute to saponin degradation. Msystems 2020, 5, e00692-19. [Google Scholar] [CrossRef] [Green Version]
- Abdelgaffar, H.; Tague, E.D.; Gonzalez, H.F.C.; Campagna, S.R.; Jurat-Fuentes, J.L. Midgut metabolomic profiling of fall armyworm (Spodoptera frugiperda) with field-evolved resistance to Cry1F corn. Insect Biochem. Mol. Biol. 2019, 106, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Bai, S.; Yao, Z.C.; Raza, M.F.; Cai, Z.H.; Zhang, H.Y. Regulatory mechanisms of microbial homeostasis in insect gut. Insect Sci. 2020, 28, 286–301. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.Y.; Saqib, H.S.A.; Chen, J.H.; Ruan, Q.Q.; Vasseur, L.; He, W.Y.; You, M.S. Differential profiles of gut microbiota and metabolites associated with host shift of Plutella xylostella. Int. J. Mol. Sci. 2020, 21, 6283. [Google Scholar] [CrossRef]
- Lauzon, C.R.; Sjogren, R.E.; Prokopy, R.J. Enzymatic capabilities of bacteria associated with apple maggot flies: A postulated role in attraction. J. Chem. Ecol. 2000, 26, 953–967. [Google Scholar] [CrossRef]
- Shao, Y.Q.; Chen, B.S.; Sun, C.; Ishida, K.; Hertweck, C.; Boland, W. Symbiont-derived antimicrobials contribute to the control of the Lepidopteran gut microbiota. Cell Chem. Biol. 2017, 24, 66–75. [Google Scholar] [CrossRef] [Green Version]
- Johnston, P.R.; Rolff, J. Host and symbiont jointly control gut microbiota during complete metamorphosis. PLoS Pathog. 2015, 11, e1005246. [Google Scholar] [CrossRef] [Green Version]
- Su, W.Z.; Liu, J.L.; Bai, P.; Ma, B.C.; Liu, W. Pathogenic fungi-induced susceptibility is mitigated by mutual Lactobacillus plantarum in the Drosophila melanogaster model. BMC Microbiol. 2019, 19, 302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, Z.H.; Yao, Z.C.; Li, Y.S.; Xi, Z.Y.; Bourtzis, K.; Zhao, Z.; Bai, S.; Zhang, H.Y. Intestinal probiotics restore the ecological fitness decline of Bactrocera dorsalis by irradiation. Evol. Appl. 2018, 11, 1946–1963. [Google Scholar] [CrossRef]
- Yong, H.S.; Song, S.L.; Chua, K.O.; Lim, P.E. High diversity of bacterial communities in developmental stages of Bactrocera carambolae (Insecta: Tephritidae) revealed by illumina miSeq sequencing of 16S rRNA gene. Curr. Microbiol. 2017, 74, 1076–1082. [Google Scholar] [CrossRef] [PubMed]
- Kyritsis, G.A.; Augustinos, A.A.; Caceres, C.; Bourtzis, K. Medfly Gut Microbiota and enhancement of the sterile insect technique: Similarities and differences of Klebsiella oxytoca and Enterobacter sp. AA26 probiotics during the larval and adult stages of the VIENNA 8(D53+) genetic sexing strain. Front. Microbiol. 2017, 8, 2064. [Google Scholar] [CrossRef]
- Cirimotich, C.M.; Dong, Y.M.; Clayton, A.M.; Sandiford, S.L.; Souza-Neto, J.A.; Mulenga, M.; Dimopoulos, G. Natural microbe-mediated refractoriness to Plasmodium infection in Anopheles gambiae. Science 2011, 332, 855–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cirimotich, C.M.; Ramirez, J.L.; Dimopoulos, G. Native microbiota shape insect vector competence for human pathogens. Cell Host Microbe. 2011, 10, 307–310. [Google Scholar] [CrossRef] [Green Version]
- Myers, C.T.; Hull, L.A.; Krawczyk, G. Early-season host plant fruit impacts on reproductive parameters of the oriental fruit moth (Lepidoptera: Tortricidae). J. Entomol. Sci. 2006, 41, 65–74. [Google Scholar] [CrossRef]
- Myers, C.T.; Hull, L.A.; Krawczyk, G. Effects of orchard host plants on the oviposition preference of the oriental fruit moth (Lepidoptera: Tortricidae). J. Econ. Entomol. 2006, 99, 1176–1183. [Google Scholar] [CrossRef]
- Neven, L.G.; Kumar, S.; Yee, W.L.; Wakie, T. Current and future potential risk of establishment of Grapholita molesta (Lepidoptera: Tortricidae) in Washington state. Environ. Entomol. 2018, 47, 448–456. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Sun, S.; Yang, X.; Cheng, J.; Wei, H.; Li, Z.; Michaud, J.P.; Liu, X. Variability of gut microbiota across the life cycle of Grapholita molesta (Lepidoptera: Tortricidae). Front. Microbiol. 2020, 11, 1366. [Google Scholar] [CrossRef]
- Chen, L.H.; Tian, K.; Xu, X.L.; Fang, A.S.; Cheng, W.N.; Wang, G.R.; Liu, W.; Wu, J.X. Detecting host-plant volatiles with odorant receptors from Grapholita molesta (Busck) (Lepidoptera: Tortricidae). J. Agric. Food Chem. 2020, 68, 2711–2717. [Google Scholar] [CrossRef]
- Tian, Z.; Li, Y.; Xing, Y.J.; Li, R.C.; Liu, J.Y. Structural insights into two representative conformations of the complex formed by Grapholita molesta (Busck) pheromone binding protein 2 and Z-8-Dodecenyl acetate. J. Agric. Food Chem. 2019, 67, 4425–4434. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.H.; Tian, K.; Wang, G.R.; Xu, X.L.; He, K.H.; Liu, W.; Wu, J.X. The general odorant receptor GmolOR9 from Grapholita molesta (Lepidoptera: Tortricidae) is mainly tuned to eight host-plant volatiles. Insect Sci. 2020, 27, 1233–1243. [Google Scholar] [CrossRef]
- Broderick, N.A.; Raffa, K.F.; Handelsman, J. Midgut bacteria required for Bacillus thuringiensis insecticidal activity. Proc. Natl. Acad. Sci. USA 2006, 103, 15196–15199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broderick, N.A.; Robinson, C.J.; Mcmahon, M.D.; Holt, J.; Handelsman, J.; Raffa, K.F. Contributions of gut bacteria to Bacillus thuringiensis-induced mortality vary across a range of Lepidoptera. BMC Biol. 2009, 7, 11. [Google Scholar] [CrossRef] [Green Version]
- Caccia, S.; Di Lelio, I.; La Storia, A.; Marinelli, A.; Varricchio, P.; Franzetti, E.; Banyuls, N.; Tettamanti, G.; Casartelli, M.; Giordana, B.; et al. Midgut microbiota and host immunocompetence underlie Bacillus thuringiensis killing mechanism. Proc. Natl. Acad. Sci. USA 2016, 113, 9486–9491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubovskiy, I.M.; Grizanova, E.V.; Whitten, M.M.; Mukherjee, K.; Greig, C.; Alikina, T.; Kabilov, M.; Vilcinskas, A.; Glupov, V.V.; Butt, T.M. Immuno-physiological adaptations confer wax moth Galleria mellonella resistance to Bacillus thuringiensis. Virulence 2016, 7, 860–870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takatsuka, J.; Kunimi, Y. Intestinal bacteria affect growth of Bacillus thuringiensis in larvae of the oriental tea tortrix, Homona magnanima diakonoff (Lepidoptera: Tortricidae). J. Invertebr. Pathol. 2000, 76, 222–226. [Google Scholar] [CrossRef]
- Martemyanov, V.V.; Belousova, I.A.; Pavlushin, S.V.; Dubovskiy, I.M.; Ershov, N.I.; Alikina, T.Y.; Kabilov, M.R.; Glupov, V.V. Phenological asynchrony between host plant and gypsy moth reduces insect gut microbiota and susceptibility to Bacillus thuringiensis. Ecol. Evol. 2016, 6, 7298–7310. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.H.; Kim, S.H.; Lee, H.Y.; Bai, J.Y.; Nam, Y.D.; Bae, J.W.; Lee, D.G.; Shin, S.C.; Ha, E.M.; Lee, W.J. Innate immune homeostasis by the homeobox gene caudal and commensal-gut mutualism in Drosophila. Science 2008, 319, 777–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.J.; Shen, Z.J.; Yu, J.M.; Li, Z.; Liu, X.X.; Xu, H.L. Comparison of gut bacterial communities and their associations with host diets in four fruit borers. Pest. Manag. Sci. 2020, 76, 1353–1362. [Google Scholar] [CrossRef]
- Zhu, Y.X.; Song, Z.R.; Song, Y.L.; Zhao, D.S.; Hong, X.Y. The microbiota in spider mite feces potentially reflects intestinal bacterial communities in the host. Insect Sci. 2020, 27, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Gong, Q.; Cao, L.J.; Sun, L.N.; Chen, J.C.; Gong, Y.J.; Pu, D.Q.; Huang, Q.; Hoffmann, A.A.; Wei, S.J. Similar gut bacterial microbiota in two fruit-feeding moth pests collected from different host species and locations. Insects 2020, 11, 840. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Du, K.; Sun, C.; Vimalanathan, A.; Liang, X.; Li, Y.; Wang, B.; Lu, X.; Li, L.; Shao, Y. Gut bacterial and fungal communities of the domesticated silkworm (Bombyx mori) and wild mulberry-feeding relatives. ISME J. 2018, 12, 2252–2262. [Google Scholar] [CrossRef] [Green Version]
- Colman, D.R.; Toolson, E.C.; Takacs-Vesbach, C.D. Do diet and taxonomy influence insect gut bacterial communities? Mol. Ecol. 2012, 21, 5124–5137. [Google Scholar] [CrossRef]
- Kudo, R.; Masuya, H.; Endoh, R.; Kikuchi, T.; Ikeda, H. Gut bacterial and fungal communities in ground-dwelling beetles are associated with host food habit and habitat. ISME J. 2019, 13, 676–685. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.X.; Jin, L.; Peng, T.; Zhang, H.Y.; Chen, Q.L.; Hua, Y.J. Identification of cultivable bacteria in the intestinal tract of Bactrocera dorsalis from three different populations and determination of their attractive potential. Pest Manag. Sci. 2014, 70, 80–87. [Google Scholar] [CrossRef]
- Zhang, Z.; Jiao, S.; Li, X.; Li, M. Bacterial and fungal gut communities of Agrilus mali at different developmental stages and fed different diets. Sci. Rep. 2018, 8, 15634. [Google Scholar] [CrossRef] [Green Version]
- Chandler, J.A.; Eisen, J.A.; Kopp, A. Yeast communities of diverse Drosophila species: Comparison of two symbiont groups in the same hosts. Appl. Environ. Microbiol. 2012, 78, 7327–7336. [Google Scholar] [CrossRef] [Green Version]
- Chandler, J.A.; Lang, J.M.; Bhatnagar, S.; Eisen, J.A.; Kopp, A. Bacterial communities of diverse Drosophila species: Ecological context of a host-microbe model system. PLoS Genet. 2011, 7, e1002272. [Google Scholar] [CrossRef]
- Chen, B.; Xie, S.; Zhang, X.; Zhang, N.; Feng, H.; Sun, C.; Lu, X.; Shao, Y. Gut microbiota metabolic potential correlates with body size between mulberry-feeding lepidopteran pest species. Pest Manag. Sci. 2020, 76, 1313–1323. [Google Scholar] [CrossRef]
- Gonzalez-Serrano, F.; Perez-Cobas, A.E.; Rosas, T.; Baixeras, J.; Latorre, A.; Moya, A. The gut microbiota composition of the moth Brithys crini reflects insect metamorphosis. Microb. Ecol. 2020, 79, 960–970. [Google Scholar] [CrossRef] [PubMed]
- Sharon, G.; Segal, D.; Ringo, J.M.; Hefetz, A.; Zilber-Rosenberg, I.; Rosenberg, E. Commensal bacteria play a role in mating preference of Drosophila melanogaster. Proc. Natl. Acad. Sci. USA 2010, 107, 20051–20056. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Li, T.P.; Zhou, C.Y.; Zhao, D.S.; Zhu, Y.X.; Bing, X.L.; Huang, H.J.; Hong, X.Y. Antibiotic exposure perturbs the bacterial community in the small brown planthopper Laodelphax striatellus. Insect Sci. 2020, 27, 895–907. [Google Scholar] [CrossRef]
- Genta, F.A.; Dillon, R.J.; Terra, W.R.; Ferreira, C. Potential role for gut microbiota in cell wall digestion and glucoside detoxification in Tenebrio molitor larvae. J. Insect Physiol. 2006, 52, 593–601. [Google Scholar] [CrossRef]
- Warnecke, F.; Luginbuhl, P.; Ivanova, N.; Ghassemian, M.; Richardson, T.H.; Stege, J.T.; Cayouette, M.; Mchardy, A.C.; Djordjevic, G.; Aboushadi, N.; et al. Metagenomic and functional analysis of hindgut microbiota of a wood-feeding higher termite. Nature 2007, 450, 560–565. [Google Scholar] [CrossRef]
- Raza, M.F.; Yao, Z.; Bai, S.; Cai, Z.; Zhang, H. Tephritidae fruit fly gut microbiome diversity, function and potential for applications. Bull. Entomol. Res. 2020, 110, 423–437. [Google Scholar] [CrossRef]
- Yun, J.H.; Jung, M.J.; Kim, P.S.; Bae, J.W. Social status shapes the bacterial and fungal gut communities of the honey bee. Sci. Rep. 2018, 8, 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.; Zhang, X.; Chen, Z.; Wang, Z.; Lu, Y.; Cheng, D. The divergence in bacterial components associated with Bactrocera dorsalis across developmental stages. Front. Microbiol. 2018, 9, 114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Yang, Y.; Wu, W.M.; Zhao, J.; Jiang, L. Evidence of polyethylene biodegradation by bacterial strains from the guts of plastic-eating waxworms. Environ. Sci. Technol. 2014, 48, 13776–13784. [Google Scholar] [CrossRef]
- Chen, B.; Teh, B.S.; Sun, C.; Hu, S.; Lu, X.; Boland, W.; Shao, Y. Biodiversity and activity of the gut microbiota across the life history of the insect herbivore Spodoptera littoralis. Sci. Rep. 2016, 6, 29505. [Google Scholar] [CrossRef]
- Chu, C.C.; Spencer, J.L.; Curzi, M.J.; Zavala, J.A.; Seufferheld, M.J. Gut bacteria facilitate adaptation to crop rotation in the western corn rootworm. Proc. Natl. Acad. Sci. USA 2013, 110, 11917–11922. [Google Scholar] [CrossRef] [Green Version]
- Suen, G.; Scott, J.J.; Aylward, F.O.; Adams, S.M.; Tringe, S.G.; Pinto-Tomas, A.A.; Foster, C.E.; Pauly, M.; Weimer, P.J.; Barry, K.W.; et al. An insect herbivore microbiome with high plant biomass-degrading capacity. PLoS Genet. 2010, 6, e1001129. [Google Scholar] [CrossRef]
- Muhammad, A.; Fang, Y.; Hou, Y.; Shi, Z. The gut entomotype of red palm weevil Rhynchophorus ferrugineus Olivier (Coleoptera: Dryophthoridae) and their effect on host nutrition metabolism. Front. Microbiol. 2017, 8, 2291. [Google Scholar] [CrossRef] [Green Version]
- Dantur, K.I.; Enrique, R.; Welin, B.; Castagnaro, A.P. Isolation of cellulolytic bacteria from the intestine of Diatraea saccharalis larvae and evaluation of their capacity to degrade sugarcane biomass. Amb. Express. 2015, 5, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben Ami, E.; Yuval, B.; Jurkevitch, E. Manipulation of the microbiota of mass-reared mediterranean fruit flies Ceratitis capitata (Diptera: Tephritidae) improves sterile male sexual performance. ISME J. 2010, 4, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Hamden, H.; Guerfali, M.M.; Fadhl, S.; Saidi, M.; Chevrier, C. Fitness improvement of mass-reared sterile males of Ceratitis capitata (Vienna 8 strain) (Diptera: Tephritidae) after gut enrichment with probiotics. J. Econ. Entomol. 2013, 106, 641–647. [Google Scholar] [CrossRef]
- Snyman, M.; Gupta, A.K.; Bezuidenhout, C.C.; Claassens, S.; Van Den Berg, J. Gut microbiota of Busseola fusca (Lepidoptera: Noctuidae). World J. Microbiol. Biotechnol. 2016, 32, 115. [Google Scholar] [CrossRef]
- Xiang, H.; Wei, G.F.; Jia, S.; Huang, J.; Miao, X.X.; Zhou, Z.; Zhao, L.P.; Huang, Y.P. Microbial communities in the larval midgut of laboratory and field populations of cotton bollworm (Helicoverpa armigera). Can. J. Microbiol. 2006, 52, 1085–1092. [Google Scholar] [CrossRef] [PubMed]
- Vilanova, C.; Baixeras, J.; Latorre, A.; Porcar, M. The generalist inside the specialist: Gut bacterial communities of two insect species feeding on toxic plants are dominated by Enterococcus sp. Front. Microbiol. 2016, 7, 1005. [Google Scholar] [CrossRef]
- Blanco, C.A.; Teran-Vargas, A.P.; Lopez, J.D.; Kauffman, J.V.; Wei, X.K. Densities of Heliothis virescens and Helicoverpa zea (Lepidoptera: Noctuidae) in three plant hosts. Fla. Entomol. 2007, 90, 742–750. [Google Scholar] [CrossRef]
- Shao, Y.; Spiteller, D.; Tang, X.; Ping, L.; Colesie, C.; Munchberg, U.; Bartram, S.; Schneider, B.; Budel, B.; Popp, J.; et al. Crystallization of alpha- and beta-carotene in the foregut of Spodoptera larvae feeding on a toxic food plant. Insect Biochem. Mol. Biol. 2011, 41, 273–281. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | Raw Reads | Clean Reads | Effective Reads | AvgLen (bp) | GC (%) | Effective (%) |
|---|---|---|---|---|---|---|
| A1 | 79884 | 67772 | 67538 | 429 | 56.69 | 84.55 |
| A2 | 79835 | 67914 | 67618 | 429 | 56.71 | 84.70 |
| A3 | 73610 | 62130 | 61705 | 429 | 56.69 | 83.83 |
| CK1 | 79980 | 70117 | 64592 | 418 | 54.89 | 80.76 |
| CK2 | 80062 | 70018 | 67177 | 423 | 55.41 | 83.91 |
| CK3 | 80384 | 69886 | 67041 | 423 | 55.73 | 83.40 |
| CP1 | 80015 | 67814 | 67101 | 429 | 56.59 | 83.86 |
| CP2 | 79585 | 68569 | 66419 | 426 | 56.53 | 83.46 |
| CP3 | 79957 | 68144 | 66329 | 426 | 56.58 | 82.96 |
| N1 | 44803 | 36668 | 36231 | 429 | 56.5 | 80.87 |
| N2 | 56666 | 36618 | 35694 | 429 | 56.49 | 62.99 |
| N3 | 69729 | 56067 | 55246 | 429 | 56.67 | 79.23 |
| P1 | 79923 | 67884 | 67687 | 429 | 56.60 | 84.69 |
| P2 | 63619 | 53983 | 53828 | 429 | 56.62 | 84.61 |
| P3 | 77451 | 65509 | 65201 | 429 | 56.59 | 84.18 |
| PL1 | 80101 | 66639 | 62056 | 413 | 55.01 | 77.47 |
| PL2 | 79776 | 66332 | 61239 | 413 | 55.22 | 76.76 |
| PL3 | 80136 | 68242 | 64313 | 421 | 55.69 | 80.25 |
| PS1 | 62578 | 52190 | 51917 | 429 | 56.58 | 82.96 |
| PS2 | 64642 | 54192 | 53968 | 429 | 56.59 | 83.49 |
| PS3 | 58473 | 49052 | 48669 | 427 | 56.47 | 83.23 |
| Sample ID | Proteobacteria | Firmicutes | Bacteroidota | Actinobacteriota |
|---|---|---|---|---|
| CK | 59.18 ± 13.26 b | 17.74 ± 4.85 b | 8.32 ± 2.56 b | 7.85 ± 3.79 a |
| A | 99.43 ± 0.07 a | 0.27 ± 0.05 c | 0.08 ± 0.01 c | 0.11 ± 0.05 b |
| CP | 94.34 ± 2.32 a | 3.02 ± 1.28 c | 0.20 ± 0.10 c | 0.67 ± 0.23 b |
| N | 98.13 ± 0.60 a | 0.96 ± 0.24 c | 0.57 ± 0.34 c | 0.07 ± 0.01 b |
| P | 99.52 ± 0.12 a | 0.28 ± 0.16 c | 0.07 ± 0.01 c | 0.09 ± 0.04 b |
| PL | 32.75 ± 18.59 b | 32.92 ± 8.90 a | 15.06 ± 4.06 a | 2.80 ± 0.86 b |
| PS | 95.63 ± 2.93 a | 0.30 ± 0.07 c | 0.12 ± 0.02 c | 0.03 ± 0.00 b |
| Sample ID | Enterobacteriaceae | Lachnospiraceae | Ruminococcaceae | Lactobacillaceae | Muribaculaceae | Oscillospiraceae | Caloramatoraceae | Moraxellaceae | Prevotellaceae | Akkermansiaceae |
|---|---|---|---|---|---|---|---|---|---|---|
| CK | 47.43 ± 16.05 b | 2.79 ± 0.46b | 2.03 ± 0.37 b | 2.14 ± 0.49 b | 2.60 ± 0.72 b | 1.33 ± 0.37 b | 1.24 ± 0.56 b | 3.84 ± 0.76 a | 1.10 ± 0.38 b | 0.61 ± 0.08 b |
| A | 99.15 ± 0.12 a | 0.06 ± 0.01b | 0.04 ± 0.01 b | 0.03 ± 0.01 c | 0.03 ± 0.01 c | 0.03 ± 0.01 c | 0.02 ± 0.01 b | 0.01 ± 0.00 b | 0.04 ± 0.00 c | 0.02 ± 0.00 b |
| CP | 92.00 ± 2.71 a | 0.42 ± 0.20b | 0.21 ± 0.11 b | 0.29 ± 0.06 c | 0.07 ± 0.04 c | 0.18 ± 0.09 c | 0.19 ± 0.09 b | 0.05 ± 0.02 b | 0.02 ± 0.01 c | 0.03 ± 0.01 b |
| N | 96.43 ± 1.04 a | 0.19 ± 0.02b | 0.11 ± 0.02 b | 0.10 ± 0.01 c | 0.06 ± 0.02 c | 0.07 ± 0.01 c | 0.05 ± 0.01 b | 0.00 ± 0.00 b | 0.04 ± 0.00 c | 0.04 ± 0.00 b |
| P | 98.72 ± 0.40 a | 0.03 ± 0.01b | 0.02 ± 0.00 b | 0.02 ± 0.00 c | 0.02 ± 0.00 c | 0.02 ± 0.00 c | 0.01 ± 0.00 b | 0.01 ± 0.00 b | 0.01 ± 0.00 c | 0.01 ± 0.00 b |
| PL | 23.54 ± 21.21 b | 8.97 ± 2.53a | 5.86 ± 1.71 a | 5.21 ± 1.21 a | 4.59 ± 1.30 a | 3.53 ± 0.87 a | 3.50 ± 0.94 a | 0.41 ± 0.15 b | 2.39 ± 0.62 a | 2.41 ± 0.69 a |
| PS | 95.35 ± 3.07 a | 0.08 ± 0.02b | 0.06 ± 0.01 b | 0.04 ± 0.01 c | 0.03 ± 0.01 c | 0.03 ± 0.01 c | 0.03 ± 0.01 b | 0.00 ± 0.00 b | 0.03 ± 0.01 c | 0.01 ± 0.00 b |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yuan, X.; Zhang, X.; Liu, X.; Dong, Y.; Yan, Z.; Lv, D.; Wang, P.; Li, Y. Comparison of Gut Bacterial Communities of Grapholita molesta (Lepidoptera: Tortricidae) Reared on Different Host Plants. Int. J. Mol. Sci. 2021, 22, 6843. https://doi.org/10.3390/ijms22136843
Yuan X, Zhang X, Liu X, Dong Y, Yan Z, Lv D, Wang P, Li Y. Comparison of Gut Bacterial Communities of Grapholita molesta (Lepidoptera: Tortricidae) Reared on Different Host Plants. International Journal of Molecular Sciences. 2021; 22(13):6843. https://doi.org/10.3390/ijms22136843
Chicago/Turabian StyleYuan, Xiangqun, Xuan Zhang, Xueying Liu, Yanlu Dong, Zizheng Yan, Dongbiao Lv, Ping Wang, and Yiping Li. 2021. "Comparison of Gut Bacterial Communities of Grapholita molesta (Lepidoptera: Tortricidae) Reared on Different Host Plants" International Journal of Molecular Sciences 22, no. 13: 6843. https://doi.org/10.3390/ijms22136843