
Stratifying Cumulus Cell Samples Based on Molecular Profiling to Help Resolve Biomarker Discrepancies and to Predict Oocyte Developmental Competence

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Clinical Characteristics of the Collected Samples
2.2. Correlation of Clinical Parameters with Both Genome Wide Expression and DNA Methylation Data
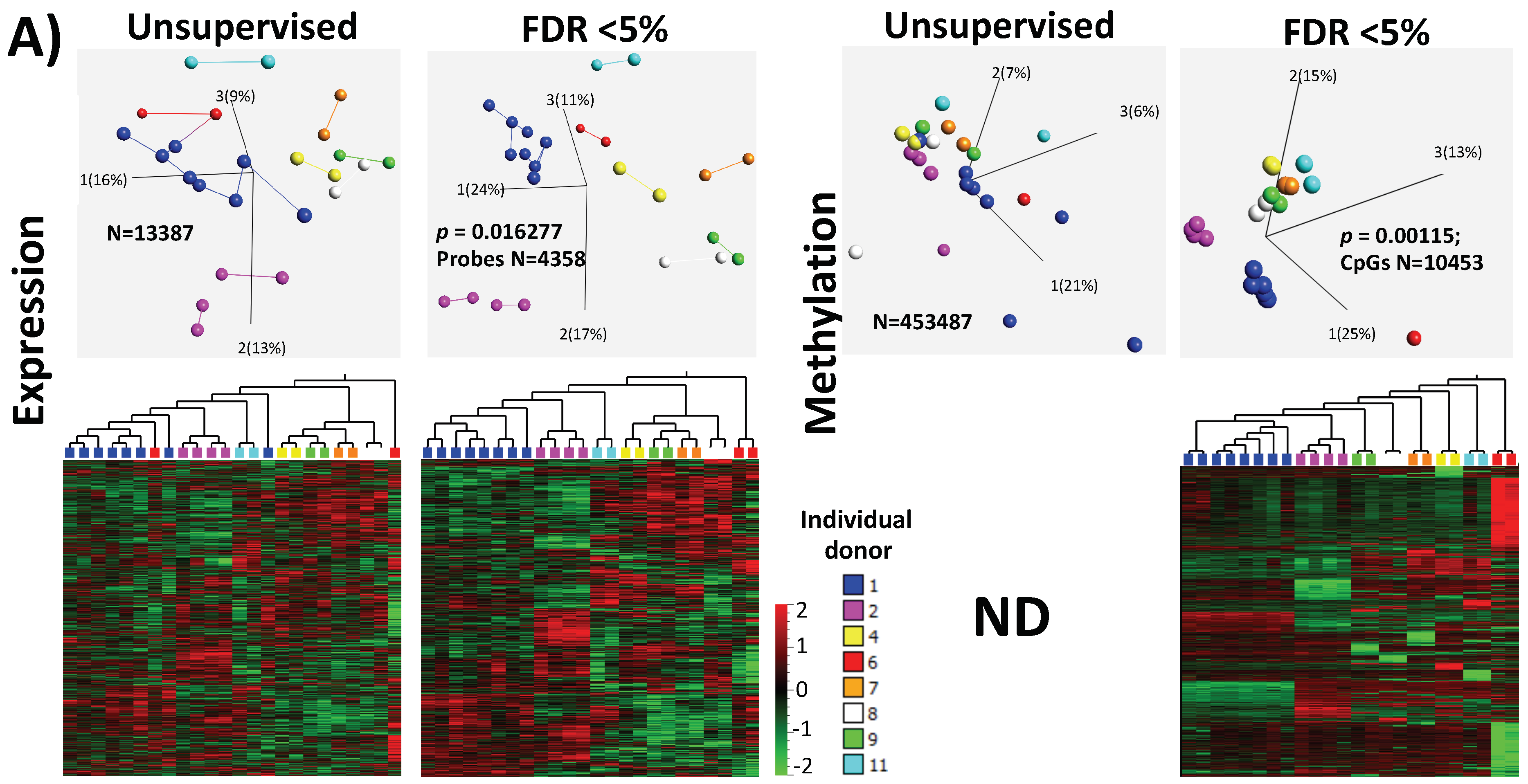
2.3. Inter-Individual Differences Order/Separate Samples According to Donor Identity Showing High Significance in Genes Related to Protein Metabolism
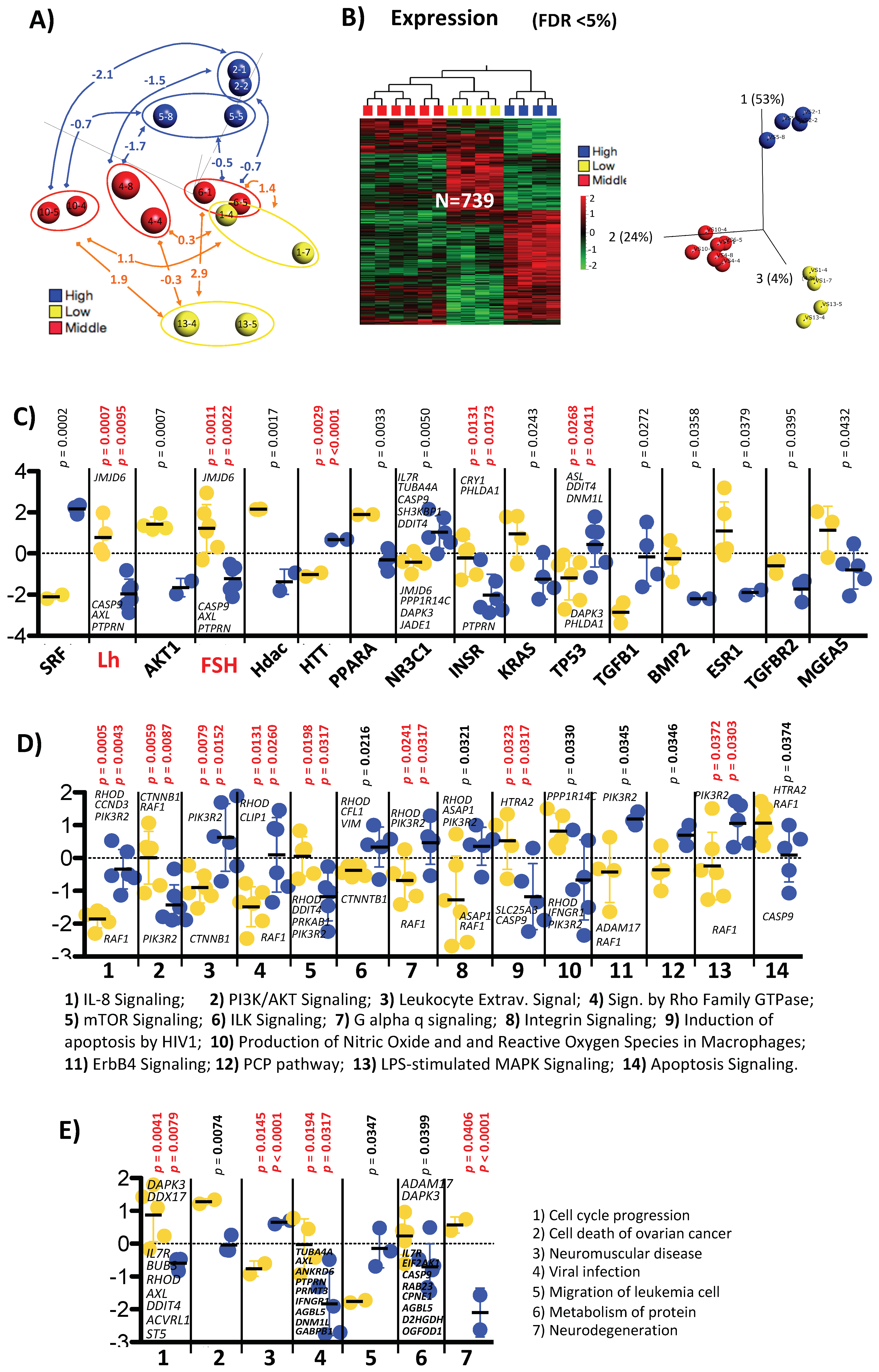
2.4. Differences between Two Treatments with the Same Protocol and the Same Individual
2.5. Differences between Long and Short Stimulation Protocols
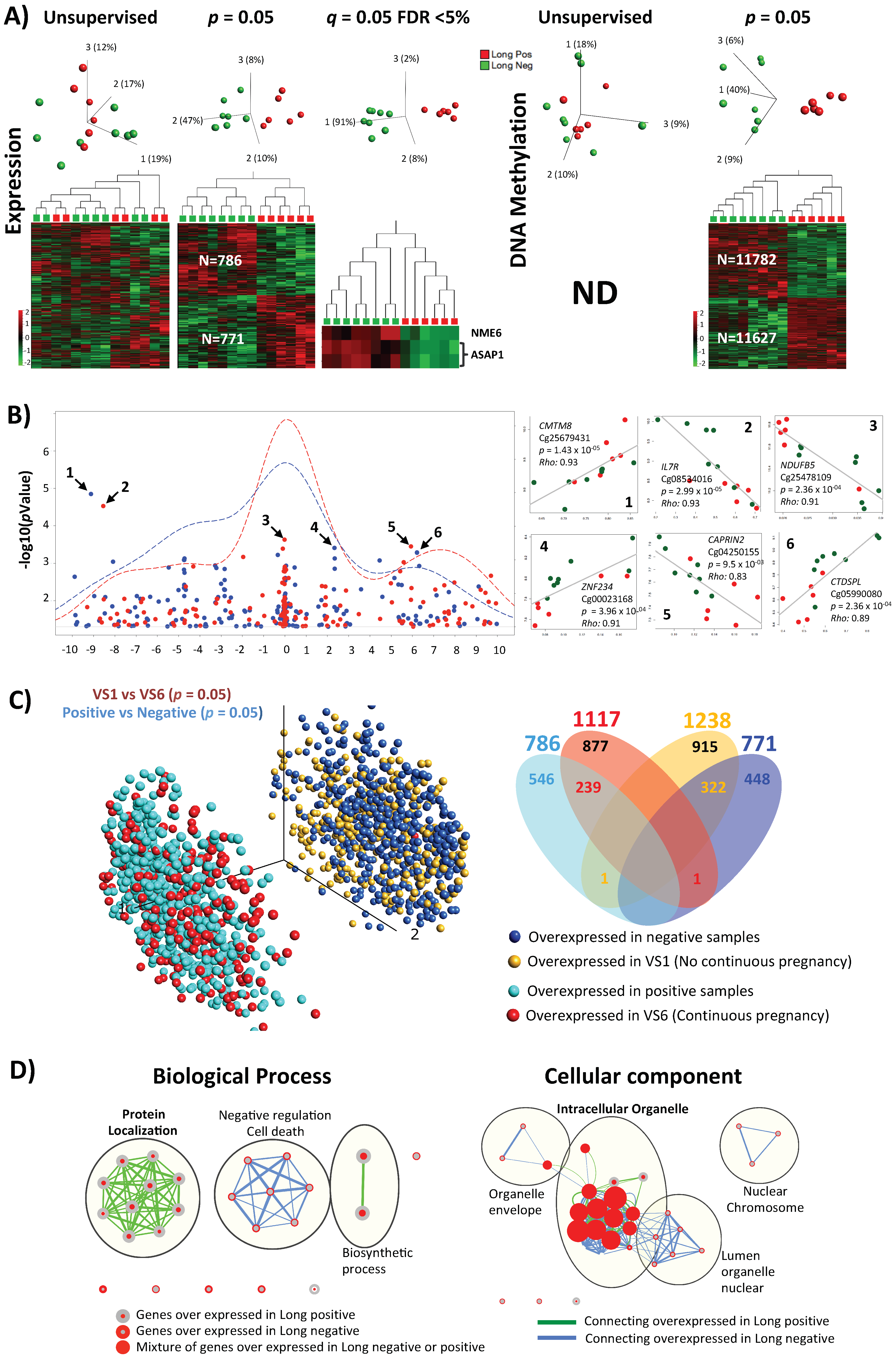
2.6. Molecular Profile of Individual Non-Pooled Cumulus Cells Derived from Positive or Negative Pregnancy Test (Long Stimulation Protocol Only)
2.7. Detailed One to One Comparison between Positive and Negative Samples
2.8. Comparing our Expression Data with Available Published Datasets
3. Discussion
4. Materials and Methods
4.1. Patient Samples and Clinical Parameters
4.2. Ovarian Hormone Stimulation Protocol and Outer Cumulus Cell Collections
4.3. Isolation of RNA and DNA
4.4. Illumina Array-Based Analysis
4.5. Processing Methylation Arrays Data and Methylation Analysis
4.6. Processing of Expression Array Data and Differential Expression Analysis
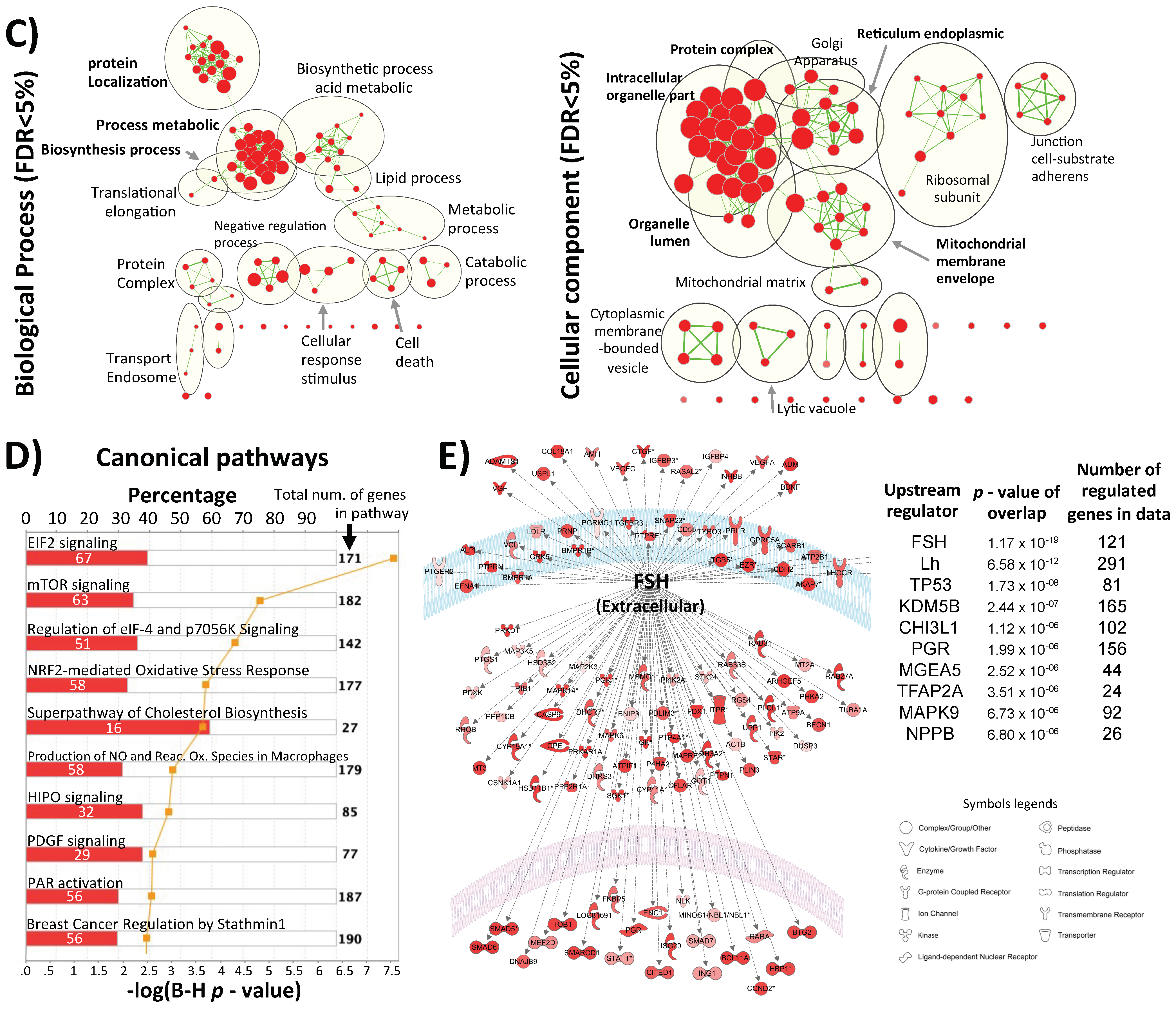
4.7. Gene Ontology and Pathway Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Inhorn, M.C.; Patrizio, P. Infertility around the globe: New thinking on gender, reproductive technologies and global movements in the 21st century. Hum. Reprod. Update 2015, 21, 411–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palemro, G.; Joris, H.; Devroey, P.; Van Steirteghem, A.C. Pregnancies after intercytoplasmic injection of single spermatozoon into an oocyte. Lancet 1992, 340, 17–18. [Google Scholar]
- Yuan, X.; Saravelos, S.H.; Wang, Q.; Xu, Y.; Li, T.C.; Zhou, C. Endometrial thickness as a predictor of pregnancy outcomes in 10787 fresh IVF-ICSI cycles. Reprod. Biomed. Online 2016, 33, 197–205. [Google Scholar] [CrossRef] [Green Version]
- Ioannidis, G.; Sacks, G.; Reddy, N.; Seyani, L.; Margara, R.; Lavery, S.; Trew, G. Day 14 maternal serum progesterone levels predict pregnancy outcome in IVF/ICSI treatment cycles: A prospective study. Hum. Reprod. 2005, 20, 741–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbara, A.; Vuong, L.N.; Ho, V.N.A.; Clarke, S.A.; Jeffers, L.; Comninos, A.N.; Salim, R.; Ho, T.M.; Kelsey, T.W.; Trew, G.H. Follicle Size on Day of Trigger Most Likely to Yield a Mature Oocyte. Front. Endocrinol. 2018, 9, 193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maman, E.; Yung, Y.; Kedem, A.; Yerushalmi, G.M.; Konopnicki, S.; Cohen, B.; Dor, J.; Hourvitz, A. High expression of luteinizing hormone receptors messenger RNA by human cumulus granulosa cells is in correlation with decreased fertilization. Fertil. Steril. 2012, 97, 592–598. [Google Scholar] [CrossRef]
- Grondahl, M.L.; Andersen, C.Y.; Bogstad, J.; Borgbo, T.; Boujida, V.H.; Borup, R. Specific genes are selectively expressed between cumulus and granulosa cells from individual human pre-ovulatory follicles. Mol. Hum. Reprod. 2012, 18, 572–584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKenzie, L.J.; Pangas, S.A.; Carson, S.A.; Kovanci, E.; Cisneros, P.; Buster, J.E.; Amato, P.; Matzuk, M.M. Human cumulus granulosa cell gene expression: A predictor of fertilization and embryo selection in women undergoing IVF. Hum. Reprod. 2004, 19, 2869–2874. [Google Scholar] [CrossRef] [Green Version]
- Hasegawa, J.; Yanaihara, A.; Iwasaki, S.; Mitsukawa, K.; Negishi, M.; Okai, T. Reduction of connexin 43 in human cumulus cells yields good embryo competence during ICSI. J. Assist. Reprod. Genet. 2007, 24, 463–466. [Google Scholar] [CrossRef] [Green Version]
- Anderson, R.A.; Sciorio, R.; Kinnell, H.; Bayne, R.A.; Thong, K.J.; de Sousa, P.A.; Pickering, S. Cumulus gene expression as a predictor of human oocyte fertilisation, embryo development and competence to establish a pregnancy. Reproduction 2009, 138, 629–637. [Google Scholar] [CrossRef] [PubMed]
- Haouzi, D.; Assou, S.; Mahmoud, K.; Hedon, B.; De Vos, J.; Dewailly, D.; Hamamah, S. LH/hCGR gene expression in human cumulus cells is linked to the expression of the extracellular matrix modifying gene TNFAIP6 and to serum estradiol levels on day of hCG administration. Hum. Reprod. 2009, 24, 2868–2878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.X.; Tong, D.; El-Gehani, F.; Tekpetey, F.R.; Kidder, G.M. Connexin expression and gap junctional coupling in human cumulus cells: Contribution to embryo quality. J. Cell. Mol. Med. 2009, 13, 972–984. [Google Scholar] [CrossRef] [PubMed]
- Adriaenssens, T.; Wathlet, S.; Segers, I.; Verheyen, G.; De Vos, A.; Van der Elst, J.; Coucke, W.; Devroey, P.; Smitz, J. Cumulus cell gene expression is associated with oocyte developmental quality and influenced by patient and treatment characteristics. Hum. Reprod. 2010, 25, 1259–1270. [Google Scholar] [CrossRef] [Green Version]
- Koks, S.; Velthut, A.; Sarapik, A.; Altmae, S.; Reinmaa, E.; Schalkwyk, L.C.; Fernandes, C.; Lad, H.V.; Soomets, U.; Jaakma, U.; et al. The differential transcriptome and ontology profiles of floating and cumulus granulosa cells in stimulated human antral follicles. Mol. Hum. Reprod. 2010, 16, 229–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Assidi, M.; Montag, M.; Van der Ven, K.; Sirard, M.A. Biomarkers of human oocyte developmental competence expressed in cumulus cells before ICSI: A preliminary study. J. Assist. Reprod. Genet. 2011, 28, 173–188. [Google Scholar] [CrossRef] [Green Version]
- Wathlet, S.; Adriaenssens, T.; Segers, I.; Verheyen, G.; Van de Velde, H.; Coucke, W.; Ron El, R.; Devroey, P.; Smitz, J. Cumulus cell gene expression predicts better cleavage-stage embryo or blastocyst development and pregnancy for ICSI patients. Hum. Reprod. 2011, 26, 1035–1051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feuerstein, P.; Puard, V.; Chevalier, C.; Teusan, R.; Cadoret, V.; Guerif, F.; Houlgatte, R.; Royere, D. Genomic assessment of human cumulus cell marker genes as predictors of oocyte developmental competence: Impact of various experimental factors. PLoS ONE 2012, 7, e40449. [Google Scholar]
- Varras, M.; Griva, T.; Kalles, V.; Akrivis, C.; Paparisteidis, N. Markers of stem cells in human ovarian granulosa cells: Is there a clinical significance in ART? J. Ovarian Res. 2012, 5, 36. [Google Scholar] [CrossRef] [Green Version]
- Wathlet, S.; Adriaenssens, T.; Segers, I.; Verheyen, G.; Van Landuyt, L.; Coucke, W.; Devroey, P.; Smitz, J. Pregnancy prediction in single embryo transfer cycles after ICSI using QPCR: Validation in oocytes from the same cohort. PLoS ONE 2013, 8, e54226. [Google Scholar] [CrossRef]
- Li, Y.; Li, R.-Q.; Song-Bang Ou, S.-B.; Zhang, N.-F.; Ren, L.; Wei, L.-N.; Zhang, Q.; Yang, D. Increased GDF9 and BMP15 mRNA levels in cumulus granulosa cells correlate with oocyte maturation, fertilization, and embryo quality in humans. Reprod. Biol. Endocrinol. RB&E 2014, 12, 1–9. [Google Scholar]
- Assidi, M.; Montag, M.; Sirard, M.A. Use of Both Cumulus Cells’ Transcriptomic Markers and Zona Pellucida Birefringence to Select Developmentally Competent Oocytes in Human Assisted Reproductive Technologies. BMC Genom. 2015, 16, 9. [Google Scholar] [CrossRef] [Green Version]
- Burnik Papler, T.; Vrtacnik Bokal, E.; Maver, A.; Lovrecic, L. Specific gene expression differences in cumulus cells as potential biomarkers of pregnancy. Reprod. Biomed. Online 2015, 30, 426–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Chen, D.; Zhang, Z.; Wei, Z.; Cao, Y. Molecular signature in human cumulus cells related to embryonic developmental potential. Reprod. Sci. 2015, 22, 173–180. [Google Scholar] [CrossRef]
- Artini, P.G.; Tatone, C.; Sperduti, S.; D’Aurora, M.; Franchi, S.; Di Emidio, G.; Ciriminna, R.; Vento, M.; di Pietro, C.; Stuppia, L.; et al. Cumulus cells surrounding oocytes with high developmental competence exhibit down-regulation of phosphoinositol 1,3 kinase/protein kinase B (PI3K/AKT) signalling genes involved in proliferation and survival. Hum. Reprod. 2017, 32, 2474–2484. [Google Scholar] [CrossRef]
- Green, K.A.; Franasiak, J.M.; Werner, M.D.; Tao, X.; Landis, J.N.; Scott, R.T., Jr.; Treff, N.R. Cumulus cell transcriptome profiling is not predictive of live birth after in vitro fertilization: A paired analysis of euploid sibling blastocysts. Fertil. Steril. 2018, 109, 460–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demiray, S.B.; Goker, E.N.T.; Tavmergen, E.; Yilmaz, O.; Calimlioglu, N.; Soykam, H.O.; Oktem, G.; Sezerman, U. Differential gene expression analysis of human cumulus cells. Clin. Exp. Reprod. Med. 2019, 46, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Faramarzi, A.; Khalili, M.A.; Jahromi, M.G. Is there any correlation between apoptotic genes expression in cumulus cells with embryo morphokinetics? Mol. Biol. Rep. 2019, 46, 3663–3670. [Google Scholar] [CrossRef] [PubMed]
- Karakaya, C.; Guzeloglu-Kayisli, O.; Uyar, A.; Kallen, A.N.; Babayev, E.; Bozkurt, N.; Unsal, E.; Karabacak, O.; Seli, E. Poor ovarian response in women undergoing in vitro fertilization is associated with altered microRNA expression in cumulus cells. Fertil. Steril. 2015, 103, 1469–1476. [Google Scholar] [CrossRef] [Green Version]
- Hamel, M.; Dufort, I.; Robert, C.; Gravel, C.; Leveille, M.C.; Leader, A.; Sirard, M. Identification of differentially expressed markers in human follicular cells associated with competent oocytes. Hum. Reprod. 2008, 23, 1118–1127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebner, T.; Balaban, B.; Moser, M.; Shebl, O.; Urman, B.; Ata, B.; Tews, G. Automatic user-independent zona pellucida imaging at the oocyte stage allows for the prediction of preimplantation development. Fertil. Steril. 2010, 94, 913–920. [Google Scholar] [CrossRef] [PubMed]
- Verpoest, W.; Staessen, C.; Bossuyt, P.M.; Goossens, V.; Altarescu, G.; Bonduelle, M.; Devesa, M.; Eldar-Geva, T.; Gianaroli, L.; Griesinger, G.; et al. Preimplantation genetic testing for aneuploidy by microarray analysis of polar bodies in advanced maternal age: A randomized clinical trial. Hum. Reprod. 2018, 33, 1767–1776. [Google Scholar] [CrossRef] [PubMed]
- Zanetti, B.F.; Braga, D.; Setti, S.; Figueira, D.C.S.; Iaconelli, A.; Borges, E. Is perivitelline space morphology of the oocyte associated with pregnancy outcome in intracytoplasmic sperm injection cycles? Eur. J. Obstet. Gynecol. Reprod. Biol. 2018, 231, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Ouandaogo, Z.G.; Frydman, N.; Hesters, L.; Assou, S.; Haouzi, D.; Dechaud, H.; Frydman, R.; Hamamah, S. Differences in transcriptomic profiles of human cumulus cells isolated from oocytes at GV, MI and MII stages after in vivo and in vitro oocyte maturation. Hum. Reprod. 2012, 27, 2438–2447. [Google Scholar] [CrossRef] [PubMed]
- Ouandaogo, Z.G.; Haouzi, D.; Assou, S.; Dechaud, H.; Kadoch, I.J.; De Vos, J.; Hamamah, S. Human cumulus cells molecular signature in relation to oocyte nuclear maturity stage. PLoS ONE 2011, 6, e27179. [Google Scholar] [CrossRef]
- Al-Edani, T.; Assou, S.; Ferrieres, A.; Bringer Deutsch, S.; Gala, A.; Lecellier, C.H.; Ait-Ahmed, O.; Hamamah, S. Female aging alters expression of human cumulus cells genes that are essential for oocyte quality. BioMed Res. Int. 2014, 2014, 964614. [Google Scholar] [CrossRef] [Green Version]
- Papler, T.B.; Bokal, E.V.; Tacer, K.F.; Juvan, P.; Virant Klun, I.; Devjak, R. Differences in cumulus cells gene expression between modified natural and stimulated in vitro fertilization cycles. J. Assist. Reprod. Genet. 2014, 31, 79–88. [Google Scholar] [CrossRef] [Green Version]
- Borgbo, T.; Povlsen, B.B.; Andersen, C.Y.; Borup, R.; Humaidan, P.; Grondahl, M.L. Comparison of gene expression profiles in granulosa and cumulus cells after ovulation induction with either human chorionic gonadotropin or a gonadotropin-releasing hormone agonist trigger. Fertil. Steril. 2013, 100, 994–1001. [Google Scholar] [CrossRef]
- Gatta, V.; Tatone, C.; Ciriminna, R.; Vento, M.; Franchi, S.; d’Aurora, M.; Sperduti, S.; Cela, V.; Borzi, P.; Palermo, R.; et al. Gene expression profiles of cumulus cells obtained from women treated with recombinant human luteinizing hormone + recombinant human follicle-stimulating hormone or highly purified human menopausal gonadotropin versus recombinant human follicle-stimulating hormone alone. Fertil. Steril. 2013, 99, 2000–2008. [Google Scholar]
- Gurgan, T.; Montjean, D.; Demirol, A.; Menezo, Y.J. Sequential (hFSH + recFSH) vs homogenous (hFSH or recFSH alone) stimulation: Clinical and biochemical (cumulus cell gene expression) aspects. J. Assist. Reprod. Genet. 2014, 31, 657–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosco, L.; Ruvolo, G.; Luparello, C.; Ferrari, S.; Valerio, D.; Santi, D.; Piomboni, P.; Sarcina, E.; Lispi, M.; Roccheri, M.C. Gene Expression and Apoptosis Levels in Cumulus Cells of Patients with Polymorphisms of FSHR and LHB Undergoing in Vitro Fertilization Program. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2017, 43, 2391–2404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karakaya, C.; Guzeloglu-Kayisli, O.; Hobbs, R.J.; Gerasimova, T.; Uyar, A.; Erdem, M.; Oktem, M.; Erdem, A.; Gumuslu, S.; Ercan, D.; et al. Follicle-stimulating hormone receptor (FSHR) alternative skipping of exon 2 or 3 affects ovarian response to FSH. Mol. Hum. Reprod. 2014, 20, 630–643. [Google Scholar] [CrossRef] [Green Version]
- Borup, R.; Thuesen, L.L.; Andersen, C.Y.; Nyboe-Andersen, A.; Ziebe, S.; Winther, O.; Grondahl, M.L. Competence Classification of Cumulus and Granulosa Cell Transcriptome in Embryos Matched by Morphology and Female Age. PLoS ONE 2016, 11, e0153562. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, M.; Sikora-Wohlfeld, W.; Beyer, A. Impact of natural genetic variation on gene expression dynamics. PLoS Genet. 2013, 9, e1003514. [Google Scholar] [CrossRef] [PubMed]
- Consortium, G.T.; Laboratory, D.A. Genetic effects on gene expression across human tissues. Nature 2017, 550, 204–213. [Google Scholar] [CrossRef]
- Wunsch, A.; Sonntag, B.; Simoni, M. Polymorphism of the FSH receptor and ovarian response to FSH. Ann. Endocrinol. 2007, 68, 160–166. [Google Scholar] [CrossRef]
- Mehus, J.G.; Deloukas, P.; Lambeth, D.O. NME6: A new member of the nm23/nucleoside diphosphate kinase gene family located on human chromosome 3p21.3. Hum. Genet. 1999, 104, 454–459. [Google Scholar] [CrossRef]
- Desvignes, T.; Fauvel, C.; Bobe, J. The NME gene family in zebrafish oogenesis and early development. Naunyn Schmiedeberg’s Arch. Pharmacol. 2011, 384, 439–449. [Google Scholar] [CrossRef] [Green Version]
- Bosnar, M.H.; Radić, M.; Ačkar, L.; Proust, B.; Ćetković, H. Subcellular Localization and Expression of Nme6, a Member of The Nme/Nm23/Ndpk Family. Hum. Tumour Cells. 2018, 3, 122. [Google Scholar]
- Gasilina, A.; Vitali, T.; Luo, R.; Jian, X.; Randazzo, P.A. The ArfGAP ASAP1 Controls Actin Stress Fiber Organization via Its N-BAR Domain. iScience 2019, 22, 166–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, R.; Reed, C.E.; Sload, J.A.; Wordeman, L.; Randazzo, P.A.; Chen, P.W. Arf GAPs and molecular motors. Small GTPases 2019, 10, 196–209. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Zhao, G.; Yang, C.; Dong, P.; Watari, H.; Zeng, L.; Pfeffer, L.M.; Yue, J. Lentiviral vector mediated-ASAP1 expression promotes epithelial to mesenchymal transition in ovarian cancer cells. Oncol. Lett. 2018, 15, 4432–4438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, T.; Stein, U.; Poletti, A.; Garzia, L.; Rothley, M.; Plaumann, D.; Thiele, W.; Bauer, M.; Galasso, A.; Schlag, P.; et al. ASAP1 promotes tumor cell motility and invasiveness, stimulates metastasis formation in vivo, and correlates with poor survival in colorectal cancer patients. Oncogene 2010, 29, 2393–2403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahraman, S.; Cetinkaya, C.P.; Cetinkaya, M.; Tufekci, M.A.; Ekmekci, C.G.; Montag, M. Is there a correlation between follicle size and gene expression in cumulus cells and is gene expression an indicator of embryo development? Reprod. Biol. Endocrinol. RB&E 2018, 16, 69. [Google Scholar]
- O’Shea, L.C.; Mehta, J.; Lonergan, P.; Hensey, C.; Fair, T. Developmental competence in oocytes and cumulus cells: Candidate genes and networks. Syst. Biol. Reprod. Med. 2012, 58, 88–101. [Google Scholar] [CrossRef] [PubMed]
- Maksimovic, J.; Gordon, L.; Oshlack, A. SWAN: Subset-quantile within array normalization for Illumina Infinium HumanMethylation450 BeadChips. Gen. Biol. 2012, 13, R44. [Google Scholar] [CrossRef] [PubMed] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
El-Maarri, O.; Jamil, M.A.; Köster, M.; Nüsgen, N.; Oldenburg, J.; Montag, M.; van der Ven, H.; van der Ven, K. Stratifying Cumulus Cell Samples Based on Molecular Profiling to Help Resolve Biomarker Discrepancies and to Predict Oocyte Developmental Competence. Int. J. Mol. Sci. 2021, 22, 6377. https://doi.org/10.3390/ijms22126377
El-Maarri O, Jamil MA, Köster M, Nüsgen N, Oldenburg J, Montag M, van der Ven H, van der Ven K. Stratifying Cumulus Cell Samples Based on Molecular Profiling to Help Resolve Biomarker Discrepancies and to Predict Oocyte Developmental Competence. International Journal of Molecular Sciences. 2021; 22(12):6377. https://doi.org/10.3390/ijms22126377
Chicago/Turabian StyleEl-Maarri, Osman, Muhammad Ahmer Jamil, Maria Köster, Nicole Nüsgen, Johannes Oldenburg, Markus Montag, Hans van der Ven, and Katrin van der Ven. 2021. "Stratifying Cumulus Cell Samples Based on Molecular Profiling to Help Resolve Biomarker Discrepancies and to Predict Oocyte Developmental Competence" International Journal of Molecular Sciences 22, no. 12: 6377. https://doi.org/10.3390/ijms22126377