
Deficits in Prenatal Serine Biosynthesis Underlie the Mitochondrial Dysfunction Associated with the Autism-Linked FMR1 Gene

 and
and
Abstract
:1. Introduction
2. Results & Discussion
2.1. Selection and Characterization of Amniotic Fluid Samples
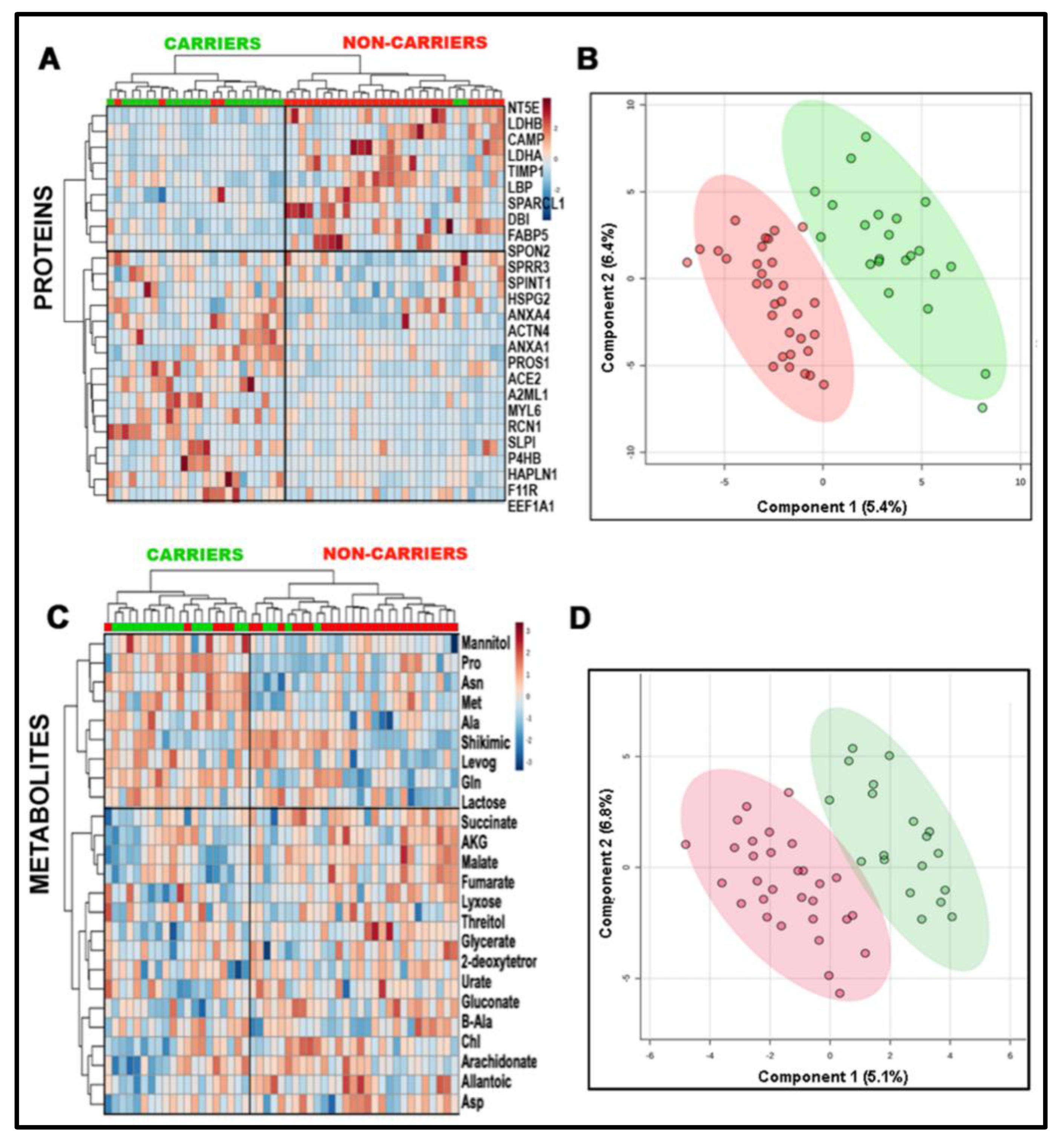
2.2. Combined Omics as a Tool to Validate the Biological Matrix as Fetal Amniotic Fluid
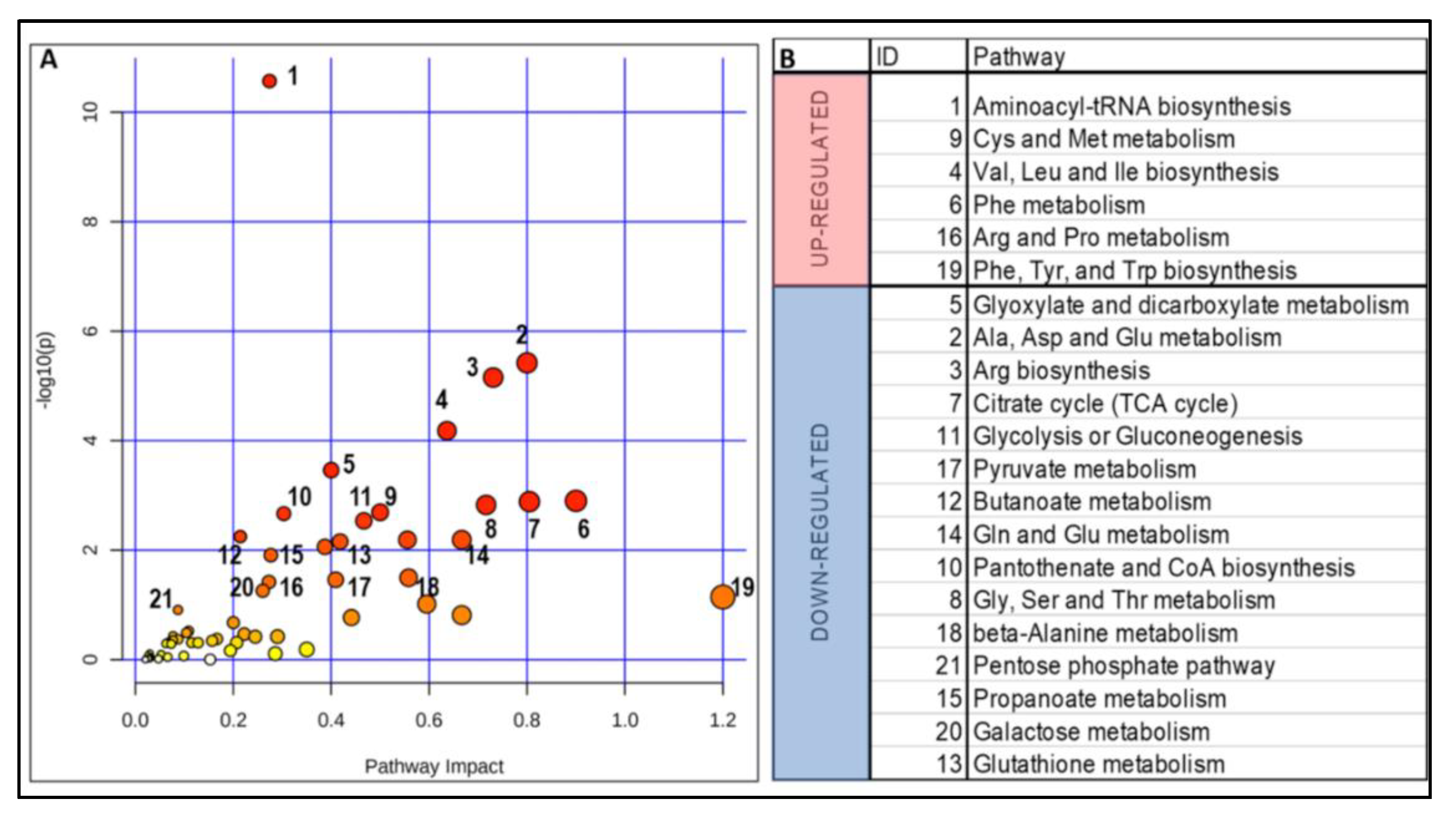
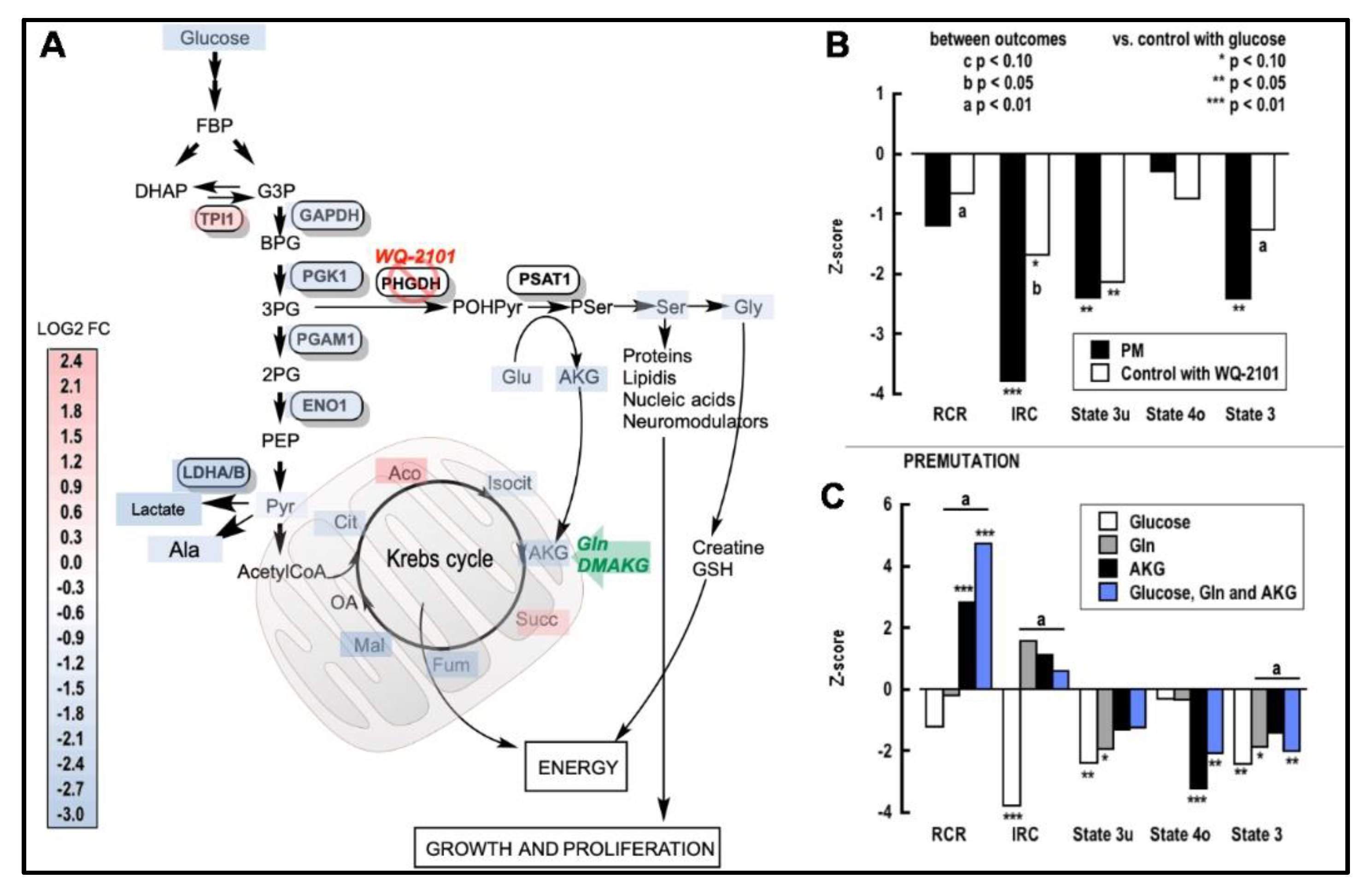
2.3. Impact of a Deficient Glycolysis-Derived Serine Biosynthetic Pathway in the Premutation
2.4. Independent Validation of the Prenatal Metabolic Footprint of the PM
2.5. Improving Ser Status Rescues Mitochondrial Function in the PM
3. Materials and Methods
3.1. Subjects’ Demographics; FMR1 Repeat Sizing and Structure
3.2. Metabolomic and Proteomic Evaluation
3.3. Cell Culture Conditions and Mitochondrial Outcomes
3.4. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- De Vries, B.B.; Wiegers, A.M.; de Graaff, E.; Verkerk, A.J.; Van Hemel, J.O.; Halley, D.J.; Fryns, J.P.; Curfs, L.M.; Niermeijer, M.F.; Oostra, B.A. Mental status and fragile X expression in relation to FMR-1 gene mutation. Eur. J. Hum. Genet. 1993, 1, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.W.; Berry-Kravis, E. Autism and fragile X syndrome. Semin. Neurol. 2014, 34, 258–265. [Google Scholar] [CrossRef]
- Nolin, S.L.; Brown, W.T.; Glicksman, A.; Houck, G.E., Jr.; Gargano, A.D.; Sullivan, A.; Biancalana, V.; Brondum-Nielsen, K.; Hjalgrim, H.; Holinski-Feder, E.; et al. Expansion of the fragile X CGG repeat in females with premutation or intermediate alleles. Am. J. Hum. Genet. 2003, 72, 454–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagerman, R.J.; Leehey, M.; Heinrichs, W.; Tassone, F.; Wilson, R.; Hills, J.; Grigsby, J.; Gage, B.; Hagerman, P.J. Intention tremor, parkinsonism, and generalized brain atrophy in male carriers of fragile X. Neurology 2001, 57, 127–130. [Google Scholar] [CrossRef]
- Berry-Kravis, E.; Abrams, L.; Coffey, S.M.; Hall, D.A.; Greco, C.; Gane, L.W.; Grigsby, J.; Bourgeois, J.A.; Finucane, B.; Jacquemont, S.; et al. Fragile X-associated tremor/ataxia syndrome: Clinical features, genetics, and testing guidelines. Mov. Disord. 2007, 22, 2018–2030. [Google Scholar] [CrossRef]
- Wheeler, A.; Raspa, M.; Hagerman, R.; Mailick, M.; Riley, C. Implications of the FMR1 premutation for children, adolescents, adults, and their families. Pediatrics 2017, 139, S172–S182. [Google Scholar] [CrossRef] [Green Version]
- Wheeler, A.C.; Sideris, J.; Hagerman, R.; Berry-Kravis, E.; Tassone, F.; Bailey, D.B., Jr. Developmental profiles of infants with an FMR1 premutation. J. Neurodev. Disord. 2016, 8, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraan, C.M.; Bui, Q.M.; Field, M.; Archibald, A.D.; Metcalfe, S.A.; Christie, L.M.; Bennetts, B.H.; Oertel, R.; Smith, M.J.; du Sart, D.; et al. FMR1 allele size distribution in 35,000 males and females: A comparison of developmental delay and general population cohorts. Genet. Med. 2018, 20, 1627–1634. [Google Scholar] [CrossRef]
- Andreux, P.A.; Houtkooper, R.H.; Auwerx, J. Pharmacological approaches to restore mitochondrial function. Nat. Rev. Drug Discov. 2013, 12, 465–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Napoli, E.; Song, G.; Wong, S.; Hagerman, R.; Giulivi, C. Altered bioenergetics in primary dermal fibroblasts from adult carriers of the FMR1 premutation before the onset of the neurodegenerative disease fragile X-associated tremor/ataxia syndrome. Cerebellum 2016, 15, 552–564. [Google Scholar] [CrossRef] [Green Version]
- Napoli, E.; Schneider, A.; Hagerman, R.; Song, G.; Wong, S.; Tassone, F.; Giulivi, C. Impact of FMR1 premutation on neurobehavior and bioenergetics in young monozygotic twins. Front. Genet. 2018, 9, 338. [Google Scholar] [CrossRef]
- Alvarez-Mora, M.I.; Rodriguez-Revenga, L.; Madrigal, I.; Guitart-Mampel, M.; Garrabou, G.; Mila, M. Impaired mitochondrial function and dynamics in the pathogenesis of FXTAS. Mol. Neurobiol. 2017, 54, 6896–6902. [Google Scholar] [CrossRef]
- Hukema, R.K.; Buijsen, R.A.; Raske, C.; Severijnen, L.A.; Nieuwenhuizen-Bakker, I.; Minneboo, M.; Maas, A.; de Crom, R.; Kros, J.M.; Hagerman, P.J.; et al. Induced expression of expanded CGG RNA causes mitochondrial dysfunction in vivo. Cell Cycle 2014, 13, 2600–2608. [Google Scholar] [CrossRef] [Green Version]
- Nobile, V.; Palumbo, F.; Lanni, S.; Ghisio, V.; Vitali, A.; Castagnola, M.; Marzano, V.; Maulucci, G.; De Angelis, C.; De Spirito, M.; et al. Altered mitochondrial function in cells carrying a premutation or unmethylated full mutation of the FMR1 gene. Hum. Genet. 2020, 139, 227–245. [Google Scholar] [CrossRef] [PubMed]
- Napoli, E.; Song, G.; Schneider, A.; Hagerman, R.; Eldeeb, M.A.; Azarang, A.; Tassone, F.; Giulivi, C. Warburg effect linked to cognitive-executive deficits in FMR1 premutation. FASEB J. 2016, 30, 3334–3351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwahashi, C.K.; Yasui, D.H.; An, H.J.; Greco, C.M.; Tassone, F.; Nannen, K.; Babineau, B.; Lebrilla, C.B.; Hagerman, R.J.; Hagerman, P.J. Protein composition of the intranuclear inclusions of FXTAS. Brain 2006, 129, 256–271. [Google Scholar] [CrossRef]
- Gardiner, E.; Beveridge, N.J.; Wu, J.Q.; Carr, V.; Scott, R.J.; Tooney, P.A.; Cairns, M.J. Imprinted DLK1-DIO3 region of 14q32 defines a schizophrenia-associated miRNA signature in peripheral blood mononuclear cells. Mol. Psychiatry 2012, 17, 827–840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, C.Y.; Yu, S.L.; Hsieh, M.H.; Chen, C.H.; Chen, H.Y.; Wen, C.C.; Huang, Y.H.; Hsiao, P.C.; Hsiao, C.K.; Liu, C.M.; et al. MicroRNA expression aberration as potential peripheral blood biomarkers for schizophrenia. PLoS ONE 2011, 6, e21635. [Google Scholar] [CrossRef] [Green Version]
- Mellios, N.; Sur, M. The emerging role of microRNAs in schizophrenia and autism spectrum disorders. Front. Psychiatry 2012, 3, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernier, F.P.; Boneh, A.; Dennett, X.; Chow, C.W.; Cleary, M.A.; Thorburn, D.R. Diagnostic criteria for respiratory chain disorders in adults and children. Neurology 2002, 59, 1406–1411. [Google Scholar] [CrossRef]
- Kuznetsov, A.V.; Veksler, V.; Gellerich, F.N.; Saks, V.; Margreiter, R.; Kunz, W.S. Analysis of mitochondrial function in situ in permeabilized muscle fibers, tissues and cells. Nat. Protoc. 2008, 3, 965–976. [Google Scholar] [CrossRef] [PubMed]
- Napoli, E.; Ross-Inta, C.; Song, G.; Wong, S.; Hagerman, R.; Gane, L.W.; Smilowitz, J.T.; Tassone, F.; Giulivi, C. Premutation in the fragile X mental retardation 1 (FMR1) gene affects maternal Zn-milk and perinatal brain bioenergetics and scaffolding. Front. Neurosci. 2016, 10, 159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross-Inta, C.; Omanska-Klusek, A.; Wong, S.; Barrow, C.; Garcia-Arocena, D.; Iwahashi, C.; Berry-Kravis, E.; Hagerman, R.J.; Hagerman, P.J.; Giulivi, C. Evidence of mitochondrial dysfunction in fragile X-associated tremor/ataxia syndrome. Biochem. J. 2010, 429, 545–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nolin, S.L.; Glicksman, A.; Tortora, N.; Allen, E.; Macpherson, J.; Mila, M.; Vianna-Morgante, A.M.; Sherman, S.L.; Dobkin, C.; Latham, G.J.; et al. Expansions and contractions of the FMR1 CGG repeat in 5,508 transmissions of normal, intermediate, and premutation alleles. Am. J. Med. Genet. A 2019, 179, 1148–1156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, T.; Zhang, J.; Sommer, K.; Bassig, B.A.; Zhang, X.; Braun, J.; Xu, S.; Boyle, P.; Zhang, B.; Shi, K.; et al. Effects of environmental exposures on fetal and childhood growth trajectories. Ann. Glob. Health 2016, 82, 41–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohn, B.R.; Fukuchi, E.Y.; Joe, B.N.; Swanson, M.G.; Kurhanewicz, J.; Yu, J.; Caughey, A.B. Calculation of gestational age in late second and third trimesters by ex vivo magnetic resonance spectroscopy of amniotic fluid. Am. J. Obstet. Gynecol. 2010, 203, 76 e1–76 e10. [Google Scholar] [CrossRef]
- Cohn, B.R.; Joe, B.N.; Zhao, S.; Kornak, J.; Zhang, V.Y.; Iman, R.; Kurhanewicz, J.; Vahidi, K.; Yu, J.; Caughey, A.B.; et al. Quantitative metabolic profiles of 2nd and 3rd trimester human amniotic fluid using (1)H HR-MAS spectroscopy. MAGMA 2009, 22, 343–352. [Google Scholar] [CrossRef] [Green Version]
- Coltart, R.M.; Seller, M.J.; Singer, J.D.; Campbell, S. Amniotic fluid concentrations of alpha-fetoprotein (AFP) in early normal pregnancy, and pregnancy complicated by neural tube defects. A review of 18 months experience. Guys Hosp. Rep. 1974, 123, 121–129. [Google Scholar] [PubMed]
- Cho, C.K.; Shan, S.J.; Winsor, E.J.; Diamandis, E.P. Proteomics analysis of human amniotic fluid. Mol. Cell. Proteom. 2007, 6, 1406–1415. [Google Scholar] [CrossRef] [Green Version]
- Engelhardt, B. Development of the blood-brain barrier. Cell Tissue Res. 2003, 314, 119–129. [Google Scholar] [CrossRef]
- Milunsky, A. Amniotic fluid. In Genetic Disorders and the Fetus, 6th ed.; Milunsky, A., Milunsky, J.M., Eds.; Wiley-Blackwell: Oxford, UK, 2010; pp. 94–137. [Google Scholar]
- Caspi, R.; Billington, R.; Ferrer, L.; Foerster, H.; Fulcher, C.A.; Keseler, I.M.; Kothari, A.; Krummenacker, M.; Latendresse, M.; Mueller, L.A.; et al. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res. 2016, 44, D471–D480. [Google Scholar] [CrossRef] [PubMed]
- Orczyk-Pawilowicz, M.; Jawien, E.; Deja, S.; Hirnle, L.; Zabek, A.; Mlynarz, P. Metabolomics of human amniotic fluid and maternal plasma during normal pregnancy. PLoS ONE 2016, 11, e0152740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beall, M.H.; van den Wijngaard, J.P.; van Gemert, M.J.; Ross, M.G. Regulation of amniotic fluid volume. Placenta 2007, 28, 824–832. [Google Scholar] [CrossRef]
- Schmidt, T.; Samaras, P.; Frejno, M.; Gessulat, S.; Barnert, M.; Kienegger, H.; Krcmar, H.; Schlegl, J.; Ehrlich, H.C.; Aiche, S.; et al. ProteomicsDB. Nucleic Acids Res. 2018, 46, D1271–D1281. [Google Scholar] [CrossRef]
- Adinolfi, A.; Adinolfi, M.; Lessof, M.H. Alpha-feto-protein during development and in disease. J. Med. Genet. 1975, 12, 138–151. [Google Scholar] [CrossRef] [Green Version]
- Buffe, D.; Rimbaut, C.; Henrion, R.; Boue, J.; Boue, A. Letter: Alpha fetoprotein in amniotic fluid and maternal serum. N. Engl. J. Med. 1976, 295, 51. [Google Scholar] [CrossRef] [PubMed]
- Chong, J.; Wishart, D.S.; Xia, J. Using MetaboAnalyst 4.0 for comprehensive and integrative metabolomics data analysis. Curr. Protoc. Bioinform. 2019, 68, e86. [Google Scholar] [CrossRef]
- Kutmon, M.; van Iersel, M.P.; Bohler, A.; Kelder, T.; Nunes, N.; Pico, A.R.; Evelo, C.T. PathVisio 3: An extendable pathway analysis toolbox. PLoS Comput. Biol. 2015, 11, e1004085. [Google Scholar] [CrossRef] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [Green Version]
- Napoli, E.; Ross-Inta, C.; Wong, S.; Omanska-Klusek, A.; Barrow, C.; Iwahashi, C.; Garcia-Arocena, D.; Sakaguchi, D.; Berry-Kravis, E.; Hagerman, R.; et al. Altered zinc transport disrupts mitochondrial protein processing/import in fragile X-associated tremor/ataxia syndrome. Hum. Mol. Genet. 2011, 20, 3079–3092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giulivi, C.; Napoli, E.; Tassone, F.; Halmai, J.; Hagerman, R. Plasma metabolic profile delineates roles for neurodegeneration, pro-inflammatory damage and mitochondrial dysfunction in the FMR1 premutation. Biochem. J. 2016, 473, 3871–3888. [Google Scholar] [CrossRef]
- Song, G.; Napoli, E.; Wong, S.; Hagerman, R.; Liu, S.; Tassone, F.; Giulivi, C. Altered redox mitochondrial biology in the neurodegenerative disorder fragile X-tremor/ataxia syndrome: Use of antioxidants in precision medicine. Mol. Med. 2016, 22, 548–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krall, A.S.; Xu, S.; Graeber, T.G.; Braas, D.; Christofk, H.R. Asparagine promotes cancer cell proliferation through use as an amino acid exchange factor. Nat. Commun. 2016, 7, 11457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Jiang, Y.; Meisenhelder, J.; Yang, W.; Hawke, D.H.; Zheng, Y.; Xia, Y.; Aldape, K.; He, J.; Hunter, T.; et al. Mitochondria-translocated PGK1 functions as a protein kinase to coordinate glycolysis and the TCA cycle in tumorigenesis. Mol. Cell 2016, 61, 705–719. [Google Scholar] [CrossRef] [Green Version]
- Qian, X.; Li, X.; Cai, Q.; Zhang, C.; Yu, Q.; Jiang, Y.; Lee, J.H.; Hawke, D.; Wang, Y.; Xia, Y.; et al. Phosphoglycerate kinase 1 phosphorylates beclin1 to induce autophagy. Mol. Cell 2017, 65, 917–931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, S.; Xiao, Y.; Li, D.; Jiang, Q.; Zhu, L.; Lin, D.; Jiang, H.; Chen, W.; Wang, L.; Liu, C.; et al. PGK1 and GRP78 overexpression correlates with clinical significance and poor prognosis in Chinese endometrial cancer patients. Oncotarget 2018, 9, 680–690. [Google Scholar] [CrossRef] [Green Version]
- Mattaini, K.R.; Sullivan, M.R.; Vander Heiden, M.G. The importance of serine metabolism in cancer. J. Cell Biol. 2016, 214, 249–257. [Google Scholar] [CrossRef] [Green Version]
- Boado, R.J.; Li, J.Y.; Nagaya, M.; Zhang, C.; Pardridge, W.M. Selective expression of the large neutral amino acid transporter at the blood-brain barrier. Proc. Natl. Acad. Sci. USA 1999, 96, 12079–12084. [Google Scholar] [CrossRef] [Green Version]
- Snell, K.; Natsumeda, Y.; Weber, G. The modulation of serine metabolism in hepatoma 3924A during different phases of cellular proliferation in culture. Biochem. J. 1987, 245, 609–612. [Google Scholar] [CrossRef] [Green Version]
- Mitoma, J.; Furuya, S.; Hirabayashi, Y. A novel metabolic communication between neurons and astrocytes: Non-essential amino acid L-serine released from astrocytes is essential for developing hippocampal neurons. Neurosci. Res. 1998, 30, 195–199. [Google Scholar] [CrossRef]
- Matsui, T.; Sekiguchi, M.; Hashimoto, A.; Tomita, U.; Nishikawa, T.; Wada, K. Functional comparison of D-serine and glycine in rodents: The effect on cloned NMDA receptors and the extracellular concentration. J. Neurochem. 1995, 65, 454–458. [Google Scholar] [CrossRef]
- Son, G.; Han, J. Roles of mitochondria in neuronal development. BMB Rep. 2018, 51, 549–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gardner, P.R. Aconitase: Sensitive target and measure of superoxide. Methods Enzymol. 2002, 349, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.J.; Levine, R.L.; Sohal, R.S. Oxidative damage during aging targets mitochondrial aconitase. Proc. Natl. Acad. Sci. USA 1997, 94, 11168–11172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, J.; Teng, X.; Liu, L.; Mattaini, K.R.; Looper, R.E.; Vander Heiden, M.G.; Rabinowitz, J.D. Human phosphoglycerate dehydrogenase produces the oncometabolite D-2-hydroxyglutarate. ACS Chem. Biol. 2015, 10, 510–516. [Google Scholar] [CrossRef]
- Intlekofer, A.M.; Dematteo, R.G.; Venneti, S.; Finley, L.W.; Lu, C.; Judkins, A.R.; Rustenburg, A.S.; Grinaway, P.B.; Chodera, J.D.; Cross, J.R.; et al. Hypoxia induces production of L-2-hydroxyglutarate. Cell Metab. 2015, 22, 304–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bleeker, S.E.; Moll, H.A.; Steyerberg, E.W.; Donders, A.R.T.; Derksen-Lubsen, G.; Grobbee, D.E.; Moons, K.G.M. External validation is necessary in, prediction research: A clinical example. J. Clin. Epidemiol. 2003, 56, 826–832. [Google Scholar] [CrossRef]
- Hutter, E.; Renner, K.; Pfister, G.; Stockl, P.; Jansen-Durr, P.; Gnaiger, E. Senescence-associated changes in respiration and oxidative phosphorylation in primary human fibroblasts. Biochem. J. 2004, 380, 919–928. [Google Scholar] [CrossRef] [Green Version]
- Reid, M.A.; Allen, A.E.; Liu, S.; Liberti, M.V.; Liu, P.; Liu, X.; Dai, Z.; Gao, X.; Wang, Q.; Liu, Y.; et al. Serine synthesis through PHGDH coordinates nucleotide levels by maintaining central carbon metabolism. Nat. Commun. 2018, 9, 5442. [Google Scholar] [CrossRef]
- Rossignol, R.; Faustin, B.; Rocher, C.; Malgat, M.; Mazat, J.P.; Letellier, T. Mitochondrial threshold effects. Biochem. J. 2003, 370, 751–762. [Google Scholar] [CrossRef] [Green Version]
- Haas, R.H.; Parikh, S.; Falk, M.J.; Saneto, R.P.; Wolf, N.I.; Darin, N.; Wong, L.J.; Cohen, B.H.; Naviaux, R.K. The in-depth evaluation of suspected mitochondrial disease. Mol. Genet. Metab. 2008, 94, 16–37. [Google Scholar] [CrossRef] [Green Version]
- Peralta, S.; Torraco, A.; Wenz, T.; Garcia, S.; Diaz, F.; Moraes, C.T. Partial complex I deficiency due to the CNS conditional ablation of Ndufa5 results in a mild chronic encephalopathy but no increase in oxidative damage. Hum. Mol. Genet. 2014, 23, 1399–1412. [Google Scholar] [CrossRef] [Green Version]
- Piruat, J.I.; Pintado, C.O.; Ortega-Saenz, P.; Roche, M.; Lopez-Barneo, J. The mitochondrial SDHD gene is required for early embryogenesis, and its partial deficiency results in persistent carotid body glomus cell activation with full responsiveness to hypoxia. Mol. Cell. Biol. 2004, 24, 10933–10940. [Google Scholar] [CrossRef] [Green Version]
- Hughes, B.G.; Hekimi, S. A mild impairment of mitochondrial electron transport has sex-specific effects on lifespan and aging in mice. PLoS ONE 2011, 6, e26116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vrbacky, M.; Kovalcikova, J.; Chawengsaksophak, K.; Beck, I.M.; Mracek, T.; Nuskova, H.; Sedmera, D.; Papousek, F.; Kolar, F.; Sobol, M.; et al. Knockout of Tmem70 alters biogenesis of ATP synthase and leads to embryonal lethality in mice. Hum. Mol. Genet. 2016, 25, 4674–4685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ingraham, C.A.; Burwell, L.S.; Skalska, J.; Brookes, P.S.; Howell, R.L.; Sheu, S.S.; Pinkert, C.A. NDUFS4: Creation of a mouse model mimicking a Complex I disorder. Mitochondrion 2009, 9, 204–210. [Google Scholar] [CrossRef] [Green Version]
- Viscomi, C.; Bottani, E.; Civiletto, G.; Cerutti, R.; Moggio, M.; Fagiolari, G.; Schon, E.A.; Lamperti, C.; Zeviani, M. In vivo correction of COX deficiency by activation of the AMPK/PGC-1alpha axis. Cell Metab. 2011, 14, 80–90. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, Y.; Kako, K.; Kashiwabara, S.; Takehara, A.; Inada, Y.; Arai, H.; Nakada, K.; Kodama, H.; Hayashi, J.; Baba, T.; et al. Mammalian copper chaperone Cox17p has an essential role in activation of cytochrome C oxidase and embryonic development. Mol. Cell. Biol. 2002, 22, 7614–7621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Brosel, S.; Acin-Perez, R.; Slavkovich, V.; Nishino, I.; Khan, R.; Goldberg, I.J.; Graziano, J.; Manfredi, G.; Schon, E.A. Analysis of mouse models of cytochrome c oxidase deficiency owing to mutations in Sco2. Hum. Mol. Genet. 2010, 19, 170–180. [Google Scholar] [CrossRef] [Green Version]
- Nonn, L.; Williams, R.R.; Erickson, R.P.; Powis, G. The absence of mitochondrial thioredoxin 2 causes massive apoptosis, exencephaly, and early embryonic lethality in homozygous mice. Mol. Cell. Biol. 2003, 23, 916–922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, T.; Hosoi, F.; Yamaguchi-Iwai, Y.; Nakamura, H.; Masutani, H.; Ueda, S.; Nishiyama, A.; Takeda, S.; Wada, H.; Spyrou, G.; et al. Thioredoxin-2 (TRX-2) is an essential gene regulating mitochondria-dependent apoptosis. EMBO J. 2002, 21, 1695–1703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jauniaux, E.; Gulbis, B.; Burton, G.J. Physiological implications of the materno-fetal oxygen gradient in human early pregnancy. Reprod. Biomed. Online 2003, 7, 250–253. [Google Scholar] [CrossRef]
- Giulivi, C. Functional implications of nitric oxide produced by mitochondria in mitochondrial metabolism. Biochem. J. 1998, 332, 673–679. [Google Scholar] [CrossRef] [Green Version]
- De Koning, T.J.; Klomp, L.W.; van Oppen, A.C.; Beemer, F.A.; Dorland, L.; van den Berg, I.; Berger, R. Prenatal and early postnatal treatment in 3-phosphoglycerate-dehydrogenase deficiency. Lancet 2004, 364, 2221–2222. [Google Scholar] [CrossRef]
- Davis, D.A.; Cox, P.A.; Banack, S.A.; Lecusay, P.D.; Garamszegi, S.P.; Hagan, M.J.; Powell, J.T.; Metcalf, J.S.; Palmour, R.M.; Beierschmitt, A.; et al. l-Serine reduces spinal cord pathology in a vervet model of preclinical ALS/MND. J. Neuropathol. Exp. Neurol. 2020, 79, 393–406. [Google Scholar] [CrossRef] [PubMed]
- Le Douce, J.; Maugard, M.; Veran, J.; Matos, M.; Jego, P.; Vigneron, P.A.; Faivre, E.; Toussay, X.; Vandenberghe, M.; Balbastre, Y.; et al. Impairment of glycolysis-derived l-serine production in astrocytes contributes to cognitive deficits in Alzheimer’s disease. Cell Metab. 2020, 31, 503–517.e8. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Non-Carrier Fetuses | Carrier Fetuses | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Maternal Age a (y) | Gestational Age (Weeks) | Maternal CGG Repeat Size | Fetal CGG Repeat Size | Fetal Repeat Structure b | Maternal Age a (y) | Gestational Age (Weeks) | Maternal CGG Repeat Size | Fetal CGG Repeat Size | Fetal Repeat Structure b |
| 25.5 | 17.3 | 29, 92 | 29 | 29 c | 17.8 | 16.4 | 37, 77 | 88 | 9A78 |
| 25.5 | 17.3 | 29, 92 | 29 | 29 c | 18.0 | 16.4 | 30, 62 | 62 | 9A9A42 |
| 25.8 | 16.7 | 29, 53 | 29 | 9A9A9 | 20.4 | 16.4 | 30, 55 | 55 | 9A45 |
| 27.4 | 18.2 | 29, 55 | 29 | 9A9A9 | 20.6 | 16.3 | 29, 60 | 66 | 66 |
| 28.5 | 18.3 | 30, 63 | 30 | 10A9A9 | 23.6 | 21.2 | 30, 56 | 57 | 9A9A37 |
| 30.0 | 15.7 | 30, 132 | 30 | 10A9A9 | 24.4 | 16.5 | 23, 57 | 58 | 10A47 |
| 30.5 | 17.5 | 26, 59 | 26 | 26 | 26.2 | 17.1 | 31, 58 | 63 | 63 |
| 30.7 | 16.3 | 30, 68 | 30 | 10A9A9 | 27.0 | 15.7 | 29, 60 | 61 | 9A51 |
| 31.2 | 16.9 | 29, 200 | 29 | 9A9A9 | 28.4 | 17.2 | 30, 60 | 63 | 9A53 |
| 31.3 | 15.9 | 29, 89 | 29 | 9A9A9 | 29.3 | 16.7 | 23, 63 | 68 | 9A58 |
| 31.5 | 16.0 | 29, 68 | 29 | 9A19 | 30.0 | 16.0 | 31, 57 | 58 | 10A47 |
| 31.6 | 20.3 | 40, 65 | 40 | 10A29 | 31.3 | 16.5 | 33, 57 | 57 | 9A9A37 |
| 31.8 | 16.6 | 20, 56 | 20 | 10A9 | 31.5 | 16.4 | 23, 67 | 78 | 9A68 |
| 31.9 | 15.5 | 30, 58 | 30 | 10A9A9 | 32.8 | 16.0 | 31, 74 | 63 | 9A53 |
| 32.0 | 17.1 | 29, 56 | 29 | 9A9A9 | 33.0 | 16.3 | 23, 59 | 67 | 67 |
| 32.8 | 16.1 | 29, 53 | 29 | 9A9A9 | 33.3 | 20.0 | 30, 57 | 59 | 10A48 |
| 33.2 | 18.2 | 29, 71 | 29 | 9A9A9 | 33.4 | 13.3 | 30, 82 | 141,157 e | 9A9A121 |
| 33.8 | 16.7 | 21, 58 | 21 | 10A10 | 33.8 | 17.0 | 30, 63 | 63 | 9A9A43 |
| 34.3 | 17.1 | 30, 73 | 30 | 10A9A9 | 35.4 | 16.0 | 20, 69 | 72 | 9A9A52 |
| 34.7 | 16.3 | 32, 60 | 32 | 9A12A9 | 35.7 | 16.5 | 31, 61 | 66 | 9A56 |
| 35.1 | 21.0 | 30, 59 | 30 | 10A9A9 | 40.7 | 15.1 | 30, 72 | 84 | 84 |
| 36.1 | 18.9 | 29, 68 | 29 | 9A19 | 42.1 | 17.7 | 30, 58 | 58 | 9A9A38 |
| 37.1 | 15.7 | 20, 105 | 20 | 10A9 d | |||||
| 37.1 | 15.7 | 20, 105 | 20 | 10A9 d | |||||
| 37.9 | 15.3 | 30, 63 | 30 | 10A9A9 | |||||
| 37.9 | 17.4 | 20, 62 | 20 | 10A9 | |||||
| 38.9 | 16.1 | 30, 75 | 30 | 10A9A9 | |||||
| 39.1 | 16.4 | 29, 59 | 29 | 9A9A9 | |||||
| 39.2 | 15.9 | 30, 58 | 30 | 10A9A9 | |||||
| 40.1 | 18.0 | 29, 56 | 29 | 9A9A9 | |||||
| 40.7 | 16.0 | 31, 44 | 44 | 9A9A24 | |||||
| Diagnosis | Non-Carrier | Carrier | Non-Carrier | Non-Carrier | Carrier | Non-Carrier | |
|---|---|---|---|---|---|---|---|
| Substrate | Gln | AKG | |||||
| Inhibitor of PHGDH | no | no | yes | no | no | yes | |
| Energy Status | [ATP]/[AMP] ratio | 2.31 | 1.88 | 2.15 | 1.18 | 1.48 | 1.23 |
| AMP | −2.73 | −1.53 | −2.30 | −1.61 | −1.10 | 1.04 | |
| Glycolysis | Glucose | −2.86 | −2.88 | −2.40 | −1.42 | 1.24 | −1.36 |
| G6P | −3.09 | −3.30 | −1.87 | 1.15 | 1.17 | 1.79 | |
| 3PG | −2.19 | −2.71 | −2.15 | 1.02 | 1.13 | 1.70 | |
| Lactate | −2.51 | −2.50 | −1.66 | −1.62 | −2.29 | −1.40 | |
| Ala | −2.61 | −1.48 | −3.04 | −1.75 | −1.35 | −1.46 | |
| TCA Cycle | Citrate | −2.42 | −2.21 | −1.84 | 1.33 | 2.77 | −1.17 |
| AKG | −3.16 | −3.20 | −3.04 | 136.28 | 51.43 | 235.19 | |
| Succinate | −1.80 | 4.68 | −1.76 | 5.90 | 12.20 | 5.26 | |
| Fumarate | −2.06 | −1.67 | −2.57 | 3.74 | 2.15 | 3.54 | |
| Malate | 1.06 | −1.57 | −1.40 | 12.65 | 2.41 | 10.00 | |
| Anaplerotic Substrates | AKG | −3.16 | −3.20 | −3.04 | 136.28 | 51.43 | 235.19 |
| Asp | −2.65 | −1.78 | −2.91 | −6.79 | −5.99 | −3.61 | |
| Glu | −2.44 | −1.57 | −1.95 | −1.11 | −1.09 | 1.29 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nolin, S.L.; Napoli, E.; Flores, A.; Hagerman, R.J.; Giulivi, C. Deficits in Prenatal Serine Biosynthesis Underlie the Mitochondrial Dysfunction Associated with the Autism-Linked FMR1 Gene. Int. J. Mol. Sci. 2021, 22, 5886. https://doi.org/10.3390/ijms22115886
Nolin SL, Napoli E, Flores A, Hagerman RJ, Giulivi C. Deficits in Prenatal Serine Biosynthesis Underlie the Mitochondrial Dysfunction Associated with the Autism-Linked FMR1 Gene. International Journal of Molecular Sciences. 2021; 22(11):5886. https://doi.org/10.3390/ijms22115886
Chicago/Turabian StyleNolin, Sarah L., Eleonora Napoli, Amanda Flores, Randi J. Hagerman, and Cecilia Giulivi. 2021. "Deficits in Prenatal Serine Biosynthesis Underlie the Mitochondrial Dysfunction Associated with the Autism-Linked FMR1 Gene" International Journal of Molecular Sciences 22, no. 11: 5886. https://doi.org/10.3390/ijms22115886