
Spatially Enriched Paralog Rearrangements Argue Functionally Diverse Ribosomes Arise during Cold Acclimation in Arabidopsis

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Results
2.1. Early Temperature Acclimation Effects on Plant Growth
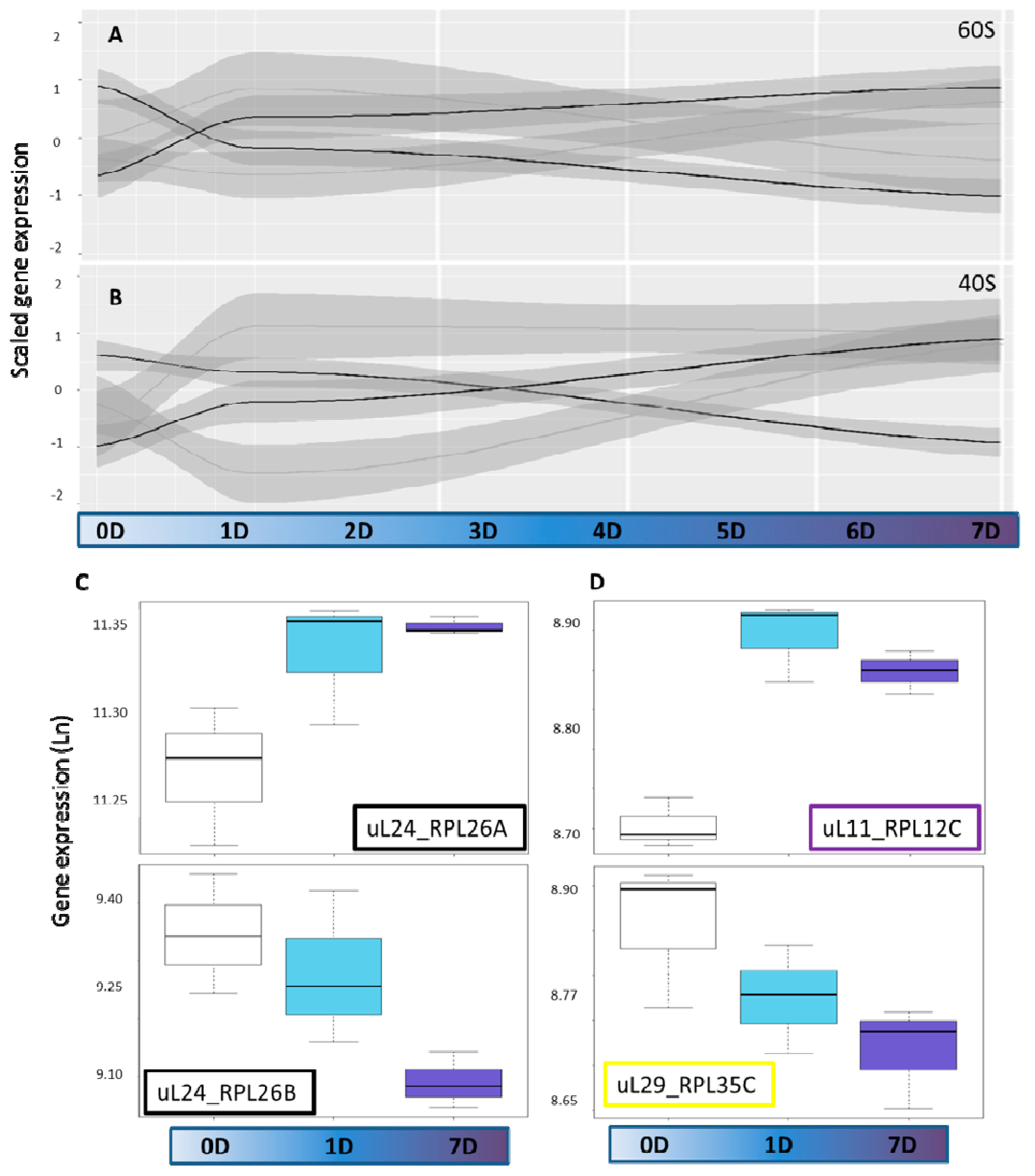
2.2. Cytosolic Ribosomal Transcriptome Reprogramming
2.3. Cytosolic Ribosomal Proteome Reprogramming
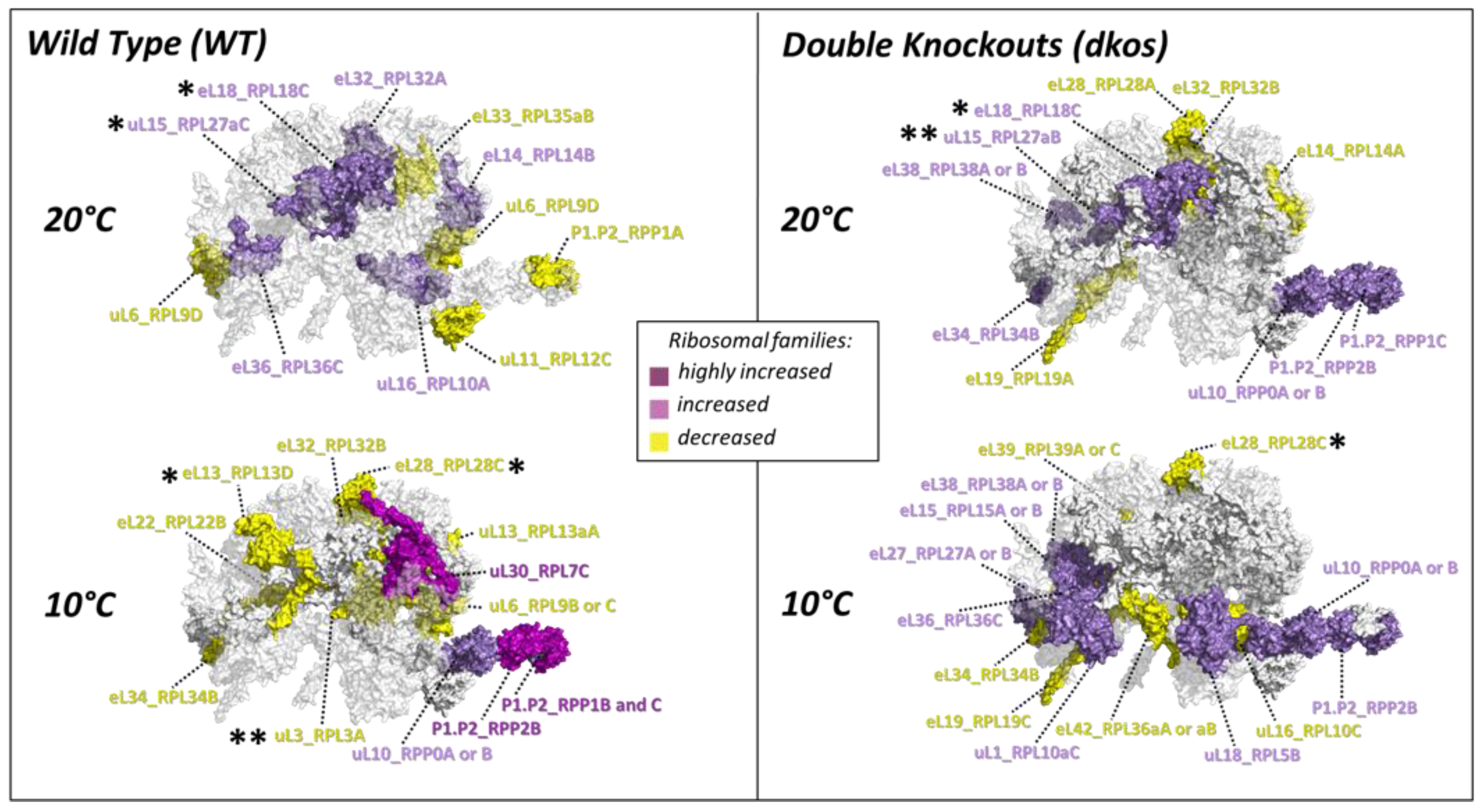
2.4. Substoichiometry in Non-Translating Versus Translating Ribosome Complexes
2.5. Cold-Induced Changes in Active Translating Polysomes
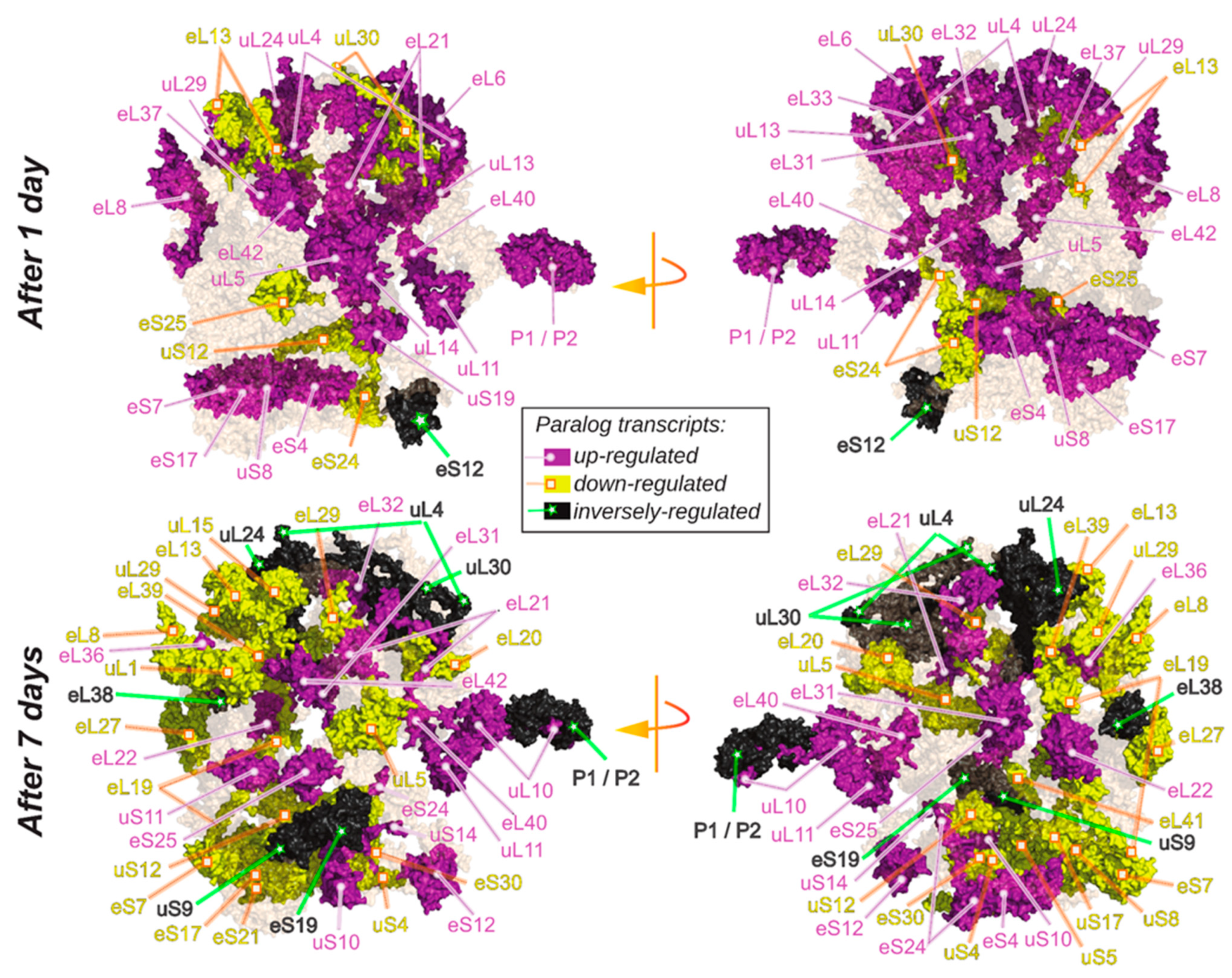
2.6. Spatially Constrained Cold-Triggered Ribosome Heterogeneity
2.7. Paralog Specific Cold Responses—uL30 Family
3. Discussion
3.1. Different Types of RAP Transcripts Mediate the Initial and Long-Term Responses to Temperature Acclimation
3.2. Cold-Triggered Reprogramming Indicates That Spatial Constraints Adjust the Ribosomal Proteome
3.3. Cold Ribosomal Protein Changes during Early Biogenesis
3.4. Cold Dynamics of Ribosomal Protein Assembly
3.5. Cold-Induced Ribosomal Protein Substoichiometry Co-Localizes With Rei1 Binding Site
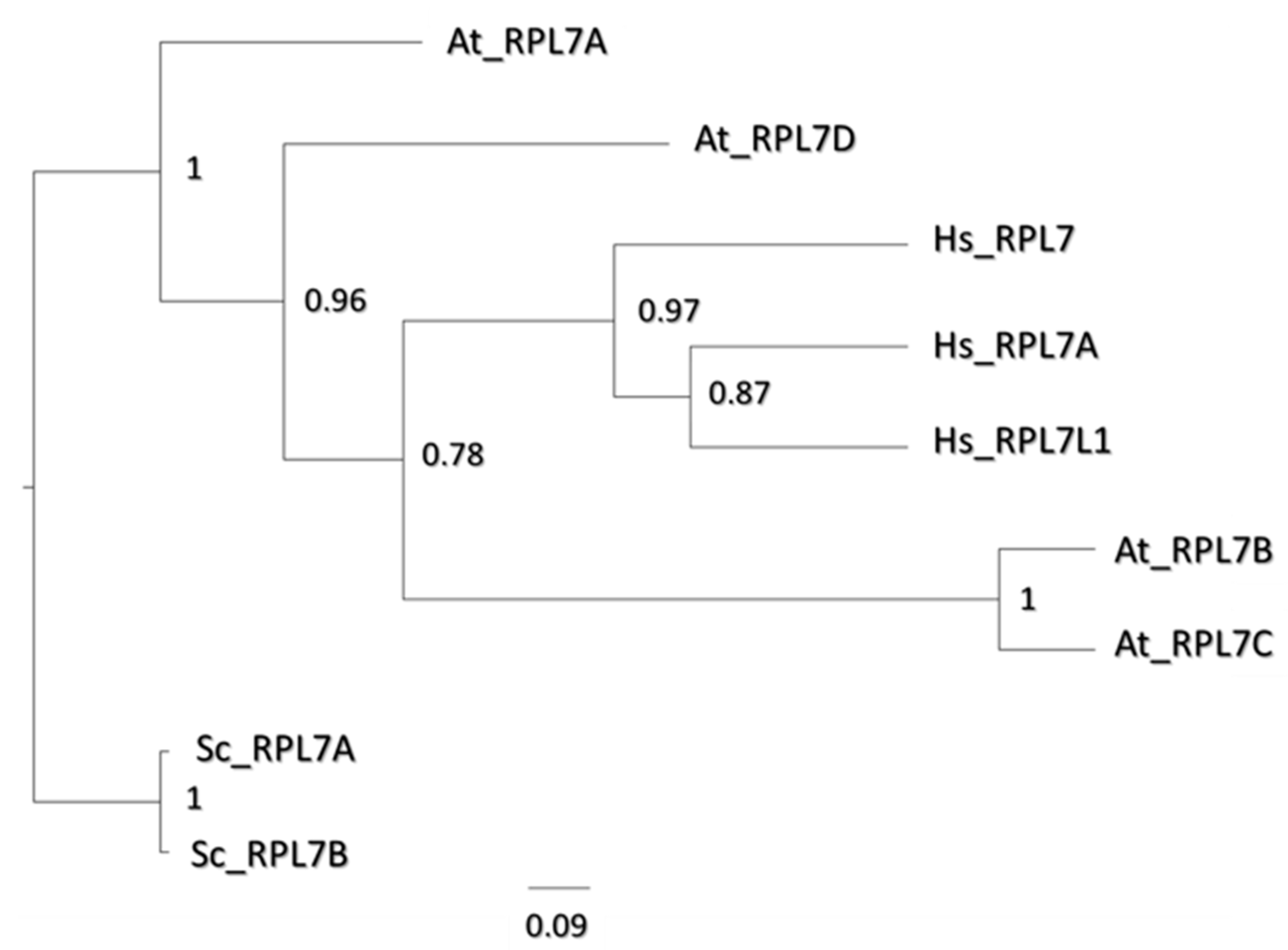
3.6. Cold Dynamics of uL30 Paralogs Could Orchestrate Spatially Constrained Rearrangements in Ribosomes
3.7. REIL Concomitant Ribosome Reprogramming Argues Potential Specialization
4. Materials and Methods
4.1. Plant Material
4.2. Growth Conditions
4.3. Dry Weight Measurements
4.4. Microarray-Based Transcriptome Analysis
4.5. Transcriptome Data Analyses
4.6. Cytosolic Ribosomal Proteome Preparation
4.7. Proteome Analysis by Liquid Chromatography—Tandem Mass Spectrometry (LC-MS/MS)
4.8. Proteome Data Analyses
4.9. Structural Analysis of Changes in Ribosome Protein or Transcript Abundance
4.10. Sequence Alignments
4.11. Software
- Ben Bolstad (2019). preprocessCore: A collection of pre-processing functions. R package version 1.46.0. Available online: https://github.com/bmbolstad/preprocessCore accessed on 30 April 2021.
- Ref. [164]. tidyverse: Easily Install and Load the ‘Tidyverse’. R package version 1.2.1.
- Ref. [165]. Reshaping Data with the reshape Package.
- Ref. [166]. stringr: Simple, Consistent Wrappers for Common String Operations. R package version 1.4.0.
- Ref. [133]. fitdistrplus: An R Package for Fitting Distributions.
- Ref. [167] ggplot2: Elegant Graphics for Data Analysis.
- Ref. [168] R package stringi: Character string processing facilities.
- Diethelm Wuertz, Tobias Setz and Yohan Chalabi (2017). timeSeries: Rmetrics—Financial Time Series Objects. R package version 3042.102. Available online: https://CRAN.R-project.org/package=timeSeries accessed on 30 April 2021.
- Ref. [169]. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics.
- Ref. [170]. Circlize implements and enhances circular visualization in R. Bioinformatics.
- Ref. [149] The igraph software package for complex network research.
- Ref. [171]. VennDiagram: Generate High-Resolution Venn and Euler Plots. R package version 1.6.20.
- Jan Graffelman (2013). calibrate: Calibration of Scatterplot and Biplot Axes. R package version 1.7.2. Available online: https://CRAN.R-project.org/package=calibrate accessed on 30 April 2021.
- Diethelm Wuertz, Tobias Setz and Yohan Chalabi (2017). fBasics: Rmetrics—Markets and Basic Statistics. R package version 3042.89. Available online: https://CRAN.R-project.org/package=fBasics accessed on 30 April 2021.
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Weis, B.L.; Kovacevic, J.; Missbach, S.; Schleiff, E. Plant-Specific Features of Ribosome Biogenesis. Trends Plant Sci. 2015, 20, 729–740. [Google Scholar] [CrossRef]
- Palm, D.; Streit, D.; Shanmugam, T.; Weis, B.L.; Ruprecht, M.; Simm, S.; Schleiff, E. Plant-specific ribosome biogenesis factors in Arabidopsis thaliana with essential function in rRNA processing. Nucleic Acids Res. 2019, 47, 1880–1895. [Google Scholar] [CrossRef] [Green Version]
- Sáez-Vásquez, J.; Delseny, M. Ribosome biogenesis in plants: From functional 45S ribosomal DNA organization to ribosome assembly factors. Plant Cell 2019, 31, 1945–1967. [Google Scholar] [CrossRef] [Green Version]
- Barakat, A.; Szick-Miranda, K.; Chang, I.F.; Guyot, R.; Blanc, G.; Cooke, R.; Delseny, M.; Bailey-Serres, J. The organization of cytoplasmic ribosomal protein genes in the Arabidopsis genome. Plant Physiol. 2001, 127, 398–415. [Google Scholar] [CrossRef]
- Delaunay, J.; Creusot, F.; Schapira, G. Evolution of ribosomal proteins. Eur. J. Biochem. 1973, 39, 305–312. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, X.; Paterson, A.H. Genome and gene duplications and gene expression divergence: A view from plants. Ann. N. Y. Acad. Sci. 2012, 1256, 1–14. [Google Scholar] [CrossRef]
- Braasch, I.; Bobe, J.; Guiguen, Y.; Postlethwait, J.H. Reply to: ‘Subfunctionalization versus neofunctionalization after whole-genome duplication’. Nat. Genet. 2018, 50, 910–911. [Google Scholar] [CrossRef]
- Gerst, J.E. Pimp My Ribosome: Ribosomal Protein Paralogs Specify Translational Control. Trends Genet. 2018, 34, 832–845. [Google Scholar] [CrossRef]
- Komili, S.; Farny, N.G.; Roth, F.P.; Silver, P.A. Functional Specificity among Ribosomal Proteins Regulates Gene Expression. Cell 2007, 131, 557–571. [Google Scholar] [CrossRef] [Green Version]
- Genuth, N.R.; Barna, M. Heterogeneity and specialized functions of translation machinery: From genes to organisms. Nat. Rev. Genet. 2018, 19, 431–452. [Google Scholar] [CrossRef]
- Bates, C.; Hubbard, S.J.; Ashe, M.P. Ribosomal flavours: An acquired taste for specific mRNAs? Biochem. Soc. Trans. 2018, 46, 1529–1539. [Google Scholar] [CrossRef]
- Shi, Z.; Fujii, K.; Kovary, K.M.; Genuth, N.R.; Röst, H.L.; Teruel, M.N.; Barna, M. Heterogeneous Ribosomes Preferentially Translate Distinct Subpools of mRNAs Genome-wide. Mol. Cell 2017, 67, 71–83.e7. [Google Scholar] [CrossRef] [Green Version]
- Hopes, T.; Agapiou, M.; Norris, K.; McCarthy, C.G.P.; O’Connell, M.J.; Fontana, J.; Aspden, J.L. Specialisation of ribosomes in gonads through paralog-switching. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Bailey-Serres, J. Selective translation of cytoplasmic mRNAs in plants. Trends Plant Sci. 1999, 4, 142–148. [Google Scholar] [CrossRef]
- Browning, K.S.; Bailey-Serres, J. Mechanism of Cytoplasmic mRNA Translation. Arab. Book 2015, 13, e0176. [Google Scholar] [CrossRef] [Green Version]
- Kawaguchi, R.; Girke, T.; Bray, E.A.; Bailey-Serres, J. Differential mRNA translation contributes to gene regulation under non-stress and dehydration stress conditions in Arabidopsis thaliana. Plant J. 2004, 38, 823–839. [Google Scholar] [CrossRef]
- Nicolaï, M.; Roncato, M.A.; Canoy, A.S.; Rouquié, D.; Sarda, X.; Freyssinet, G.; Robaglia, C. Large-scale analysis of mRNA translation states during sucrose starvation in Arabidopsis cells identifies cell proliferation and chromatin structure as targets of translational control. Plant Physiol. 2006, 141, 663–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moeller, J.R.; Moscou, M.J.; Bancroft, T.; Skadsen, R.W.; Wise, R.P.; Whitham, S.A. Differential accumulation of host mRNAs on polyribosomes during obligate pathogen-plant interactions. Mol. Biosyst. 2012, 8, 2153–2165. [Google Scholar] [CrossRef] [Green Version]
- Ueda, K.; Matsuura, H.; Yamaguchi, M.; Demura, T.; Kato, K. Genome-wide analyses of changes in translation state caused by elevated temperature in Oryza sativa. Plant Cell Physiol. 2012, 53, 1481–1491. [Google Scholar] [CrossRef] [Green Version]
- Matsuura, H.; Ishibashi, Y.; Shinmyo, A.; Kanaya, S.; Kato, K. Genome-wide analyses of early translational responses to elevated temperature and high salinity in arabidopsis thaliana. Plant Cell Physiol. 2010, 51, 448–462. [Google Scholar] [CrossRef]
- Branco-Price, C.; Kaiser, K.A.; Jang, C.J.H.; Larive, C.K.; Bailey-Serres, J. Selective mRNA translation coordinates energetic and metabolic adjustments to cellular oxygen deprivation and reoxygenation in Arabidopsis thaliana. Plant J. 2008, 56, 743–755. [Google Scholar] [CrossRef]
- Branco-Price, C.; Kawaguchi, R.; Ferreira, R.B.; Bailey-Serres, J. Genome-wide analysis of transcript abundance and translation in arabidopsis seedlings subjected to oxygen deprivation. Ann. Bot. 2005, 96, 647–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mustroph, A.; Zanetti, M.E.; Jang, C.J.H.; Holtan, H.E.; Repetti, P.P.; Galbraith, D.W.; Girke, T.; Bailey-Serres, J. Profiling translatomes of discrete cell populations resolves altered cellular priorities during hypoxia in Arabidopsis. Proc. Natl. Acad. Sci. USA 2009, 106, 18843–18848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juntawong, P.; Bailey-Serres, J. Dynamic light regulation of translation status in Arabidopsis thaliana. Front. Plant Sci. 2012, 3, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, S.H.; Liu, M.J.; Wu, S.H.; Chen, H.M. Widespread translational control contributes to the regulation of Arabidopsis photomorphogenesis. Mol. Syst. Biol. 2012, 8, 566. [Google Scholar] [CrossRef] [Green Version]
- Liberman, N.; O’Brown, Z.K.; Earl, A.S.; Boulias, K.; Gerashchenko, M.V.; Wang, S.Y.; Fritsche, C.; Fady, P.E.; Dong, A.; Gladyshev, V.N.; et al. N6-adenosine methylation of ribosomal RNA affects lipid oxidation and stress resistance. Sci. Adv. 2020, 6, eaaz4370. [Google Scholar] [CrossRef] [Green Version]
- Jha, S.; Rollins, M.G.; Fuchs, G.; Procter, D.J.; Hall, E.A.; Cozzolino, K.; Sarnow, P.; Savas, J.N.; Walsh, D. Trans-kingdom mimicry underlies ribosome customization by a poxvirus kinase. Nature 2017, 546, 651–655. [Google Scholar] [CrossRef]
- Thompson, M.K.; Rojas-Duran, M.F.; Gangaramani, P.; Gilbert, W.V. The ribosomal protein Asc1/RACK1 is required for efficient translation of short mRNAs. eLife 2016, 5, e11154. [Google Scholar] [CrossRef]
- Sormani, R.; Delannoy, E.; Lageix, S.; Bitton, F.; Lanet, E.; Saez-Vasquez, J.; Deragon, J.M.; Renou, J.P.; Robaglia, C. Sublethal cadmium intoxication in arabidopsis thaliana impacts translation at multiple levels. Plant Cell Physiol. 2011, 52, 436–447. [Google Scholar] [CrossRef] [Green Version]
- Simsek, D.; Tiu, G.C.; Flynn, R.A.; Byeon, G.W.; Leppek, K.; Xu, A.F.; Chang, H.Y.; Barna, M. The Mammalian Ribo-interactome Reveals Ribosome Functional Diversity and Heterogeneity. Cell 2017, 169, 1051–1065.e18. [Google Scholar] [CrossRef] [Green Version]
- Boex-Fontvieille, E.; Daventure, M.; Jossier, M.; Zivy, M.; Hodges, M.; Tcherkez, G. Photosynthetic Control of Arabidopsis Leaf Cytoplasmic Translation Initiation by Protein Phosphorylation. PLoS ONE 2013, 8, e70692. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Molina, A.; Kleine, T.; Schneider, K.; Mühlhaus, T.; Lehmann, M.; Leister, D. Translational Components Contribute to Acclimation Responses to High Light, Heat, and Cold in Arabidopsis. iScience 2020, 23, 101331. [Google Scholar] [CrossRef]
- Martinez-Seidel, F.; Beine-Golovchuk, O.; Hsieh, Y.C.; Kopka, J. Systematic review of plant ribosome heterogeneity and specialization. Front. Plant Sci. 2020, 11, 948. [Google Scholar] [CrossRef]
- Beine Golovchuk, O.; Firmino, A.A.P.; Dąbrowska, A.; Schmidt, S.; Erban, A.; Walther, D.; Zuther, E.; Hincha, D.K.; Kopka, J. Plant temperature acclimation and growth rely on cytosolic ribosome biogenesis factor homologs. Plant Physiol. 2018, 176, 2251–2276. [Google Scholar] [CrossRef] [Green Version]
- Calixto, C.P.G.; Guo, W.; James, A.B.; Tzioutziou, N.A.; Entizne, J.C.; Panter, P.E.; Knight, H.; Nimmo, H.G.; Zhang, R.; Brown, J.W.S. Rapid and dynamic alternative splicing impacts the arabidopsis cold response transcriptome. Plant Cell 2018, 30, 1424–1444. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Seidel, F.; Suwanchaikasem, P.; Nie, S.; Leeming, M.G.; Firmino, A.A.P.; Williamson, N.A.; Kopka, J.; Roessner, U.; Boughton, B.A. Membrane-Enriched Proteomics Link Ribosome Accumulation and Proteome Reprogramming With Cold Acclimation in Barley Root Meristems. Front. Plant Sci. 2021, 12. [Google Scholar] [CrossRef]
- Seki, M.; Narusaka, M.; Ishida, J.; Nanjo, T.; Fujita, M.; Oono, Y.; Kamiya, A.; Nakajima, M.; Enju, A.; Sakurai, T.; et al. Monitoring the expression profiles of 7000 Arabidopsis genes under drought, cold and high-salinity stresses using a full-length cDNA microarray. Plant J. 2002, 31, 279–292. [Google Scholar] [CrossRef]
- Cheong, B.E.; Beine-Golovchuk, O.; Gorka, M.; Ho, W.W.H.; Martinez-Seidel, F.; Firmino, A.A.P.; Skirycz, A.; Roessner, U.; Kopka, J. Arabidopsis REI-LIKE proteins activate ribosome biogenesis during cold acclimation. Sci. Rep. 2021, 11, 1–25. [Google Scholar] [CrossRef]
- Schmidt, S.; Dethloff, F.; Beine-Golovchuk, O.; Kopka, J. The REIL1 and REIL2 proteins of Arabidopsis thaliana are required for leaf growth in the cold. Plant Physiol. 2013, 163, 1623–1639. [Google Scholar] [CrossRef] [Green Version]
- Beine-Golovchuk, O. Characterization and Functional Complementation of the Arabidopsis Ribosomal Reil1-1Reil2-1 Double Mutant. Ph.D. Thesis, University of Potsdam, Potsdam, Germany, 2016. [Google Scholar]
- Greber, B.J.; Gerhardy, S.; Leitner, A.; Leibundgut, M.; Salem, M.; Boehringer, D.; Leulliot, N.; Aebersold, R.; Panse, V.G.; Ban, N. Insertion of the Biogenesis Factor Rei1 Probes the Ribosomal Tunnel during 60S Maturation. Cell 2016, 164, 91–102. [Google Scholar] [CrossRef]
- Meyer, A.E.; Hoover, L.A.; Craig, E.A. The cytosolic J-protein, Jjj1, and Rei1 function in the removal of the Pre-60 S subunit factor Arx1. J. Biol. Chem. 2010, 285, 961–968. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Kong, X.; Huang, H.; Wu, W.; Park, J.; Yun, D.J.; Lee, B.H.; Shi, H.; Zhu, J.K. STCH4/REIL2 Confers Cold Stress Tolerance in Arabidopsis by Promoting rRNA Processing and CBF Protein Translation. Cell Rep. 2020, 30, 229–242.e5. [Google Scholar] [CrossRef] [Green Version]
- Klingauf-Nerurkar, P.; Gillet, L.C.; Portugal-Calisto, D.; Oborská-Oplová, M.; Jäger, M.; Schubert, O.T.; Pisano, A.; Peña, C.; Rao, S.; Altvater, M.; et al. The gtpase nog1 co-ordinates the assembly, maturation and quality control of distant ribosomal functional centers. eLife 2020, 9, e52474. [Google Scholar] [CrossRef]
- Zhou, Y.; Musalgaonkar, S.; Johnson, A.W.; Taylor, D.W. Tightly-orchestrated rearrangements govern catalytic center assembly of the ribosome. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gamalinda, M.; Jakovljevic, J.; Babiano, R.; Talkish, J.; De La Cruz, J.; Woolford, J.L., Jr. Yeast polypeptide exit tunnel ribosomal proteins L17, L35 and L37 are necessary to recruit late-assembling factors required for 27SB pre-rRNA processing. Nucleic Acids Res. 2013, 41, 1965–1983. [Google Scholar] [CrossRef] [Green Version]
- Wilson, D.M.; Li, Y.; LaPeruta, A.; Gamalinda, M.; Gao, N.; Woolford, J.L. Structural insights into assembly of the ribosomal nascent polypeptide exit tunnel. Nat. Commun. 2020, 11, 1–15. [Google Scholar] [CrossRef]
- Nakata, M.T.; Sato, M.; Wakazaki, M.; Sato, N.; Kojima, K.; Sekine, A.; Nakamura, S.; Shikanai, T.; Toyooka, K.; Tsukaya, H.; et al. Plastid translation is essential for lateral root stem cell patterning in Arabidopsis thaliana. Biol. Open 2018, 7. [Google Scholar] [CrossRef] [Green Version]
- Firmino, A.A.P.; Gorka, M.; Graf, A.; Skirycz, A.; Martinez-Seidel, F.; Zander, K.; Kopka, J.; Beine-Golovchuk, O. Separation and paired proteome profiling of plant chloroplast and cytoplasmic ribosomes. Plants 2020, 9, 892. [Google Scholar] [CrossRef]
- Erban, A.; Martinez-Seidel, F.; Rajarathinam, Y.; Dethloff, F.; Orf, I.; Fehrle, I.; Alpers, J.; Beine-Golovchuk, O.; Kopka, J. Multiplexed Profiling and Data Processing Methods to Identify Temperature-Regulated Primary Metabolites Using Gas Chromatography Coupled to Mass Spectrometry. In Methods in Molecular Biology; Hincha, D.K., Zuther, E., Eds.; Springer: New York, NY, USA, 2020; Volume 2156, pp. 203–239. ISBN 978-1-0716-0660-5. [Google Scholar]
- Ashraf, M.A.; Rahman, A. Cold stress response in Arabidopsis thaliana is mediated by GNOM ARF-GEF. Plant J. 2019, 97, 500–516. [Google Scholar] [CrossRef] [Green Version]
- Kaspar, T.C.; Bland, W.L. Soil temperature and root growth. Soil Sci. 1992, 154, 290–299. [Google Scholar] [CrossRef]
- Levitt, J. Responses of Plants to Environmental Stresses: Chilling, Freezing, and High Temperature Stresses; Academic Press: New York, NY, USA; London, UK, 1980; Volume 1. [Google Scholar]
- Hincha, D.K.; Zuther, E. Introduction: Plant Cold Acclimation and Winter Survival. In Methods in Molecular Biology; Hincha, D.K., Zuther, E., Eds.; Springer: New York, NY, USA, 2020; Volume 2156, pp. 1–7. ISBN 978-1-0716-0660-5. [Google Scholar]
- Boyes, D.C.; Zayed, A.M.; Ascenzi, R.; McCaskill, A.J.; Hoffman, N.E.; Davis, K.R.; Görlach, J. Growth Stage-Based Phenotypic Analysis of Arabidopsis: A Model for High Throughput Functional Genomics in Plants. Plant Cell 2001, 13, 1499–1510. [Google Scholar] [CrossRef] [Green Version]
- Williams, M.E.; Sussex, I.M. Developmental regulation of ribosomal protein L16 genes in Arabidopsis thaliana. Plant J. 1995, 8, 65–76. [Google Scholar] [CrossRef]
- Horiguchi, G.; Van Lijsebettens, M.; Candela, H.; Micol, J.L.; Tsukaya, H. Ribosomes and translation in plant developmental control. Plant Sci. 2012, 191–192, 24–34. [Google Scholar] [CrossRef]
- Hernández-Hermenegildo, R.A.; Bernal, L.; Jiménez-Pérez, L.V.; Bernal-Lugo, I.; de Jiménez, E.S. Ribosomal Heterogeneity of Maize Tissues: Insights of Biological Relevance. Plant Mol. Biol. Rep. 2018, 36, 491–499. [Google Scholar] [CrossRef]
- Armache, J.P.; Jarasch, A.; Anger, A.M.; Villa, E.; Becker, T.; Bhushan, S.; Jossinet, F.; Habeck, M.; Dindar, G.; Franckenberg, S.; et al. Localization of eukaryote-specific ribosomal proteins in a 5.5-Å cryo-EM map of the 80S eukaryotic ribosome. Proc. Natl. Acad. Sci. USA 2010, 107, 19754–19759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piques, M.; Schulze, W.X.; Höhne, M.; Usadel, B.; Gibon, Y.; Rohwer, J.; Stitt, M. Ribosome and transcript copy numbers, polysome occupancy and enzyme dynamics in Arabidopsis. Mol. Syst. Biol. 2009, 5, 314. [Google Scholar] [CrossRef] [PubMed]
- Maldonado-Bonilla, L.D. Composition and function of P bodies in Arabidopsis thaliana. Front. Plant Sci. 2014, 5, 201. [Google Scholar] [CrossRef] [Green Version]
- Jang, G.J.; Yang, J.Y.; Hsieh, H.L.; Wu, S.H. Processing bodies control the selective translation for optimal development of Arabidopsis young seedlings. Proc. Natl. Acad. Sci. USA 2019, 116, 6451–6456. [Google Scholar] [CrossRef] [Green Version]
- Eraslan, B.; Wang, D.; Gusic, M.; Prokisch, H.; Hallström, B.M.; Uhlén, M.; Asplund, A.; Pontén, F.; Wieland, T.; Hopf, T.; et al. Quantification and discovery of sequence determinants of protein-per-mRNA amount in 29 human tissues. Mol. Syst. Biol. 2019, 15, e8513. [Google Scholar] [CrossRef]
- Zimmermann, P.; Hirsch-Hoffmann, M.; Hennig, L.; Gruissem, W. GENEVESTIGATOR. Arabidopsis microarray database and analysis toolbox. Plant Physiol. 2004, 136, 2621–2632. [Google Scholar] [CrossRef] [Green Version]
- McKown, R.; Kuroki, G.; Warren, G. Cold responses of Arabidopsis mutants impaired in freezing tolerance. J. Exp. Bot. 1996, 47, 1919–1925. [Google Scholar] [CrossRef] [Green Version]
- Lee, B.H.; Henderson, D.A.; Zhu, J.K. The Arabidopsis cold-responsive transcriptome and its regulation by ICE1. Plant Cell 2005, 17, 3155–3175. [Google Scholar] [CrossRef] [Green Version]
- Hannah, M.A.; Wiese, D.; Freund, S.; Fiehn, O.; Heyer, A.G.; Hincha, D.K. Natural genetic variation of freezing tolerance in arabidopsis. Plant Physiol. 2006, 142, 98–112. [Google Scholar] [CrossRef] [Green Version]
- Chiba, Y.; Mineta, K.; Hirai, M.Y.; Suzuki, Y.; Kanaya, S.; Takahashi, H.; Onouchi, H.; Yamaguchi, J.; Naito, S. Changes in mRNA stability associated with cold stress in arabidopsis cells. Plant Cell Physiol. 2013, 54, 180–194. [Google Scholar] [CrossRef] [Green Version]
- Maeda, H.; DellaPenna, D. Tocopherol functions in photosynthetic organisms. Curr. Opin. Plant Biol. 2007, 10, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Hannah, M.A.; Heyer, A.G.; Hincha, D.K. A global survey of gene regulation during cold acclimation in Arabidopsis thaliana. PLoS Genet. 2005, 1, e26. [Google Scholar] [CrossRef]
- Kilian, J.; Whitehead, D.; Horak, J.; Wanke, D.; Weinl, S.; Batistic, O.; D’Angelo, C.; Bornberg-Bauer, E.; Kudla, J.; Harter, K. The AtGenExpress global stress expression data set: Protocols, evaluation and model data analysis of UV-B light, drought and cold stress responses. Plant J. 2007, 50, 347–363. [Google Scholar] [CrossRef] [PubMed]
- Vogel, J.T.; Zarka, D.G.; Van Buskirk, H.A.; Fowler, S.G.; Thomashow, M.F. Roles of the CBF2 and ZAT12 transcription factors in configuring the low temperature transcriptome of Arabidopsis. Plant J. 2005, 41, 195–211. [Google Scholar] [CrossRef]
- Juntawong, P.; Sorenson, R.; Bailey-Serres, J. Cold shock protein 1 chaperones mRNAs during translation in Arabidopsis thaliana. Plant J. 2013, 74, 1016–1028. [Google Scholar] [CrossRef]
- Rihan, H.Z.; Al-Issawi, M.; Fuller, M.P. Advances in physiological and molecular aspects of plant cold tolerance. J. Plant Interact. 2017, 12, 143–157. [Google Scholar] [CrossRef]
- Missbach, S.; Weis, B.L.; Martin, R.; Simm, S.; Bohnsack, M.T.; Schleiff, E. 40S Ribosome Biogenesis Co-Factors Are Essential for Gametophyte and Embryo Development. PLoS ONE 2013, 8, e54084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palm, D.; Simm, S.; Darm, K.; Weis, B.L.; Ruprecht, M.; Schleiff, E.; Scharf, C. Proteome distribution between nucleoplasm and nucleolus and its relation to ribosome biogenesis in Arabidopsis thaliana. RNA Biol. 2016, 13, 441–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gasse, L.; Flemming, D.; Hurt, E. Coordinated Ribosomal ITS2 RNA Processing by the Las1 Complex Integrating Endonuclease, Polynucleotide Kinase, and Exonuclease Activities. Mol. Cell 2015, 60, 808–815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hang, R.; Wang, Z.; Deng, X.; Liu, C.; Yan, B.; Yang, C.; Song, X.; Mo, B.; Cao, X. Ribosomal RNA Biogenesis and Its Response to Chilling Stress in Oryza sativa. Plant Physiol. 2018, 177, 381–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, G.; Yan, P.; Du, Q.; Wang, Y.; Guo, Y.; Fu, Z.; Wang, H.; Tang, J. Pre-rRNA processing and its response to temperature stress in maize. J. Exp. Bot. 2020, 71, 1363–1374. [Google Scholar] [CrossRef]
- Jiang, J.; Seo, H.; Chow, C.S. Post-transcriptional Modifications Modulate rRNA Structure and Ligand Interactions. Acc. Chem. Res. 2016, 49, 893–901. [Google Scholar] [CrossRef]
- Yin, C.; Yau, S.S.T. A coevolution analysis for identifying protein-protein interactions by Fourier transform. PLoS ONE 2017, 12, e0174862. [Google Scholar] [CrossRef] [Green Version]
- Maekawa, S.; Yanagisawa, S. Nucleolar stress and sugar response in plants. Plant Signal. Behav. 2018, 13, e1442975. [Google Scholar] [CrossRef] [Green Version]
- Ben-Shem, A.; De Loubresse, N.G.; Melnikov, S.; Jenner, L.; Yusupova, G.; Yusupov, M. The structure of the eukaryotic ribosome at 3.0 Å resolution. Science 2011, 334, 1524–1529. [Google Scholar] [CrossRef] [Green Version]
- Woolford, J.L.; Baserga, S.J. Ribosome biogenesis in the yeast Saccharomyces cerevisiae. Genetics 2013, 195, 643–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ezkurdia, I.; Graña, O.; Izarzugaza, J.M.G.; Tress, M.L. Assessment of domain boundary predictions and the prediction of intramolecular contacts in CASP8. Proteins Struct. Funct. Bioinform. 2009, 77, 196–209. [Google Scholar] [CrossRef]
- Lury, D.A. Statistical Methods for Research Workers. J. R. Stat. Soc. Ser. D Stat. 1972, 21, 229. [Google Scholar] [CrossRef]
- Fisher, R.A. Statistical Methods for Research Workers. Biometrics 1971, 27, 1106. [Google Scholar] [CrossRef]
- Bland, J.M.; Altman, D.G. Multiple significance tests: The Bonferroni method. BMJ 1995, 310, 170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timsit, Y.; Sergeant-Perthuis, G.; Bennequin, D. Evolution of ribosomal protein network architectures. Sci. Rep. 2021, 11, 1–13. [Google Scholar] [CrossRef]
- Timsit, Y.; Bennequin, D. Nervous-like circuits in the ribosome facts, hypotheses and perspectives. Int. J. Mol. Sci. 2019, 20, 2911. [Google Scholar] [CrossRef] [Green Version]
- Poirot, O.; Timsit, Y. Neuron-Like Networks between Ribosomal Proteins Within the Ribosome. Sci. Rep. 2016, 6, 26485. [Google Scholar] [CrossRef] [Green Version]
- Rhodin, M.H.J.; Dinman, J.D. An extensive network of information flow through the B1b/c intersubunit bridge of the yeast ribosome. PLoS ONE 2011, 6, e20048. [Google Scholar] [CrossRef] [Green Version]
- Meskauskas, A.; Dinman, J.D. A molecular clamp ensures allosteric coordination of peptidyltransfer and ligand binding to the ribosomal A-site. Nucleic Acids Res. 2010, 38, 7800–7813. [Google Scholar] [CrossRef] [Green Version]
- Armache, J.P.; Anger, A.M.; Márquez, V.; Franckenberg, S.; Fröhlich, T.; Villa, E.; Berninghausen, O.; Thomm, M.; Arnold, G.J.; Beckmann, R.; et al. Promiscuous behaviour of archaeal ribosomal proteins: Implications for eukaryotic ribosome evolution. Nucleic Acids Res. 2013, 41, 1284–1293. [Google Scholar] [CrossRef]
- Bowman, J.C.; Petrov, A.S.; Frenkel-Pinter, M.; Penev, P.I.; Williams, L.D. Root of the Tree: The Significance, Evolution, and Origins of the Ribosome. Chem. Rev. 2020, 120, 4848–4878. [Google Scholar] [CrossRef] [PubMed]
- Bernier, C.R.; Petrov, A.S.; Kovacs, N.A.; Penev, P.I.; Williams, L.D. Translation: The universal structural core of life. Mol. Biol. Evol. 2018, 35, 2065–2076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, M.S.; Sah, R.; Balasubramaniam, A.; Sallee, F.R.; Park, E.A.; Parker, S.L. On the expansion of ribosomal proteins and RNAs in eukaryotes. Amino Acids 2014, 46, 1589–1604. [Google Scholar] [CrossRef]
- Parker, M.S.; Balasubramaniam, A.; Parker, S.L. On the segregation of protein ionic residues by charge type. Amino Acids 2012, 43, 2231–2247. [Google Scholar] [CrossRef]
- Melnikov, S.; Manakongtreecheep, K.; Söll, D. Revising the structural diversity of ribosomal proteins across the three domains of life. Mol. Biol. Evol. 2018, 35, 1588–1598. [Google Scholar] [CrossRef] [Green Version]
- Evangelisti, A.M.; Conant, G.C. Nonrandom survival of gene conversions among yeast ribosomal proteins duplicated through genome doubling. Genome Biol. Evol. 2010, 2, 826–834. [Google Scholar] [CrossRef] [Green Version]
- Petibon, C.; Parenteau, J.; Catala, M.; Elela, S.A. Introns regulate the production of ribosomal proteins by modulating splicing of duplicated ribosomal protein genes. Nucleic Acids Res. 2016, 44, 3878–3891. [Google Scholar] [CrossRef] [Green Version]
- Briggs, J.W.; Dinman, J.D. Subtractional Heterogeneity: A Crucial Step toward Defining Specialized Ribosomes. Mol. Cell 2017, 67, 3–4. [Google Scholar] [CrossRef] [Green Version]
- Agozzino, L.; Dill, K.A. Protein evolution speed depends on its stability and abundance and on chaperone concentrations. Proc. Natl. Acad. Sci. USA 2018, 115, 9092–9097. [Google Scholar] [CrossRef] [Green Version]
- Privalov, P.L. Cold denaturation of protein. Crit. Rev. Biochem. Mol. Biol. 1990, 25, 281–306. [Google Scholar] [CrossRef] [PubMed]
- Graziano, G. On the mechanism of cold denaturation. Phys. Chem. Chem. Phys. 2014, 16, 21755–21767. [Google Scholar] [CrossRef]
- Dias, C.L.; Ala-Nissila, T.; Wong-ekkabut, J.; Vattulainen, I.; Grant, M.; Karttunen, M. The hydrophobic effect and its role in cold denaturation. Cryobiology 2010, 60, 91–99. [Google Scholar] [CrossRef]
- Sanfelice, D.; Temussi, P.A. Cold denaturation as a tool to measure protein stability. Biophys. Chem. 2016, 208, 4–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balcerak, A.; Trebinska-Stryjewska, A.; Konopinski, R.; Wakula, M.; Grzybowska, E.A. RNA-protein interactions: Disorder, moonlighting and junk contribute to eukaryotic complexity. Open Biol. 2019, 9, 190096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timsit, Y.; Acosta, Z.; Allemand, F.; Chiaruttini, C.; Springer, M. The role of disordered ribosomal protein extensions in the early steps of eubacterial 50 S ribosomal subunit assembly. Int. J. Mol. Sci. 2009, 10, 817–834. [Google Scholar] [CrossRef] [Green Version]
- Tutuncuoglu, B.; Jakovljevic, J.; Wu, S.; Gao, N.; Woolford, J.L. The N-terminal extension of yeast ribosomal protein L8 is involved in two major remodeling events during late nuclear stages of 60S ribosomal subunit assembly. RNA 2016, 22, 1386–1399. [Google Scholar] [CrossRef] [Green Version]
- Zengel, J.M.; Jerauld, A.; Walker, A.; Wahl, M.C.; Lindahl, L. The extended loops of ribosomal proteins L4 and L22 are not required for ribosome assembly or L4-mediated autogenous control. RNA 2003, 9, 1188–1197. [Google Scholar] [CrossRef] [Green Version]
- Greber, B.J.; Boehringer, D.; Montellese, C.; Ban, N. Cryo-EM structures of Arx1 and maturation factors Rei1 and Jjj1 bound to the 60S ribosomal subunit. Nat. Struct. Mol. Biol. 2012, 19, 1228–1233. [Google Scholar] [CrossRef] [PubMed]
- Lo, K.Y.; Li, Z.; Bussiere, C.; Bresson, S.; Marcotte, E.M.; Johnson, A.W. Defining the pathway of cytoplasmic maturation of the 60S ribosomal subunit. Mol. Cell 2010, 39, 196–208. [Google Scholar] [CrossRef] [Green Version]
- Mageeney, C.M.; Ware, V.C. Specialized eRpL22 paralogue-specific ribosomes regulate specific mRNA translation in spermatogenesis in Drosophila melanogaster. Mol. Biol. Cell 2019, 30, 2240–2253. [Google Scholar] [CrossRef]
- Maitra, N.; He, C.; Blank, H.M.; Tsuchiya, M.; Schilling, B.; Kaeberlein, M.; Aramayo, R.; Kennedy, B.K.; Polymenis, M. Translational control of methionine and serine metabolic pathways underpin the paralog-specific phenotypes of Rpl22 ribosomal protein mutants in cell division and replicative longevity. bioRxiv 2020. [Google Scholar] [CrossRef]
- Yu, P.; Wang, S.; Ma, C.; Luo, X.; Xing, Z.; Wu, X.; Clyne, R.K.; Hwang, G.-J. Affirmation of Distinctive Ribosomal Protein Paralog-Specific Ribosomes. SSRN Electron. J. 2019, 3334430. [Google Scholar] [CrossRef]
- Ghulam, M.M.; Catala, M.; Elela, S.A. Differential expression of duplicated ribosomal protein genes modifies ribosome composition in response to stress. Nucleic Acids Res. 2020, 48, 1954–1968. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; De Croos, J.N.A.; Storey, K.B. Cold acclimation-induced up-regulation of the ribosomal protein L7 gene in the freeze tolerant wood frog, Rana sylvatica. Gene 2008, 424, 48–55. [Google Scholar] [CrossRef]
- Gadal, O.; Strauss, D.; Petfalski, E.; Gleizes, P.E.; Gas, N.; Tollervey, D.; Hurt, E. Rlp7p is associated with 60S preribosomes, restricted to the granular component of the nucleolus, and required for pre-rRNA processing. J. Cell Biol. 2002, 157, 941–952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.; Zhao, J.; Jia, M.; Xu, N.; Liang, S.; Shao, J.; Qi, Y.; Liu, X.; An, L.; Yu, F. Balance between cytosolic and chloroplast translation affects leaf variegation. Plant Physiol. 2018, 176, 804–818. [Google Scholar] [CrossRef] [Green Version]
- De Las Heras-Rubio, A.; Perucho, L.; Paciucci, R.; Vilardell, J.; Lleonart, M.E. Ribosomal proteins as novel players in tumorigenesis. Cancer Metastasis Rev. 2014, 33, 115–141. [Google Scholar] [CrossRef]
- Neumann, F.; Krawinkel, U. Constitutive expression of human ribosomal protein L7 arrests the cell cycle in G1 and induces apoptosis in Jurkat T-lymphoma cells. Exp. Cell Res. 1997, 230, 252–261. [Google Scholar] [CrossRef]
- Von Arnim, A.G.; Jia, Q.; Vaughn, J.N. Regulation of plant translation by upstream open reading frames. Plant Sci. 2014, 214, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Jakovljevic, J.; Ohmayer, U.; Gamalinda, M.; Talkish, J.; Alexander, L.; Linnemann, J.; Milkereit, P.; Woolford, J.L. Ribosomal proteins L7 and L8 function in concert with six A3 assembly factors to propagate assembly of domains I and II of 25S rRNA in yeast 60S ribosomal subunits. RNA 2012, 18, 1805–1822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charneski, C.A.; Hurst, L.D. Positively Charged Residues Are the Major Determinants of Ribosomal Velocity. PLoS Biol. 2013, 11, e1001508. [Google Scholar] [CrossRef] [Green Version]
- Diaconu, M.; Kothe, U.; Schlünzen, F.; Fischer, N.; Harms, J.M.; Tonevitsky, A.G.; Stark, H.; Rodnina, M.V.; Wahl, M.C. Structural basis for the function of the ribosomal L7/12 stalk in factor binding and GTpase activation. Cell 2005, 121, 991–1004. [Google Scholar] [CrossRef] [Green Version]
- Bourne, H.R.; Sanders, D.A.; McCormick, F. The GTPase superfamily: Conserved structure and molecular mechanism. Nature 1991, 349, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Rosso, M.G.; Li, Y.; Strizhov, N.; Reiss, B.; Dekker, K.; Weisshaar, B. An Arabidopsis thaliana T-DNA mutagenized population (GABI-Kat) for flanking sequence tag-based reverse genetics. Plant Mol. Biol. 2003, 53, 247–259. [Google Scholar] [CrossRef]
- Murashige, T.; Skoog, F. A Revised Medium for Rapid Growth and Bio Assays with Tobacco Tissue Cultures. Physiol. Plant. 1962, 15, 473–497. [Google Scholar] [CrossRef]
- QIAGEN. RNeasy Mini Handbook. HB-0435-005 10/2019. Available online: http://www.qiagen.com (accessed on 30 April 2021).
- Hummel, M.; Dobrenel, T.; Cordewener, J.J.H.G.; Davanture, M.; Meyer, C.; Smeekens, S.J.C.M.; Bailey-Serres, J.; America, T.A.H.P.; Hanson, J. Proteomic LC-MS analysis of Arabidopsis cytosolic ribosomes: Identification of ribosomal protein paralogs and re-annotation of the ribosomal protein genes. J. Proteom. 2015, 128, 436–449. [Google Scholar] [CrossRef] [Green Version]
- Johnson, W.E.; Li, C.; Rabinovic, A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 2007, 8, 118–127. [Google Scholar] [CrossRef]
- Delignette-Muller, M.L.; Dutang, C. fitdistrplus: An R package for fitting distributions. J. Stat. Softw. 2015, 64, 1–34. [Google Scholar] [CrossRef] [Green Version]
- Smyth, G.K. Limma: Linear Models for Microarray Data. In Bioinformatics and Computational Biology Solutions Using R Bioconductor; Springer: New York, NY, USA, 2005; pp. 397–420. [Google Scholar] [CrossRef] [Green Version]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Hartigan, J.A.; Wong, M.A. Algorithm AS 136: A K-Means Clustering Algorithm. J. R. Stat. Soc. Ser. C Appl. Stat. 1979, 28, 100. [Google Scholar] [CrossRef]
- Kawaguchi, R.; Williams, A.J.; Bray, E.A.; Bailey-Serres, J. Water-deficit-induced translational control in Nicotiana tabacum. Plant, Cell Environ. 2003, 26, 221–229. [Google Scholar] [CrossRef]
- Reynoso, M.A.; Juntawong, P.; Lancia, M.; Blanco, F.A.; Bailey-Serres, J.; Zanetti, M.E. Translating ribosome affinity purification (TRAP) followed by RNA sequencing technology (TRAP-SEQ) for quantitative assessment of plant translatomes. In Plant Functional Genomics: Methods in Molecular Biology; Humana Press: New York, NY, USA, 2015; pp. 185–207. ISBN 9781493924448. [Google Scholar]
- Lebowitz, J.; Lewis, M.S.; Schuck, P. Modern analytical ultracentrifugation in protein science: A tutorial review. Protein Sci. 2009, 11, 2067–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erde, J.; Loo, R.R.O.; Loo, J.A. Enhanced FASP (eFASP) to increase proteome coverage and sample recovery for quantitative proteomic experiments. J. Proteome Res. 2014, 13, 1885–1895. [Google Scholar] [CrossRef]
- Swart, C.; Martínez-Jaime, S.; Gorka, M.; Zander, K.; Graf, A. Hit-Gel: Streamlining in-gel protein digestion for high-throughput proteomics experiments. Sci. Rep. 2018, 8, 8582. [Google Scholar] [CrossRef] [PubMed]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhang, J.; Xu, C.; Li, N.; Liu, H.; Ma, J.; Zhu, Y.; Xie, H. LFQuant: A label-free fast quantitative analysis tool for high-resolution LC-MS/MS proteomics data. Proteomics 2012, 12, 3475–3484. [Google Scholar] [CrossRef]
- The UniProt Consortium. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [Google Scholar] [CrossRef] [Green Version]
- Armache, J.P.; Jarasch, A.; Anger, A.M.; Villa, E.; Becker, T.; Bhushan, S.; Jossinet, F.; Habeck, M.; Dindar, G.; Franckenberg, S.; et al. Cryo-EM structure and rRNA model of a translating eukaryotic 80S ribosome at 5.5-Å resolution. Proc. Natl. Acad. Sci. USA 2010, 107, 19748–19753. [Google Scholar] [CrossRef] [Green Version]
- Rose, P.W.; Bi, C.; Bluhm, W.F.; Christie, C.H.; Dimitropoulos, D.; Dutta, S.; Green, R.K.; Goodsell, D.S.; Prlić, A.; Quesada, M.; et al. The RCSB Protein Data Bank: New resources for research and education. Nucleic Acids Res. 2013, 41, D475–D482. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Ban, N.; Beckmann, R.; Cate, J.H.D.; Dinman, J.D.; Dragon, F.; Ellis, S.R.; Lafontaine, D.L.J.; Lindahl, L.; Liljas, A.; Lipton, J.M.; et al. A new system for naming ribosomal proteins. Curr. Opin. Struct. Biol. 2014, 24, 165–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Csardi, G.; Nepusz, T. The igraph software package for complex network research. InterJ. Complex Syst. 2006, 1695, 1–9. [Google Scholar]
- Virtanen, P.; Gommers, R.; Oliphant, T.E.; Haberland, M.; Reddy, T.; Cournapeau, D.; Burovski, E.; Peterson, P.; Weckesser, W.; Bright, J.; et al. SciPy 1.0: Fundamental algorithms for scientific computing in Python. Nat. Methods 2020, 17, 261–272. [Google Scholar] [CrossRef] [Green Version]
- Sela, I.; Ashkenazy, H.; Katoh, K.; Pupko, T. GUIDANCE2: Accurate detection of unreliable alignment regions accounting for the uncertainty of multiple parameters. Nucleic Acids Res. 2015, 43, W7–W14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landan, G.; Graur, D. Local reliability measures from sets of co-optimal multiple sequence alignments. In Proceedings of the Pacific Symposium on Biocomputing 2008, PSB 2008, Kohala Coast, HI, USA, 4–8 January 2008; pp. 15–24. [Google Scholar]
- Penn, O.; Privman, E.; Ashkenazy, H.; Landan, G.; Graur, D.; Pupko, T. GUIDANCE: A web server for assessing alignment confidence scores. Nucleic Acids Res. 2010, 38, W23–W28. [Google Scholar] [CrossRef] [Green Version]
- Bouckaert, R.; Heled, J.; Kühnert, D.; Vaughan, T.; Wu, C.H.; Xie, D.; Suchard, M.A.; Rambaut, A.; Drummond, A.J. BEAST 2: A Software Platform for Bayesian Evolutionary Analysis. PLoS Comput. Biol. 2014, 10, e1003537. [Google Scholar] [CrossRef] [Green Version]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef] [Green Version]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. JModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [Green Version]
- Guindon, S.; Gascuel, O. A Simple, Fast, and Accurate Algorithm to Estimate Large Phylogenies by Maximum Likelihood. Syst. Biol. 2003, 52, 696–704. [Google Scholar] [CrossRef] [Green Version]
- Steel, M.; McKenzie, A. Properties of phylogenetic trees generated by yule-type speciation models. Math. Biosci. 2001, 170, 91–112. [Google Scholar] [CrossRef]
- Drummond, A.J.; Nicholls, G.K.; Rodrigo, A.G.; Solomon, W. Estimating mutation parameters, population history and genealogy simultaneously from temporally spaced sequence data. Genetics 2002, 161, 1307–1320. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeLano, W.L. The PyMOL Molecular Graphics System, Version 1.8; Schrödinger LLC: New York, NY, USA, 2014; Available online: http://www.pymol.org (accessed on 30 April 2021).
- Ihaka, R.; Gentleman, R. R: A Language for Data Analysis and Graphics. J. Comput. Graph. Stat. 1996, 5, 299–314. [Google Scholar] [CrossRef]
- R Development Core Team 3.0.1. A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2013; Volume 2. [Google Scholar]
- Wickham, H.; Averick, M.; Bryan, J.; Chang, W.; McGowan, L.; François, R.; Grolemund, G.; Hayes, A.; Henry, L.; Hester, J.; et al. Welcome to the Tidyverse. J. Open Source Softw. 2019, 4, 1686. [Google Scholar] [CrossRef]
- Wickham, H. Reshaping data with the reshape package. J. Stat. Softw. 2007, 21, 1–20. [Google Scholar] [CrossRef]
- Hadley Wickham Stringr: Simple, Consistent Wrappers for Common String Operations. R Package Version 1.3.1. R Packag. version2018. Available online: http://stringr.tidyverse.org and https://github.com/tidyverse/stringr (accessed on 30 April 2021).
- Ginestet, C. ggplot2: Elegant Graphics for Data Analysis. J. R. Stat. Soc. Ser. A Stat. Soc. 2011, 174, 245–246. [Google Scholar] [CrossRef]
- Gagolewski, M. Package “stringi”: Character String Processing Facilities. Cran 2016. Available online: http://www.gagolewski.com/software/stringi/ (accessed on 30 April 2021).
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [Green Version]
- Gu, Z.; Gu, L.; Eils, R.; Schlesner, M.; Brors, B. Circlize implements and enhances circular visualization in R. Bioinformatics 2014, 30, 2811–2812. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Boutros, P.C. VennDiagram: A package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinform. 2011, 12, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Temperature | Genotypes | More Abundant in 60S | More Abundant in Polysomes |
|---|---|---|---|
| 20 °C | Genotype-independent * | uL15_RPL27aB, uL15_RPL27aC, eL18_RPL18C | |
| 10 °C | uL3_RPL3A, eL28_RPL28C, eL13_RPL13D | ||
| 20 °C | WT | eL36_RPL36C, eL18_RPL18C, eL32_RPL32A, uL15_RPL27aC, eL14_RPL14B, uL16_RPL10A | P1.P2_RPP1A, uL11_RPL12C, uL6_RPL9D, eL27_RPL27A or B, eL33_RPL35aB |
| dkos | eL34_RPL34B, eL38_RPL38A or B, uL10_RPP0A or B, P1.P2_RPP1C, P1.P2_RPP2B | eL19_RPL19A, eL14_RPL14A, eL28_RPL28A, eL32_RPL32B | |
| 10 °C | WT | P1.P2_RPP2B, uL10_RPP0A or B, uL30_RPL7C, P1.P2_RPP1C, P1.P2_RPP1B, P1.P2_RPP2A | eL22_RPL22B, eL32_RPL32B, uL13_RPL13aA, uL6_RPL9B or C |
| dkos | eL15_RPL15A or B, P1.P2_RPP2A, uL18_RPL5B, eL36_RPL36C, eL38_RPL38A or B, uL10_RPP0A or B, uL1_RPL10aC, eL27_RPL27A or B | uL16_RPL10C, eL42_RPL36aA or aB, eL39_RPL39A or C, eL19_RPL19C |
| Regions | Ribosomal Protein Families | Region Identifiers | p Values (Fisher) | Q Values (Bonferroni) |
|---|---|---|---|---|
| Transcriptomics | 4v7e_dt8_IN20_WL10 | |||
| 1T—A | eL39 eL37 uL24 uL23 uL4 uL29 eL8 uL30 eL27 eL30 eL34 | LSU-PET | 0.07 | 0.80 |
| 1T—A | uL11 P1 P2 uL10 | LSU-P-Stalk | 0.05 | 0.51 |
| 2T—B | uS15 uS12 uS8 uS3 eS17 uS2 eS7 eS21 eS27 uS5 eL27 eL30 eL34 | SSU-LSU-RibosomalHead.1 | 0.00 | 0.02 |
| 2T—B | uS15 uS12 eS17 eL19 eS7 uS2 uS17 eS21 eS27 uS5 uS8 | SSU-LSU-RibosomalHead.2 | 0.00 | 0.00 |
| 2T—B | uL11 P1 P2 uL10 | LSU-P-Stalk | 0.00 | 0.05 |
| 3T—C | uL13 eL24 eS8 uL14 uL3 | SSU-LSU-uL3 region | 0.01 | 0.18 |
| 3T—C | uL11 P1 P2 uL10 | LSU-P-Stalk | 0.02 | 0.36 |
| Proteomics | 4v7e_dt8_IN20_WL10 | |||
| 1P—D | eL13 eL15 uS7 uL15 eL18 uL29 eS25 uL23 uL5 eL36 eL42 eL8 uL30 uS13 eL21 uS11 | SSU-LSU-uL30 region | 0.03 | 0.42 |
| 1P—D | uL15 uL13 eL24 eS8 uL14 uL3 | SSU-LSU-uL3 region | 0.03 | 0.39 |
| 3P—E | eL13 eL15 eL18 uL15 eL39 eL37 uL4 uL24 eL6 uL23 uL29 eL36 eL8 uL30 eL21 uL18 | LSU-PET | 0.00 | 0.04 |
| 3P—E | eL32 eL13 eL18 uL15 eL33 uL4 eL14 eL20 eL6 uL13 uL30 eL21 uL3 eL28 | LSU-uL30-uL3 region | 0.00 | 0.05 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martinez-Seidel, F.; Beine-Golovchuk, O.; Hsieh, Y.-C.; Eshraky, K.E.; Gorka, M.; Cheong, B.-E.; Jimenez-Posada, E.V.; Walther, D.; Skirycz, A.; Roessner, U.; et al. Spatially Enriched Paralog Rearrangements Argue Functionally Diverse Ribosomes Arise during Cold Acclimation in Arabidopsis. Int. J. Mol. Sci. 2021, 22, 6160. https://doi.org/10.3390/ijms22116160
Martinez-Seidel F, Beine-Golovchuk O, Hsieh Y-C, Eshraky KE, Gorka M, Cheong B-E, Jimenez-Posada EV, Walther D, Skirycz A, Roessner U, et al. Spatially Enriched Paralog Rearrangements Argue Functionally Diverse Ribosomes Arise during Cold Acclimation in Arabidopsis. International Journal of Molecular Sciences. 2021; 22(11):6160. https://doi.org/10.3390/ijms22116160
Chicago/Turabian StyleMartinez-Seidel, Federico, Olga Beine-Golovchuk, Yin-Chen Hsieh, Kheloud El Eshraky, Michal Gorka, Bo-Eng Cheong, Erika V. Jimenez-Posada, Dirk Walther, Aleksandra Skirycz, Ute Roessner, and et al. 2021. "Spatially Enriched Paralog Rearrangements Argue Functionally Diverse Ribosomes Arise during Cold Acclimation in Arabidopsis" International Journal of Molecular Sciences 22, no. 11: 6160. https://doi.org/10.3390/ijms22116160