
Targeting Glycolysis in Macrophages Confers Protection Against Pancreatic Ductal Adenocarcinoma

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
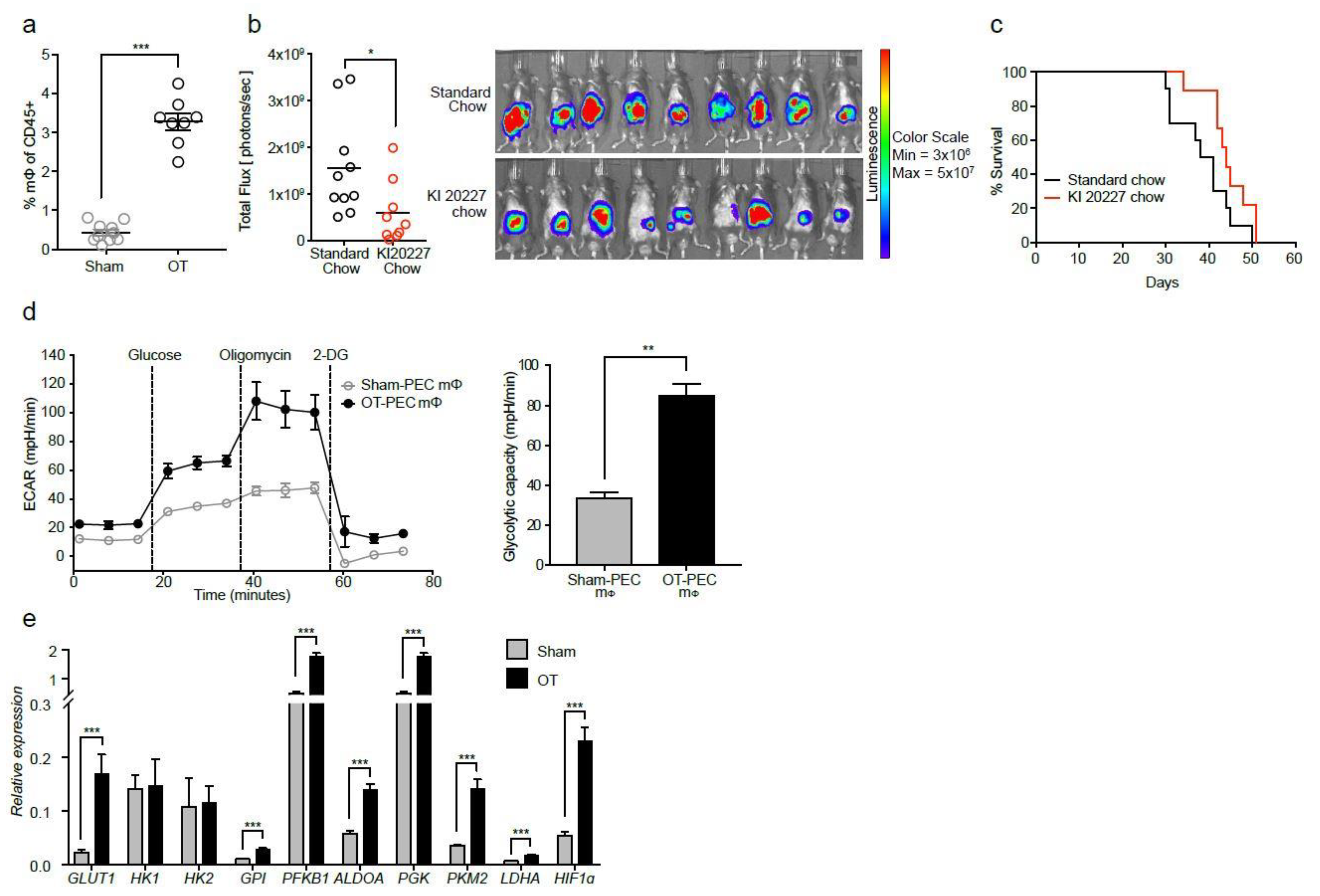
2.1. Macrophages from Tumor-Bearing Mice Are Highly Glycolytic
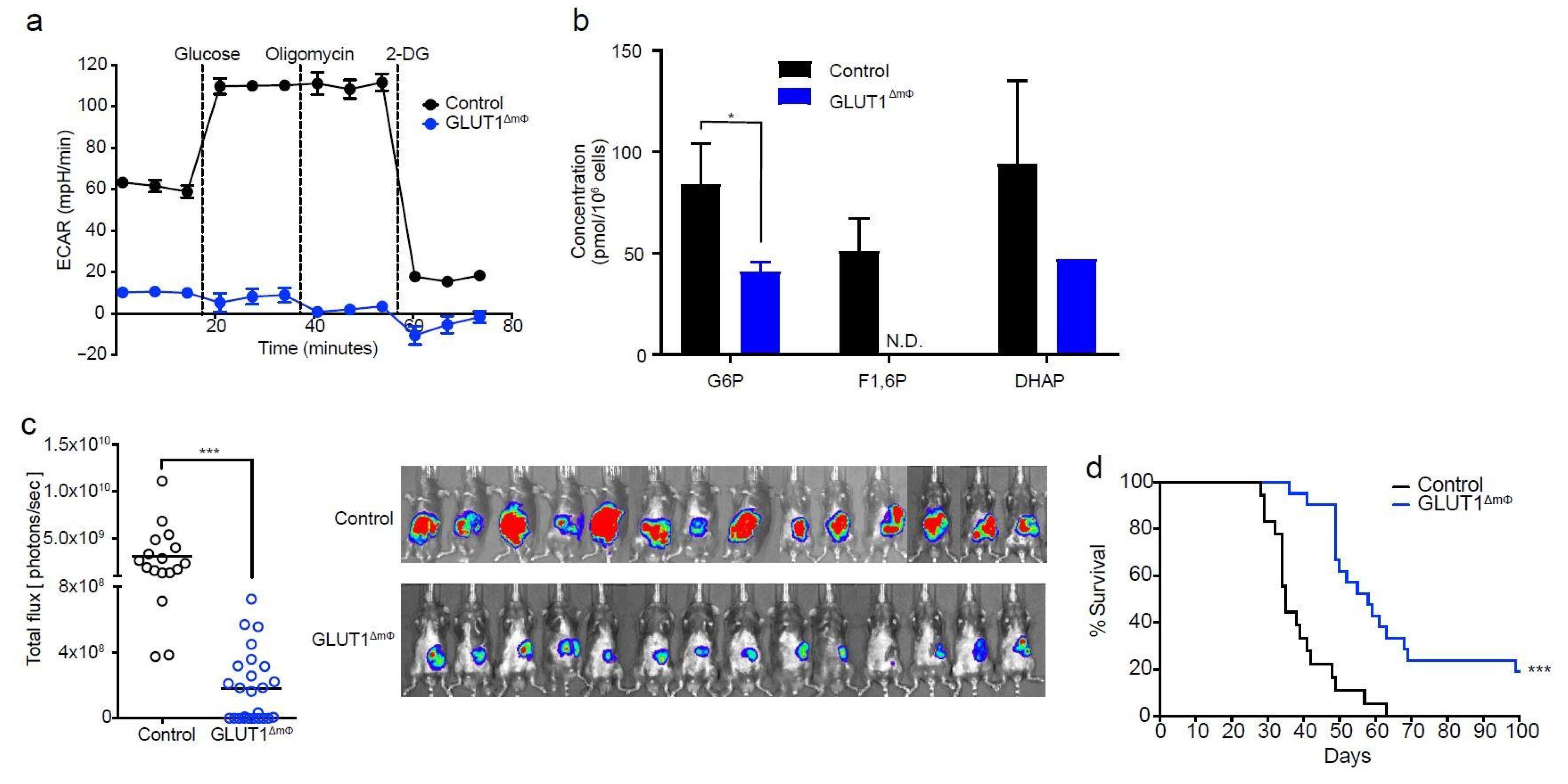
2.2. Macrophage-Specific Deletion of GLUT1 Confers Resistance to Tumor Growth
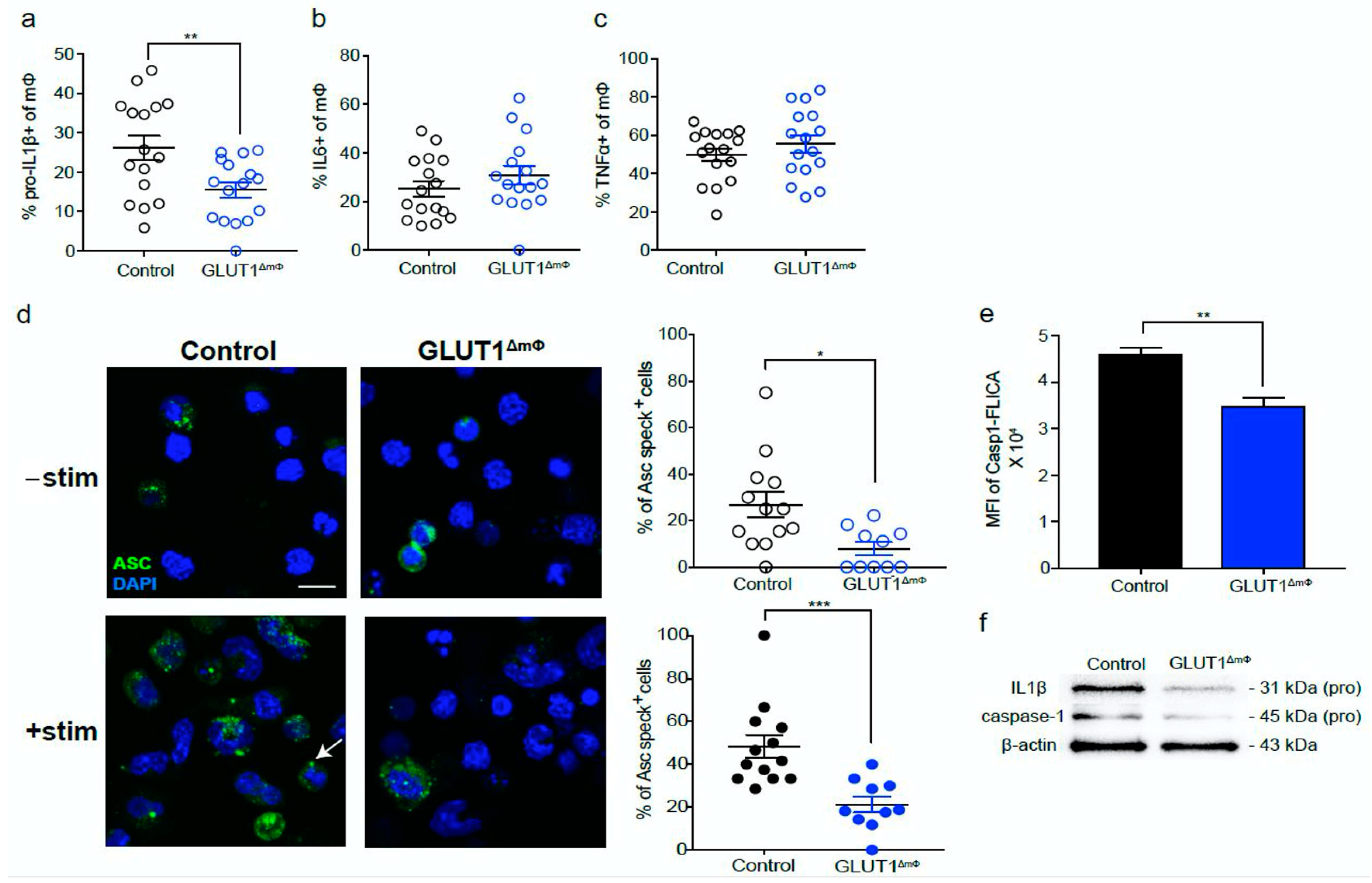
2.3. The pro-Inflammatory NLRP3-IL1β Axis Is Suppressed in GLUT1ΔmΦ Macrophages
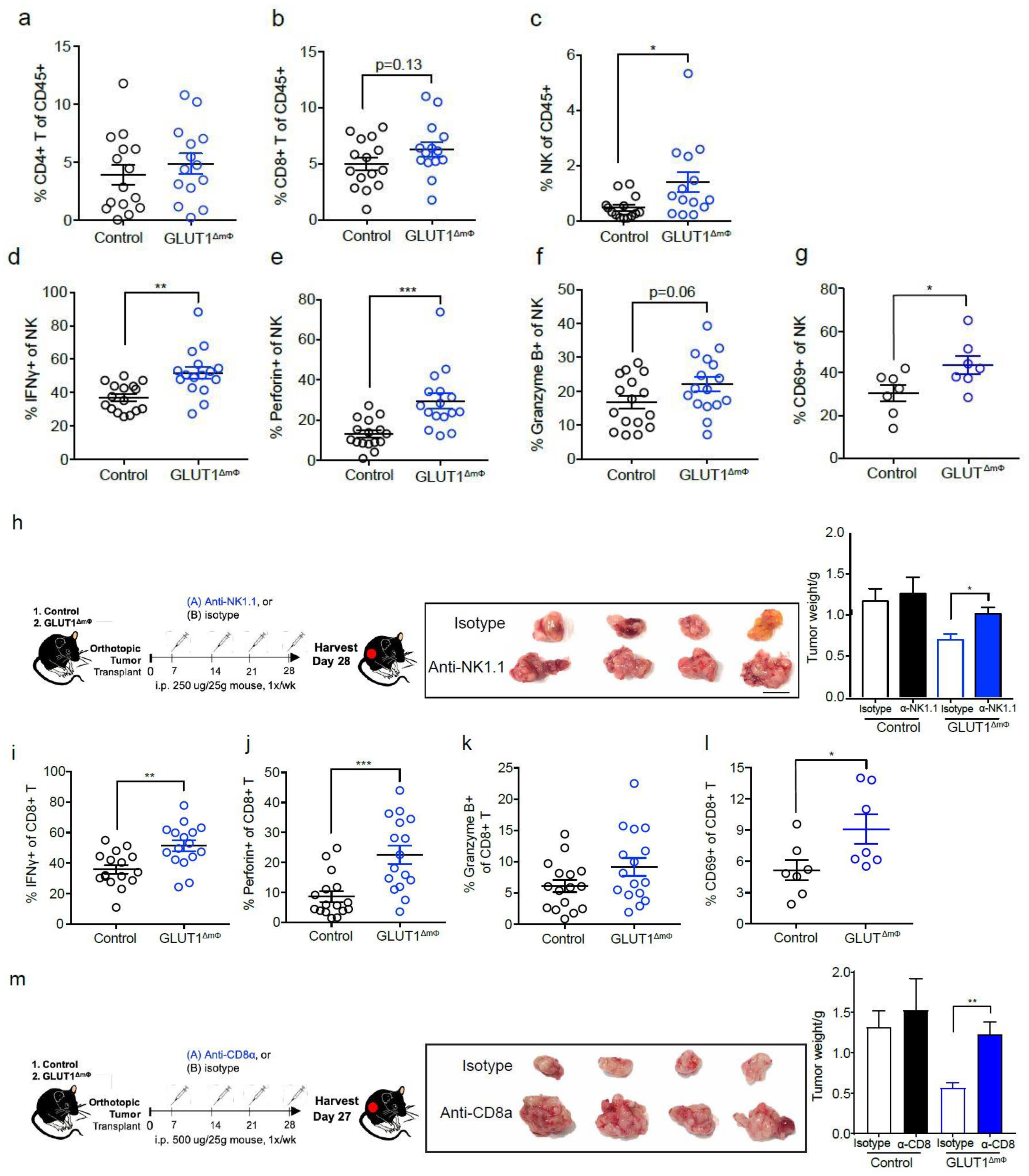
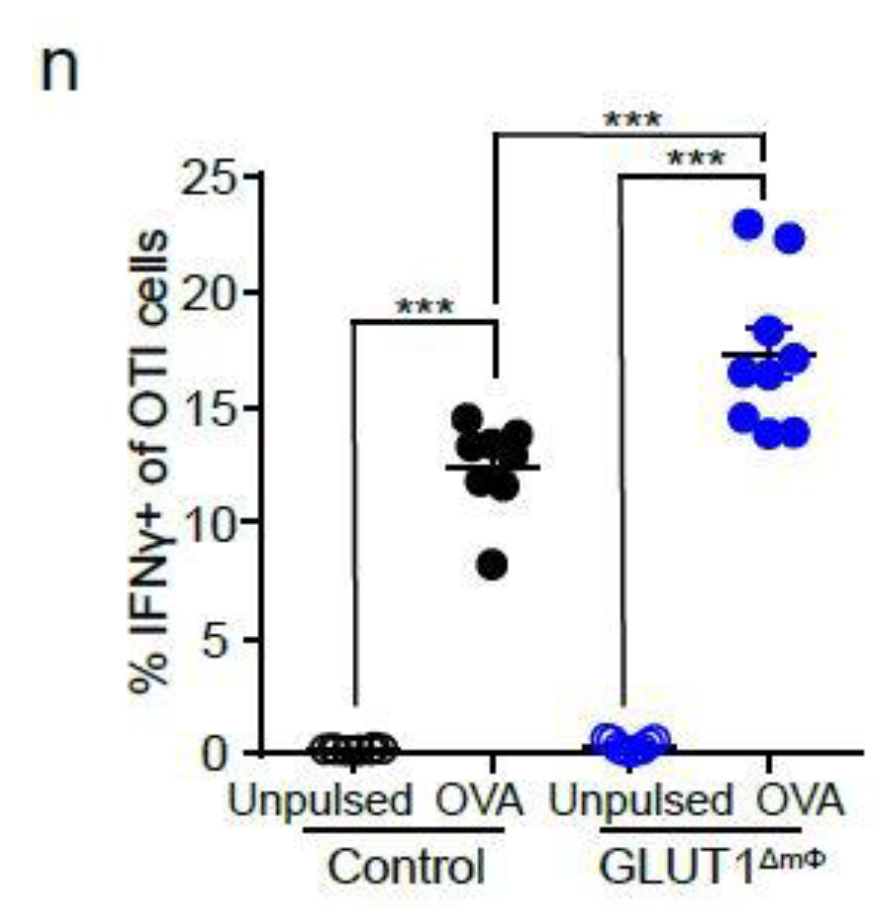
2.4. NK Cells and CTLs Mediate Anti-Tumor Immunity in GLUT1ΔmΦ Mice
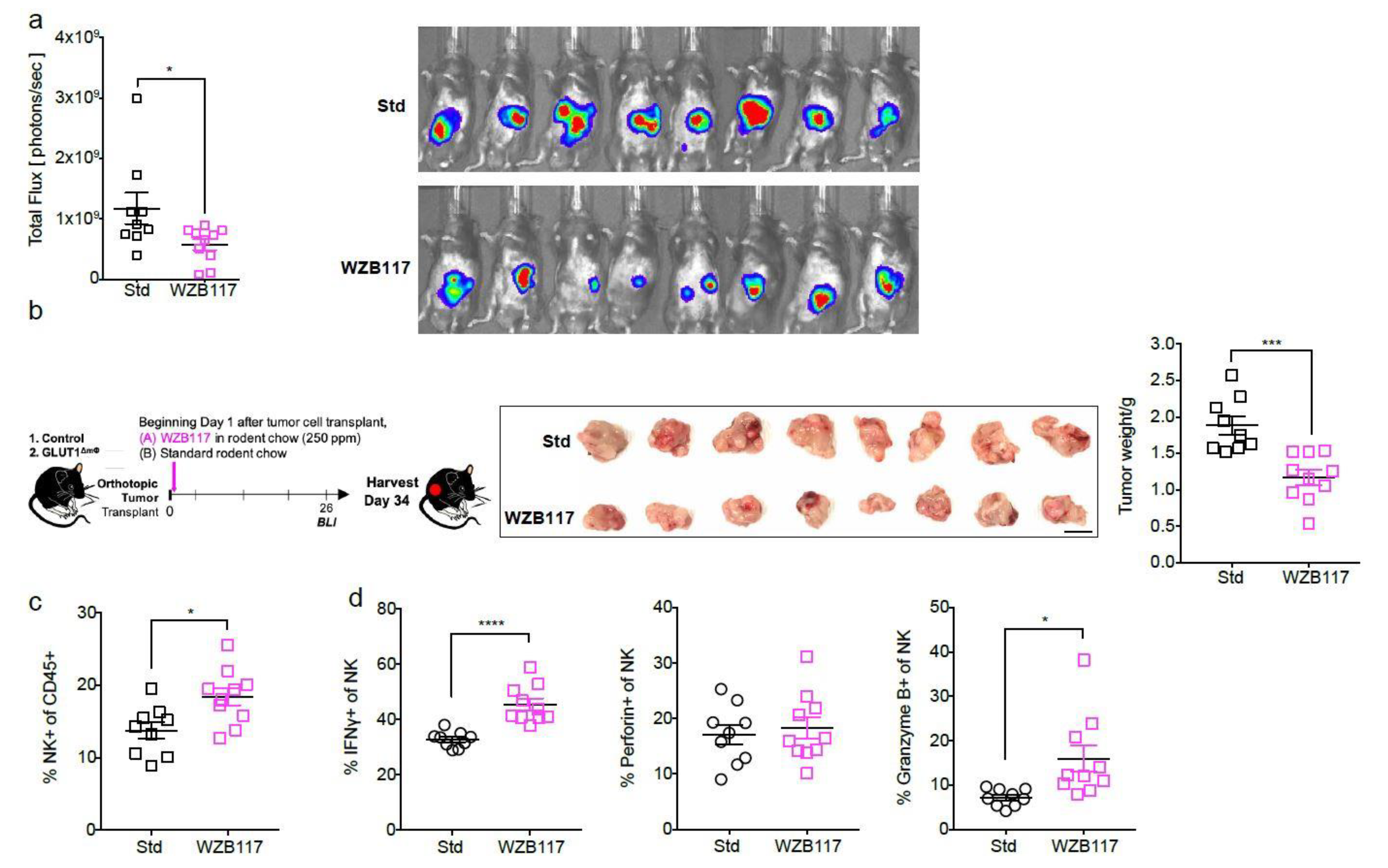
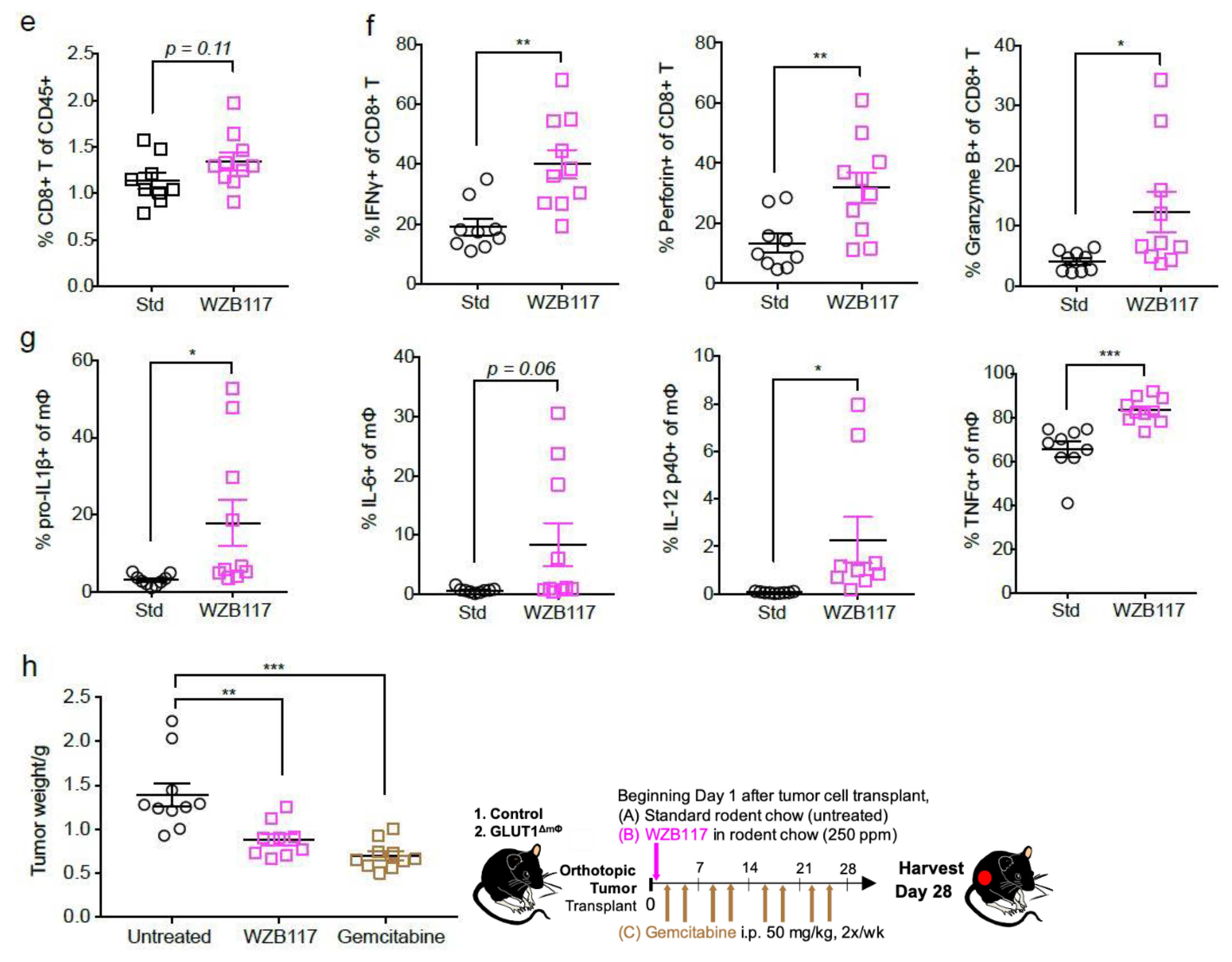
2.5. The GLUT1 Inhibitor WZB117 Attenuates Tumor Burden In Vivo
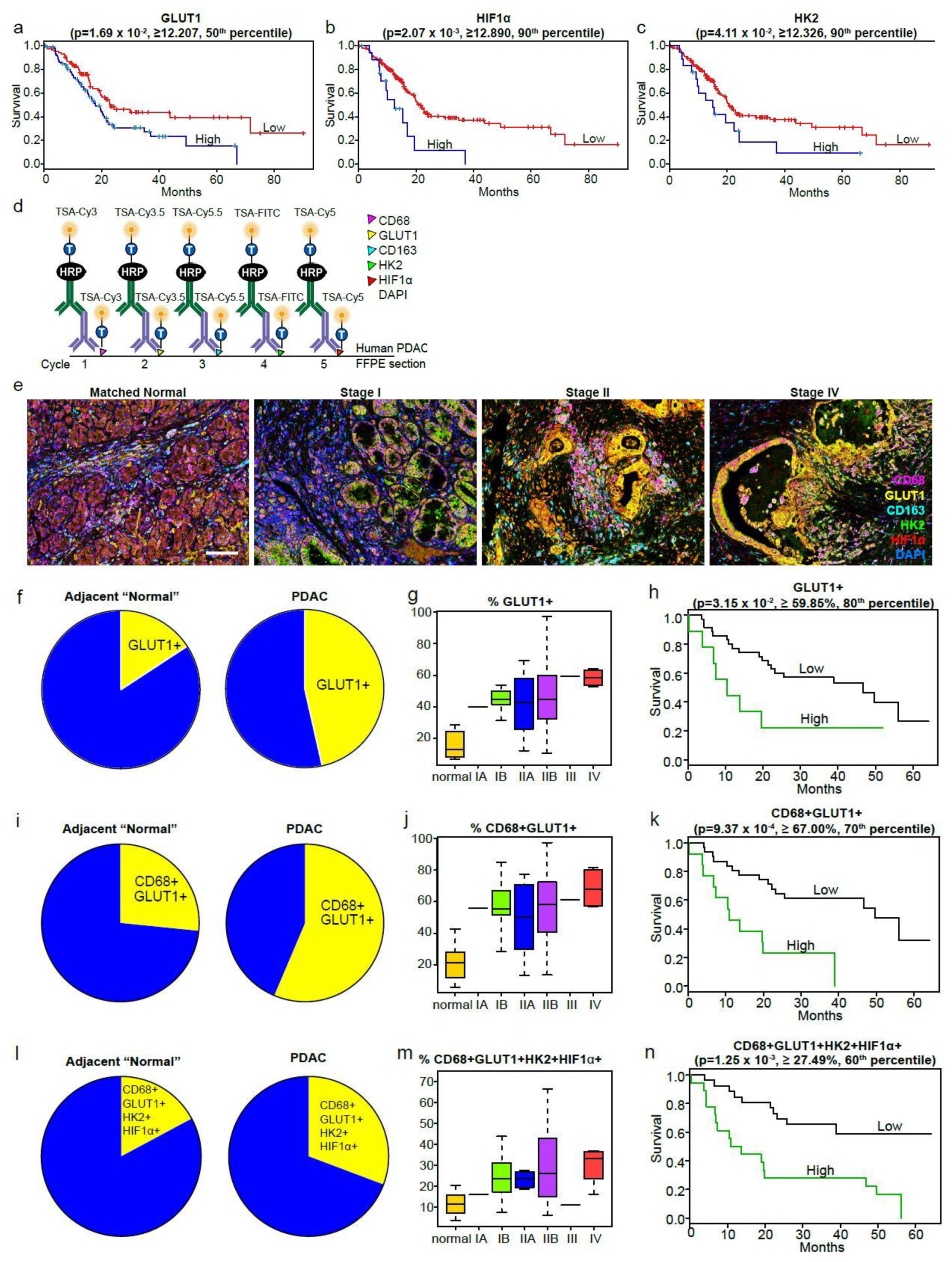
2.6. The Macrophage Glycolytic Signature Predicts Poor Survival in Human PDAC
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. Generation of Luciferase STable 1242L Cell Line
4.3. Generation of Orthotopic Pancreatic tumors
4.4. In Vivo Bioluminescent Imaging (BLI)
4.5. Tissue Dissociation
4.6. Flow Cytometry
4.7. Seahorse Live Metabolic Flux Assay
4.8. Metabolomics Analysis
4.9. Western Blot
4.10. RT-qPCR
4.11. Immunofluorescence of ASC
4.12. In Vitro OTI T Cells Stimulation
4.13. Publicly Available PDAC Data (TCGA and QCMG Cohorts)
4.14. Multiplex Quantitative Immunofluorescence (MQIF) Staining and Analysis
4.15. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zambirinis, C.; Miller, G. Cancer manipulation of host physiology: Lessons from pancreatic cancer. Trends Mol. Med. 2017, 23, 465–481. [Google Scholar] [CrossRef] [Green Version]
- Mielgo, A.; Schmid, M.C. Impact of tumour associated macrophages in pancreatic cancer. BMB Rep. 2013, 46, 131–138. [Google Scholar] [CrossRef] [Green Version]
- Greer, J.B.; Whitcomb, D.C. Inflammation and pancreatic cancer: An evidence-based review. Curr. Opin. Pharmacol. 2009, 9, 411–418. [Google Scholar] [CrossRef]
- Yadav, D.; Lowenfels, A.B. The epidemiology of pancreatitis and pancreatic cancer. Gastroenterology 2013, 144, 1252–1261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerra, C.; Schuhmacher, A.J.; Cañamero, M.; Grippo, P.J.; Verdaguer, L.; Pérez-Gallego, L.; Dubus, P.; Sandgren, E.P.; Barbacid, M. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by k-ras oncogenes in adult mice. Cancer Cell 2007, 11, 291–302. [Google Scholar] [CrossRef] [Green Version]
- Evans, A.; Costello, E. The role of inflammatory cells in fostering pancreatic cancer cell growth and invasion. Front. Physiol. 2012, 3, 270. [Google Scholar] [PubMed] [Green Version]
- Cantor, J.R.; Sabatini, D.M. Cancer cell metabolism: One hallmark, many faces. Cancer Discov. 2012, 2, 881–898. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelloff, G.J.; Hoffman, J.M.; Johnson, B.; Scher, H.I.; Siegel, B.A.; Cheng, E.Y.; Cheson, B.D.; O’Shaughnessy, J.; Guyton, K.Z.; Mankoff, D.A.; et al. Progress and promise of FDG-PET imaging for cancer patient management and oncologic drug development. Clin. Cancer Res. 2005, 11, 2785–2808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gambhir, S.S. Molecular imaging of cancer with positron emission tomography. Nat. Rev. Cancer 2002, 2, 683–693. [Google Scholar] [CrossRef]
- O’Neill, L.A.J.; Kishton, R.J.; Rathmell, R.J.K.J. A guide to immunometabolism for immunologists. Nat. Rev. Immunol. 2016, 16, 553–565. [Google Scholar] [CrossRef] [Green Version]
- Pearce, E.L.; Pearce, E.J. Metabolic pathways in immune cell activation and quiescence. Immunity 2013, 38, 633–643. [Google Scholar] [CrossRef] [Green Version]
- Ryan, D.G.; O’Neill, L.A. Krebs cycle reborn in macrophage immunometabolism. Annu. Rev. Immunol. 2020, 38, 289–313. [Google Scholar] [CrossRef] [Green Version]
- Bossche, J.V.D.; O’Neill, L.A.; Menon, D. Macrophage Immunometabolism: Where are we (going)? Trends Immunol. 2017, 38, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Caputa, G.; Flachsmann, L.J.; Cameron, A.M. Macrophage metabolism: A wound-healing perspective. Immunol. Cell Biol. 2019, 97, 268–278. [Google Scholar] [CrossRef] [PubMed]
- Groh, L.; Keating, S.T.; Joosten, L.A.B.; Netea, M.G.; Riksen, N.P. Monocyte and macrophage immunometabolism in atherosclerosis. Semin. Immunopathol. 2018, 40, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-McDermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate is an inflammatory signal that induces IL-1[bgr] through HIF-1[agr]. Nature 2013, 496, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, J.A.; Vairo, G.; Lingelbach, S.R. CSF-1 stimulates glucose uptake in murine bone marrow-derived macrophages. Biochem. Biophys. Res. Commun. 1986, 138, 445–454. [Google Scholar] [CrossRef]
- Freemerman, A.J.; Johnson, A.R.; Sacks, G.N.; Milner, J.J.; Kirk, E.L.; Troester, M.A.; Macintyre, A.N. Goraksha-Hicks, P.; Rathmell, J.C.; Makowski, L. Metabolic reprogramming of macrophages: Glucose transporter 1 (glut1)-mediated glucose metabolism drives a proinflammatory phenotypE. J. Biol. Chem. 2014, 289, 7884–7896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michl, J.; Ohlbaum, D.J.; Silverstein, S.C. 2-Deoxyglucose selectively inhibits Fc and complement receptor-mediated phagocytosis in mouse peritoneal macrophages. I. Description of the inhibitory effect. J. Exp. Med. 1976, 144, 1465–1483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Y.; Maianu, L.; Melbert, B.R.; Garvey, W.T. Facilitative glucose transporter gene expression in human lymphocytes, monocytes, and macrophages: A role for GLUT isoforms 1, 3, and 5 in the immune response and foam cell formation. Blood Cells Mol. Dis. 2004, 32, 182–190. [Google Scholar] [CrossRef]
- Fukuzumi, M.; Shinomiya, H.; Shimizu, Y.; Ohishi, K.; Utsumi, S. Endotoxin-induced enhancement of glucose influx into murine peritoneal macrophages via GLUT. Infect. Immun. 1996, 64, 108–112. [Google Scholar] [CrossRef] [Green Version]
- Bentley, J.; Itchayanan, D.; Barnes, K.; McIntosh, E.; Tang, X.; Downes, C.; Holman, G.; Whetton, A.D.; Owen-Lynch, P.J.; Baldwin, S.A. Interleukin-3-mediated cell survival signals include phosphatidylinositol 3-kinase-dependent translocation of the glucose transporter GLUT1 to the cell surface. J. Biol. Chem. 2003, 278, 39337–39348. [Google Scholar] [CrossRef] [Green Version]
- Buller, C.L.; Loberg, R.D.; Fan, M.H.; Zhu, Q.; Park, J.L.; Vesely, E.; Inoki, K.; Brosius FC 3rd Guan, K.L. A GSK-3/TSC2/mTOR pathway regulates glucose uptake and GLUT1 glucose transporter expression. Am. J. Physiol. Cell Physiol. 2008, 295, 836–843. [Google Scholar] [CrossRef] [Green Version]
- Michalek, R.D.; Gerriets, V.A.; Jacobs, S.R.; Macintyre, A.; MacIver, N.; Mason, E.F.; Sullivan, S.A.; Nichols, A.G.; Rathmell, J.C. Cutting edge: Distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J. Immunol. 2011, 186, 3299–3303. [Google Scholar] [CrossRef] [Green Version]
- Penny, H.L.; Sieow, J.L.; Adriani, G.; Yeap, W.H.; Ee, P.S.C.; Luis, B.S.; Lee, B.; Lee, T.; Mak, S.Y.; Ho, Y.S.; et al. Warburg metabolism in tumor-conditioned macrophages promotes metastasis in human pancreatic ductal adenocarcinoma. OncoImmunology 2016, 5, e1191731. [Google Scholar] [CrossRef] [PubMed]
- Zambirinis, C.; Levie, E.; Nguy, S.; Avanzi, A.; Barilla, R.; Xu, Y.; Seifert, L.; Daley, D.; Greco, S.H.; Deutsch, M.; et al. TLR9 ligation in pancreatic stellate cells promotes tumorigenesis. J. Exp. Med. 2015, 212, 2077–2094. [Google Scholar] [CrossRef] [PubMed]
- Pyonteck, S.M.; Akkari, L.; Schuhmacher, A.J.; Bowman, R.L.; Sevenich, L.; Quail, D.F.; Olson, O.; Quick, M.L.; Huse, J.T.; Teijeiro, V.; et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 2013, 19, 1264–1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeNardo, D.G.; Brennan, D.J.; Rexhepaj, E.; Ruffell, B.; Shiao, S.L.; Madden, S.F.; Gallagher, W.M.; Wadhwani, N.; Keil, S.D.; Junaid, S.A.; et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov. 2011, 1, 54–67. [Google Scholar] [CrossRef] [Green Version]
- Daley, D.; Mani, V.R.; Mohan, N.; Akkad, N.; Pandian, G.S.B.; Savadkar, S.; Lee, K.B.; Torres-Hernandez, A.; Aykut, B.; Diskin, B.; et al. NLRP3 signaling drives macrophage-induced adaptive immune suppression in pancreatic carcinoma. J. Exp. Med. 2017, 214, 1711–1724. [Google Scholar] [CrossRef] [Green Version]
- Hruz, P.W.; Mueckler, M.M. Structural analysis of the GLUT1 facilitative glucose transporter (review). Mol. Membr. Biol. 2001, 18, 183–193. [Google Scholar] [CrossRef]
- Kunkel, M.; Reichert, T.E.; Benz, P.; Lehr, H.-A.; Jeong, J.; Wieand, S.; Bartenstein, P.; Wagner, W.; Whiteside, T.L. Overexpression of Glut-1 and increased glucose metabolism in tumors are associated with a poor prognosis in patients with oral squamous cell carcinoma. Cancer 2003, 97, 1015–1024. [Google Scholar] [CrossRef] [PubMed]
- Manolescu, A.R.; Witkowska, K.; Kinnaird, A.; Cessford, T.; Cheeseman, C. Facilitated hexose transporters: New perspectives on form and function. Physiology 2007, 22, 234–240. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.Q.; Keating, A.F. Functional properties and genomics of glucose transporters. Curr. Genom. 2007, 8, 113–128. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, W.; Cao, Y.; Liu, Y.; Bergmeier, S.; Chen, X. Small compound inhibitors of basal glucose transport inhibit cell proliferation and induce apoptosis in cancer cells via glucose-deprivation-like mechanisms. Cancer Lett. 2010, 298, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Yip-Schneider, M.T.; Wu, H.; Stantz, K.; Agaram, N.; Crooks, P.; Schmidt, C.M. Dimethylaminoparthenolide and gemcitabine: A survival study using a genetically engineered mouse model of pancreatic cancer. BMC Cancer 2013, 13, 194. [Google Scholar] [CrossRef] [Green Version]
- Kolb, A.; Rieder, S.; Born, D.; Giese, N.A.; Giese, T.; Rudofsky, G.; Werner, J.; Büchler, M.W.; Friess, H.; Esposito, I.; et al. Glucagon/insulin ratio as a potential biomarker for pancreatic cancer in patients with new-onset diabetes mellitus. Cancer Biol. Ther. 2009, 8, 1527–1533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ying, H.; Zhang, H.; Coloff, J.L.; Yan, H.; Wang, W.; Chen, S.; Viale, A.; Zheng, H.; Paik, J.-H.; Lim, C.; et al. Abstract A101: Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Tumor Biol. 2012, 72, A101. [Google Scholar] [CrossRef]
- Munn, D.H. Expression of indoleamine 2,3-dioxygenase by plasmacytoid dendritic cells in tumor-draining lymph nodes. J. Clin. Investig. 2004, 114, 280–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kishton, R.J.; Barnes, C.E.; Nichols, A.G.; Cohen, S.; Gerriets, V.A.; Siska, P.J.; Macintyre, A.; Goraksha-Hicks, P.; de Cubas, A.A.; Liu, T.; et al. AMPK is essential to balance glycolysis and mitochondrial metabolism to control T-ALL cell stress and survival. Cell Metab. 2016, 23, 649–662. [Google Scholar] [CrossRef] [Green Version]
- Weinlich, G.; Murr, C.; Richardsen, L.; Winkler, C.; Fuchs, D. Decreased serum tryptophan concentration predicts poor prognosis in malignant melanoma patients. Dermatology 2006, 214, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Holmgaard, R.B.; Zamarin, D.; Munn, D.H.; Wolchok, J.D.; Allison, J.P. Indoleamine 2,3-dioxygenase is a critical resistance mechanism in antitumor T cell immunotherapy targeting CTLA-. J. Exp. Med. 2013, 210, 1389–1402. [Google Scholar] [CrossRef]
- Bhattacharya, N.; Yuan, R.; Prestwood, T.R.; Penny, H.L.; DiMaio, M.A.; Reticker-Flynn, N.; Krois, C.R.; Kenkel, J.A.; Pham, T.D.; Carmi, Y.; et al. Normalizing microbiota-induced retinoic acid deficiency stimulates protective CD8 + T Cell-mediated immunity in colorectal cancer. Immunity 2016, 45, 641–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penny, H.L.; Prestwood, T.R.; Bhattacharya, N.; Sun, F.; Kenkel, J.A.; Davidson, M.G.; Shen, L.; Zuniga, L.A.; Seeley, E.S.; Pai, R.; et al. Restoring retinoic acid attenuates intestinal inflammation and tumorigenesis in APCMin/+ Mice. Cancer Immunol. Res. 2016, 4, 917–926. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Prados, J.C. Substrate fate in activated macrophages: A comparison between innate, classic, and alternative acti-vation. J. Immunol. 2010, 185, 605–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palsson-McDermott, E.M.; Curtis, A.M.; Goel, G.; Lauterbach, M.A.; Sheedy, F.J.; Gleeson, L.E. Pyruvate kinase M2 regulates Hif-1α activity and IL-1β induction and is a critical de-terminant of the warburg effect in LPS-activated macrophages. Cell Metab. 2015, 21, 65–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, W.; Hu, H.; Chang, R.; Zhong, J.; Knabel, M.; O’Meally, R.; Cole, R.N.; Pandey, A.; Semenza, G.L. Pyruvate Kinase M2 Is a PHD3-stimulated coactivator for hypoxia-inducible factor. Cell 2011, 145, 732–744. [Google Scholar] [CrossRef] [Green Version]
- Shirai, T.; Nazarewicz, R.R.; Wallis, B.B.; Yanes, R.E.; Watanabe, R.; Hilhorst, M.; Tian, L.; Harrison, D.G.; Giacomini, J.C.; Assimes, T.L.; et al. The glycolytic enzyme PKM2 bridges metabolic and inflammatory dysfunction in coronary artery disease. J. Exp. Med. 2016, 213, 337–354. [Google Scholar] [CrossRef] [Green Version]
- Moon, J.-S.; Hisata, S.; Park, M.-A.; DeNicola, G.M.; Ryter, S.W.; Nakahira, K.; Choi, A.M. mTORC1-induced HK1-Dependent glycolysis regulates NLRP3 inflammasome activation. Cell Rep. 2015, 12, 102–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freemerman, A.J.; Zhao, L.; Pingili, A.K.; Teng, B.; Cozzo, A.J.; Fuller, A.M.; Johnson, A.R.; Milner, J.J.; Lim, M.F.; Galanko, J.A.; et al. MyeloidSlc2a1-deficient murine model revealed macrophage activation and metabolic phenotype are fueled by GLUT. J. Immunol. 2019, 202, 1265–1286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esteve, M.I.; Marselli, L.; Da Cunha, D.A.; Ladrière, L.; Ortis, F.; Grieco, F.A.; Dotta, F.; Weir, G.C.; Marchetti, P.; Eizirik, D.L.; et al. Palmitate induces a pro-inflammatory response in human pancreatic islets that mimics CCL2 expression by beta cells in type 2 diabetes. Diabetologia 2010, 53, 1395–1405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shoelson, S.E.; Lee, J.; Goldfine, A.B. Inflammation and insulin resistance. J. Clin. Investig. 2006, 116, 1793–1801. [Google Scholar] [CrossRef]
- Lusis, A.J. Atherosclerosis. Nature 2000, 407, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, K.L.H.; Van Der Veen, C.; Taylor, S.E.; Hardwick, S.J.; Clare, K.; Hegyi, L.; Mitchinson, M.J. Macrophages, lipid oxidation, ceroid accumulation and alpha-tocopherol depletion in human athero-sclerotic lesions. Gerontology 1995, 41, 53–67. [Google Scholar] [CrossRef] [PubMed]
- Freigang, S.; Ampenberger, F.; Weiss, A.; Kanneganti, T.D.; Iwakura, Y.; Hersberger, M.; Kopf, M. Fatty acid-induced mitochondrial uncoupling elicits inflammasome-independent IL-1alpha and sterile vas-cular inflammation in atherosclerosis. Nat. Immunol. 2013, 14, 1045–1053. [Google Scholar] [CrossRef]
- Wallace, C.; Keast, D. Glutamine and macrophage function. Metabolism 1992, 41, 1016–1020. [Google Scholar] [CrossRef]
- Yassad, A.; Lavoinne, A.; Bion, A.; Daveau, M.; Husson, A. Glutamine accelerates interleukin-6 production by rat peritoneal macrophages in culture. FEBS Lett. 1997, 413, 81–84. [Google Scholar] [CrossRef] [Green Version]
- Liang, M.; Wang, X.; Yuan, Y.; Zhou, Q.; Tong, C.; Jiang, W. Different effect of glutamine on macrophage tumor necrosis factor-alpha release and heat shock protein 72 expression in vitro and in vivo. Acta Biochim. Biophys. Sin. 2009, 41, 171–177. [Google Scholar] [CrossRef]
- Murphy, C.; Newsholme, P. Macrophage-mediated lysis of a beta-cell line, tumour necrosis factor-alpha release from bacillus Calmette-Guérin (BCG)-activated murine macrophages and interleukin-8 release from human monocytes are dependent on extracellular glutamine concentration and glutamine metabolism. Clin. Sci. 1999, 96, 89–97. [Google Scholar]
- Palmieri, E.M.; Menga, A.; Martín-Pérez, R.; Quinto, A.; Riera-Domingo, C.; De Tullio, G.; Hooper, D.C.; Lamers, W.H.; Ghesquière, B.; McVicar, D.W.; et al. Pharmacologic or genetic targeting of glutamine synthetase skews macrophages toward an m1-like phenotype and inhibits tumor metastasis. Cell Rep. 2017, 20, 1654–1666. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.-H.; Qiu, J.; O’Sullivan, D.; Buck, M.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; Van Der Windt, G.J.; et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Dillon, C.P.; Shi, L.Z.; Milasta, S.; Carter, R.; Finkelstein, D.; McCormick, L.L.; Fitzgerald, P.; Chi, H.; Munger, J.; et al. The transcription factor myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 2011, 35, 871–882. [Google Scholar] [CrossRef] [Green Version]
- Macintyre, A.; Gerriets, V.A.; Nichols, A.G.; Michalek, R.D.; Rudolph, M.C.; DeOliveira, D.; Anderson, S.M.; Abel, E.D.; Chen, B.J.; Hale, L.P.; et al. The glucose transporter glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab. 2014, 20, 61–72. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Cao, Y.; Zhang, W.; Bergmeier, S.; Qian, Y.; Akbar, H.; Colvin, R.; Ding, J.; Tong, L.; Wu, S.; et al. A small-molecule inhibitor of glucose transporter 1 downregulates glycolysis, induces cell-cycle arrest, and inhibits cancer cell growth in vitro and in vivo. Mol. Cancer Ther. 2012, 11, 1672–1682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogawa, H.; Nagano, H.; Konno, M.; Eguchi, H.; Koseki, J.; Kawamoto, K.; Nishida, N.; Colvin, H.; Tomokuni, A.; Tomimaru, Y.; et al. The combination of the expression of hexokinase 2 and pyruvate kinase M2 is a prognostic marker in pa-tients with pancreatic cancer. Mol. Clin. Oncol. 2015, 3, 563–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, A.C.; Mori, R.; Vallbohmer, D.; Brabender, J.; Klein, E.; Drebber, U.; Baldus, S.E.; Cooc, J.; Azuma, M.; Metzger, R.; et al. High expression of HIF1a is a predictor of clinical outcome in patients with pancreatic ductal ade-nocarcinomas and correlated to PDGFA, VEGF, and bFGF. Neoplasia 2008, 10, 674–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basturk, O.; Singh, R.; Kaygusuz, E.; Balci, S.; Dursun, N.; Culhaci, N.; Adsay, N.V. GLUT-1 expression in pancreatic neoplasia: Implications in pathogenesis, diagnosis, and prognosis. Pancreas 2011, 40, 187–192. [Google Scholar] [CrossRef] [Green Version]
- Sharen, G.; Peng, Y.; Cheng, H.; Liu, Y.; Shi, Y.; Zhao, J. Prognostic value of GLUT-1 expression in pancreatic cancer: Results from 538 patients. Oncotarget 2017, 8, 19760–19767. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, D.-T.; Liu, X.G. mTOR signaling in T cell immunity and autoimmunity. Int. Rev. Immunol. 2014, 34, 50–66. [Google Scholar] [CrossRef]
- Weichhart, T.; Hengstschläger, M.; Linke, M. Regulation of innate immune cell function by mTOR. Nat. Rev. Immunol. 2015, 15, 599–614. [Google Scholar] [CrossRef]
- O’Neill, L.A.J.; Hardie, D.G. Metabolism of inflammation limited by AMPK and pseudo-starvation. Nat. Cell Biol. 2013, 493, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Wang, X.; Chen, X. Inhibitors of glucose transport and glycolysis as novel anticancer therapeutics. World J. Transl. Med. 2014, 3, 37–57. [Google Scholar] [CrossRef]
- Heiden, M.G.V. Targeting cancer metabolism: A therapeutic window opens. Nat. Rev. Drug Discov. 2011, 10, 671–684. [Google Scholar] [CrossRef] [Green Version]
- Yin, Y.; Choi, S.-C.; Xu, Z.; Perry, D.; Seay, H.; Croker, B.P.; Sobel, E.S.; Brusko, T.M.; Morel, L. Normalization of CD4 + T cell metabolism reverses lupus. Sci. Transl. Med. 2015, 7, 274ra18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.-F.; Lo, Y.-C.; Cheng, C.-H.; Furtmüller, G.J.; Oh, B.; Andrade-Oliveira, V.; Thomas, A.G.; Bowman, C.E.; Slusher, B.S.; Wolfgang, M.J.; et al. Preventing allograft rejection by targeting immune metabolism. Cell Rep. 2015, 13, 760–770. [Google Scholar] [CrossRef] [Green Version]
- Ohashi, Y.; Hirayama, A.; Ishikawa, T.; Nakamura, S.; Shimizu, K.; Ueno, Y.; Tomita, M.; Soga, T. Depiction of metabolome changes in histidine-starved Escherichia coli by CE-TOFMS. Mol. Biosyst. 2008, 4, 135–147. [Google Scholar] [CrossRef]
- Ooga, T.; Sato, H.; Nagashima, A.; Sasaki, K.; Tomita, M.; Soga, T.; Ohashi, Y. Metabolomic anatomy of an animal model revealing homeostatic imbalances in dyslipidaemia. Mol. BioSyst. 2011, 7, 1217–1223. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, M.; Wong, D.T.; Hirayama, A.; Soga, T.; Tomita, M. Capillary electrophoresis mass spectrometry-based saliva metabolomics identified oral, breast and pancreatic cancer-specific profiles. Metabolomics 2009, 6, 78–95. [Google Scholar] [CrossRef] [Green Version]
- Junker, B.H.; Klukas, C.; Schreiber, F. VANTED: A system for advanced data analysis and visualization in the context of biological networks. BMC Bioinform. 2006, 7, 109. [Google Scholar] [CrossRef] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Penny, H.L.; Sieow, J.L.; Gun, S.Y.; Lau, M.C.; Lee, B.; Tan, J.; Phua, C.; Toh, F.; Nga, Y.; Yeap, W.H.; et al. Targeting Glycolysis in Macrophages Confers Protection Against Pancreatic Ductal Adenocarcinoma. Int. J. Mol. Sci. 2021, 22, 6350. https://doi.org/10.3390/ijms22126350
Penny HL, Sieow JL, Gun SY, Lau MC, Lee B, Tan J, Phua C, Toh F, Nga Y, Yeap WH, et al. Targeting Glycolysis in Macrophages Confers Protection Against Pancreatic Ductal Adenocarcinoma. International Journal of Molecular Sciences. 2021; 22(12):6350. https://doi.org/10.3390/ijms22126350
Chicago/Turabian StylePenny, Hweixian Leong, Je Lin Sieow, Sin Yee Gun, Mai Chan Lau, Bernett Lee, Jasmine Tan, Cindy Phua, Florida Toh, Yvonne Nga, Wei Hseun Yeap, and et al. 2021. "Targeting Glycolysis in Macrophages Confers Protection Against Pancreatic Ductal Adenocarcinoma" International Journal of Molecular Sciences 22, no. 12: 6350. https://doi.org/10.3390/ijms22126350