
P2Y2R Deficiency Ameliorates Hepatic Steatosis by Reducing Lipogenesis and Enhancing Fatty Acid β-Oxidation through AMPK and PGC-1α Induction in High-Fat Diet-Fed Mice

 ,
,  , and
, and
Abstract
:1. Introduction
2. Results
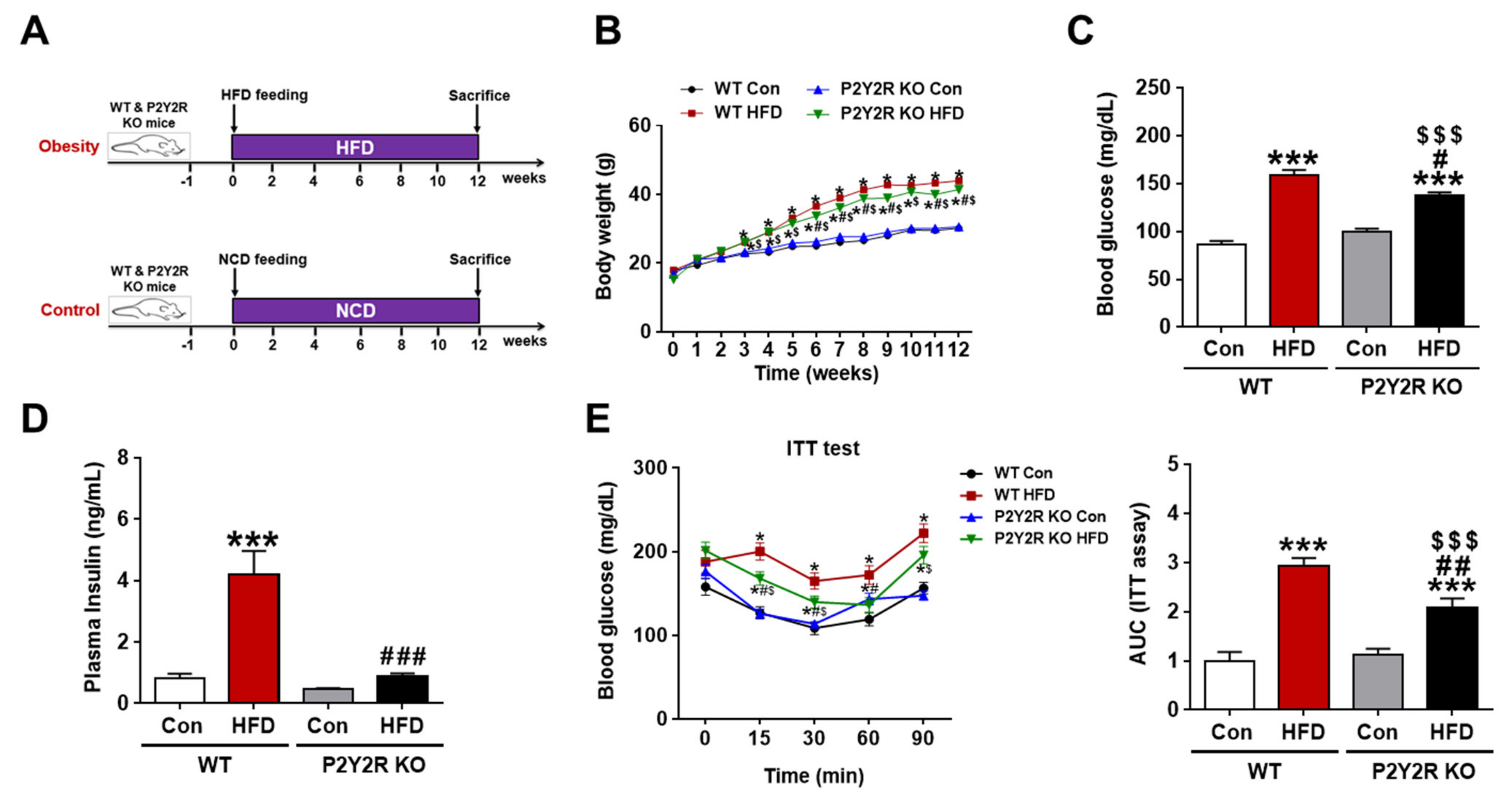
2.1. P2Y2R Deficiency Reduced Insulin Resistance in HFD-Fed Mice
2.2. P2Y2R Deficiency Attenuated Hepatic Steatosis and Cellular Injury in HFD-Fed Mice
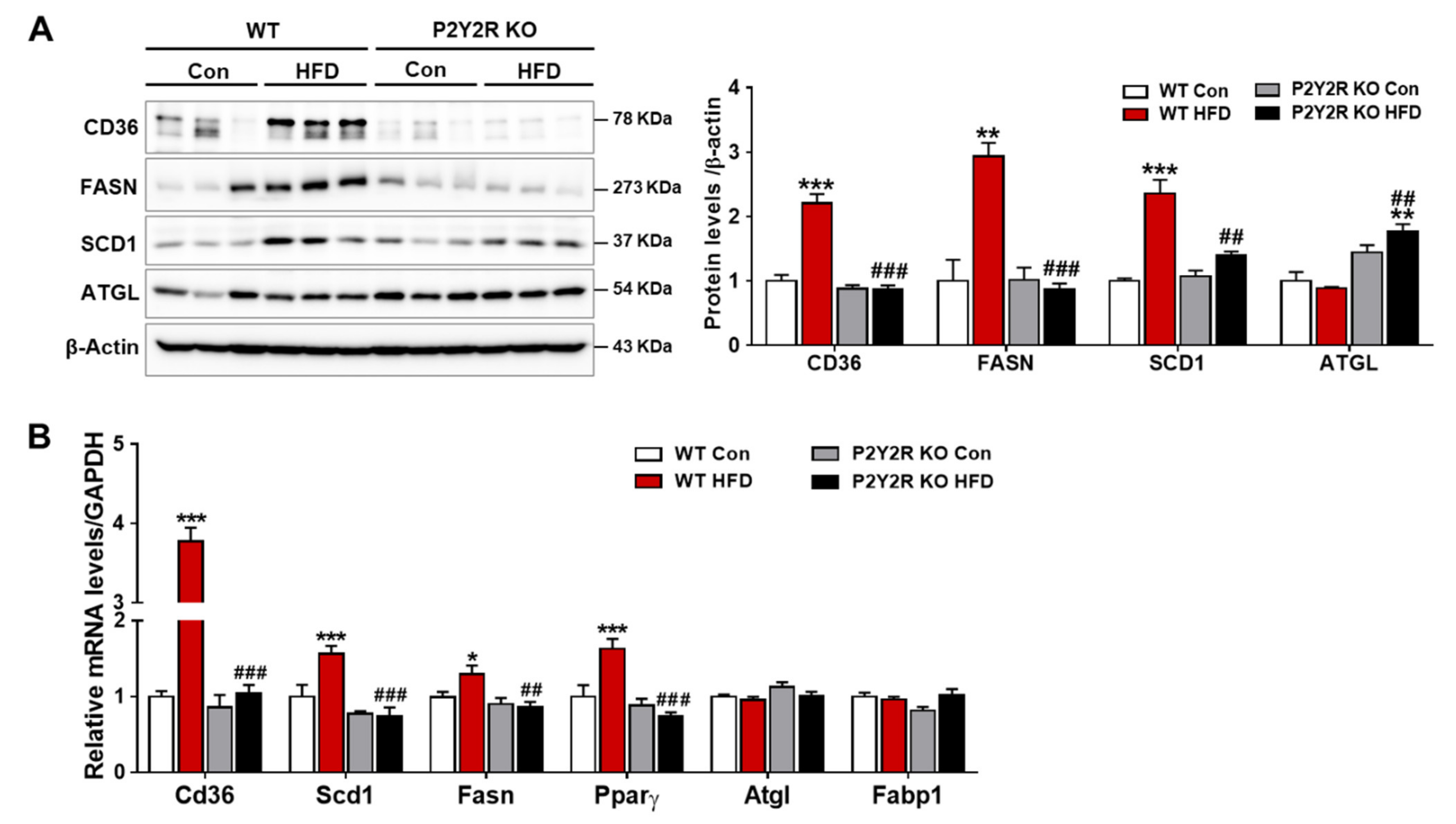
2.3. P2Y2R Deficiency Decreased De Novo Lipogenesis in HFD-Fed Mice
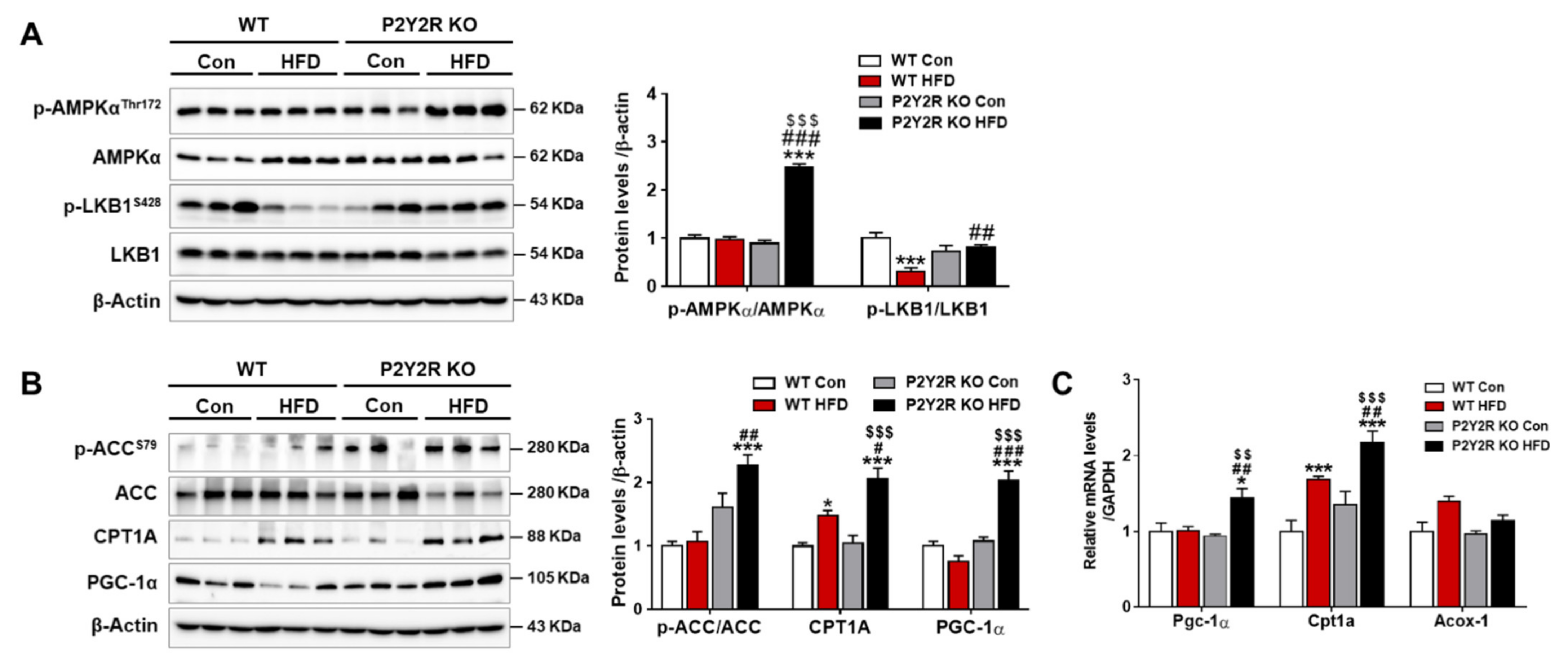
2.4. P2Y2R Deficiency Enhanced Mitochondrial Fatty Acid β-Oxidation in HFD-Fed Mice
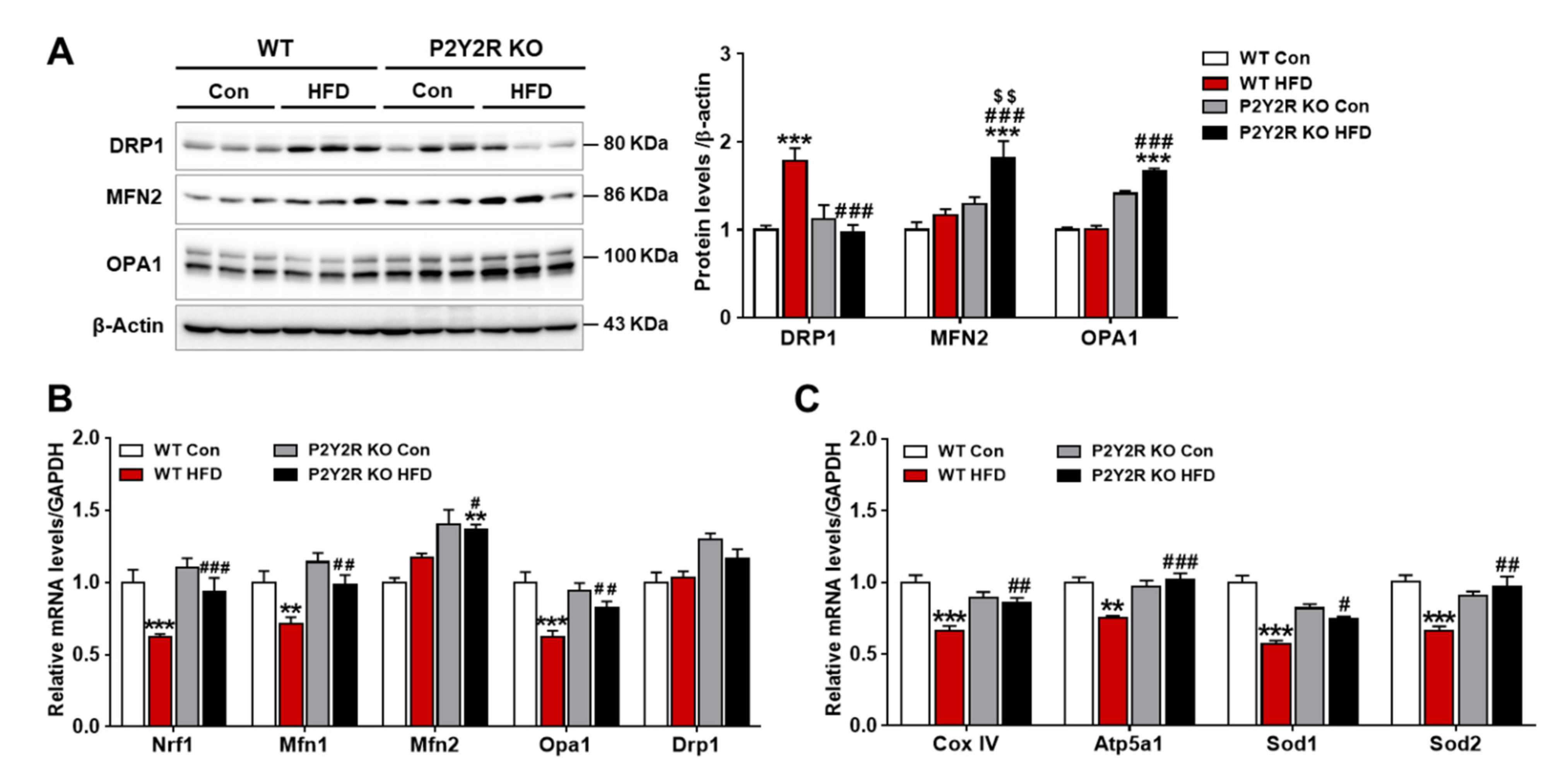
2.5. P2Y2R Deficiency Improved Hepatic Mitochondrial Function in HFD-Fed Mice
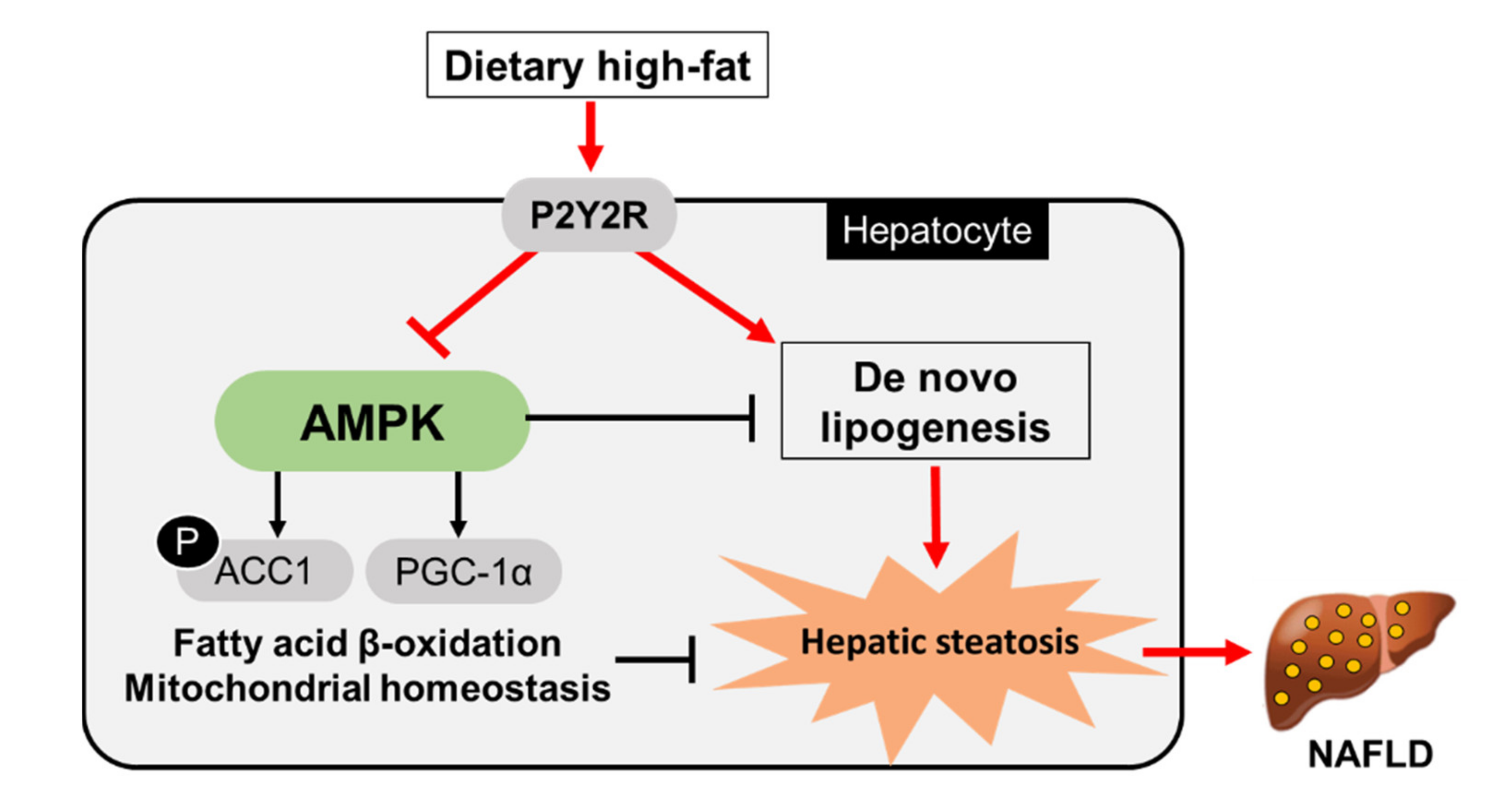
3. Discussion
4. Materials and Methods
4.1. Experimental Animals
4.2. NAFLD Mouse Model
4.3. Biochemical Assays
4.4. Hepatic Triglyceride Assay
4.5. Histological Examination
4.6. Western Blot Analysis
4.7. Quantitative Real-Time Polymerase Chain Reaction (PCR) Analysis
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef]
- Edmunds, L.R.; Xie, B.; Mills, A.M.; Huckestein, B.R.; Undamatla, R.; Murali, A.; Pangburn, M.M.; Martin, J.; Sipula, I.; Kaufman, B.A.; et al. Liver-specific Prkn knockout mice are more susceptible to diet-induced hepatic steatosis and insulin resistance. Mol. Metab. 2020, 41, 101051. [Google Scholar] [CrossRef]
- Kwon, E.Y.; Jung, U.J.; Park, T.; Yun, J.W.; Choi, M.S. Luteolin attenuates hepatic steatosis and insulin resistance through the interplay between the liver and adipose tissue in mice with diet-induced obesity. Diabetes 2015, 64, 1658–1669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, J.; Han, C.J.; Zhang, J.Z.; He, W.Z.; Zhao, G.J.; Cheng, X.; Zhang, L.; Deng, K.Q.; Liu, Y.; Fan, H.F.; et al. Hepatic Interferon Regulatory Factor 6 Alleviates Liver Steatosis and Metabolic Disorder by Transcriptionally Suppressing Peroxisome Proliferator-Activated Receptor gamma in Mice. Hepatology 2019, 69, 2471–2488. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.W.; Adams, L.A. Nonalcoholic fatty liver disease and diabetes mellitus: Pathogenesis and treatment. Nat. Rev. Endocrinol. 2011, 7, 456–465. [Google Scholar] [CrossRef]
- Tilg, H.; Moschen, A.R.; Roden, M. NAFLD and diabetes mellitus. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Begriche, K.; Massart, J.; Robin, M.A.; Bonnet, F.; Fromenty, B. Mitochondrial adaptations and dysfunctions in nonalcoholic fatty liver disease. Hepatology 2013, 58, 1497–1507. [Google Scholar] [CrossRef] [PubMed]
- Schlaepfer, I.R.; Joshi, M. CPT1A-mediated Fat Oxidation, Mechanisms, and Therapeutic Potential. Endocrinology 2020, 161. [Google Scholar] [CrossRef] [PubMed]
- Monsenego, J.; Mansouri, A.; Akkaoui, M.; Lenoir, V.; Esnous, C.; Fauveau, V.; Tavernier, V.; Girard, J.; Prip-Buus, C. Enhancing liver mitochondrial fatty acid oxidation capacity in obese mice improves insulin sensitivity independently of hepatic steatosis. J. Hepatol. 2012, 56, 632–639. [Google Scholar] [CrossRef]
- Wu, L.; Zhang, L.; Li, B.; Jiang, H.; Duan, Y.; Xie, Z.; Shuai, L.; Li, J.; Li, J. AMP-Activated Protein Kinase (AMPK) Regulates Energy Metabolism through Modulating Thermogenesis in Adipose Tissue. Front. Physiol. 2018, 9, 122. [Google Scholar] [CrossRef] [Green Version]
- Foretz, M.; Even, P.C.; Viollet, B. AMPK Activation Reduces Hepatic Lipid Content by Increasing Fat Oxidation In Vivo. Int. J. Mol. Sci. 2018, 19, 2826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mottillo, E.P.; Desjardins, E.M.; Crane, J.D.; Smith, B.K.; Green, A.E.; Ducommun, S.; Henriksen, T.I.; Rebalka, I.A.; Razi, A.; Sakamoto, K.; et al. Lack of Adipocyte AMPK Exacerbates Insulin Resistance and Hepatic Steatosis through Brown and Beige Adipose Tissue Function. Cell Metab. 2016, 24, 118–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, D.; Hellberg, K.; Chaix, A.; Wallace, M.; Herzig, S.; Badur, M.G.; Lin, T.; Shokhirev, M.N.; Pinto, A.F.M.; Ross, D.S.; et al. Genetic Liver-Specific AMPK Activation Protects against Diet-Induced Obesity and NAFLD. Cell Rep. 2019, 26, 192–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jager, S.; Handschin, C.; St-Pierre, J.; Spiegelman, B.M. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc. Natl. Acad. Sci. USA 2007, 104, 12017–12022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, C.F.; Ku, H.C.; Lin, H. PGC-1alpha as a Pivotal Factor in Lipid and Metabolic Regulation. Int. J. Mol. Sci. 2018, 19, 3447. [Google Scholar] [CrossRef] [Green Version]
- Morris, E.M.; Meers, G.M.; Booth, F.W.; Fritsche, K.L.; Hardin, C.D.; Thyfault, J.P.; Ibdah, J.A. PGC-1alpha overexpression results in increased hepatic fatty acid oxidation with reduced triacylglycerol accumulation and secretion. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 303, G979–G992. [Google Scholar] [CrossRef] [Green Version]
- Aharoni-Simon, M.; Hann-Obercyger, M.; Pen, S.; Madar, Z.; Tirosh, O. Fatty liver is associated with impaired activity of PPARgamma-coactivator 1alpha (PGC1alpha) and mitochondrial biogenesis in mice. Lab. Investig. 2011, 91, 1018–1028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piccinin, E.; Villani, G.; Moschetta, A. Metabolic aspects in NAFLD, NASH and hepatocellular carcinoma: The role of PGC1 coactivators. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 160–174. [Google Scholar] [CrossRef]
- Wang, P.; Jia, J.; Zhang, D. Purinergic signalling in liver diseases: Pathological functions and therapeutic opportunities. JHEP. Rep. 2020, 2, 100165. [Google Scholar] [CrossRef]
- Burnstock, G.; Vaughn, B.; Robson, S.C. Purinergic signalling in the liver in health and disease. Purinergic Signal. 2014, 10, 51–70. [Google Scholar] [CrossRef] [Green Version]
- Kishore, B.K.; Carlson, N.G.; Ecelbarger, C.M.; Kohan, D.E.; Muller, C.E.; Nelson, R.D.; Peti-Peterdi, J.; Zhang, Y. Targeting renal purinergic signalling for the treatment of lithium-induced nephrogenic diabetes insipidus. Acta Physiol. 2015, 214, 176–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Ecelbarger, C.M.; Lesniewski, L.A.; Muller, C.E.; Kishore, B.K. P2Y2 Receptor Promotes High-Fat Diet-Induced Obesity. Front. Endocrinol. 2020, 11, 341. [Google Scholar] [CrossRef]
- Tackett, B.C.; Sun, H.; Mei, Y.; Maynard, J.P.; Cheruvu, S.; Mani, A.; Hernandez-Garcia, A.; Vigneswaran, N.; Karpen, S.J.; Thevananther, S. P2Y2 purinergic receptor activation is essential for efficient hepatocyte proliferation in response to partial hepatectomy. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 307, G1073–G1087. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.B.; Turner, J.J.O.; Fountain, S.J. Constitutive P2Y2 receptor activity regulates basal lipolysis in human adipocytes. J. Cell. Sci. 2018, 131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merz, J.; Albrecht, P.; von Garlen, S.; Ahmed, I.; Dimanski, D.; Wolf, D.; Hilgendorf, I.; Hardtner, C.; Grotius, K.; Willecke, F.; et al. Purinergic receptor Y2 (P2Y2)- dependent VCAM-1 expression promotes immune cell infiltration in metabolic syndrome. Basic Res. Cardiol. 2018, 113, 45. [Google Scholar] [CrossRef]
- Guilherme, A.; Virbasius, J.V.; Puri, V.; Czech, M.P. Adipocyte dysfunctions linking obesity to insulin resistance and type 2 diabetes. Nat. Rev. Mol. Cell. Biol. 2008, 9, 367–377. [Google Scholar] [CrossRef] [Green Version]
- Longo, M.; Zatterale, F.; Naderi, J.; Parrillo, L.; Formisano, P.; Raciti, G.A.; Beguinot, F.; Miele, C. Adipose Tissue Dysfunction as Determinant of Obesity-Associated Metabolic Complications. Int. J. Mol. Sci. 2019, 20, 2358. [Google Scholar] [CrossRef] [Green Version]
- Dumas, K.; Ayachi, C.; Gilleron, J.; Lacas-Gervais, S.; Pastor, F.; Favier, F.B.; Peraldi, P.; Vaillant, N.; Yvan-Charvet, L.; Bonnafous, S.; et al. REDD1 deficiency protects against nonalcoholic hepatic steatosis induced by high-fat diet. FASEB J. 2020, 34, 5046–5060. [Google Scholar] [CrossRef]
- Ipsen, D.H.; Lykkesfeldt, J.; Tveden-Nyborg, P. Molecular mechanisms of hepatic lipid accumulation in non-alcoholic fatty liver disease. Cell. Mol. Life Sci. 2018, 75, 3313–3327. [Google Scholar] [CrossRef] [Green Version]
- Sozio, M.S.; Liangpunsakul, S.; Crabb, D. The role of lipid metabolism in the pathogenesis of alcoholic and nonalcoholic hepatic steatosis. Semin. Liver Dis. 2010, 30, 378–390. [Google Scholar] [CrossRef]
- Xia, M.F.; Bian, H.; Gao, X. NAFLD and Diabetes: Two Sides of the Same Coin? Rationale for Gene-Based Personalized NAFLD Treatment. Front. Pharmacol. 2019, 10, 877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tak, E.; Jun, D.Y.; Kim, S.H.; Park, G.C.; Lee, J.; Hwang, S.; Song, G.W.; Lee, S.G. Upregulation of P2Y2 nucleotide receptor in human hepatocellular carcinoma cells. J. Int. Med. Res. 2016, 44, 1234–1247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayata, C.K.; Ganal, S.C.; Hockenjos, B.; Willim, K.; Vieira, R.P.; Grimm, M.; Robaye, B.; Boeynaems, J.M.; di Virgilio, F.; Pellegatti, P.; et al. Purinergic P2Y(2) receptors promote neutrophil infiltration and hepatocyte death in mice with acute liver injury. Gastroenterology 2012, 143, 1620–1629. [Google Scholar] [CrossRef] [PubMed]
- Velazquez-Miranda, E.; Molina-Aguilar, C.; Gonzalez-Gallardo, A.; Vazquez-Martinez, O.; Diaz-Munoz, M.; Vazquez-Cuevas, F.G. Increased Purinergic Responses Dependent on P2Y2 Receptors in Hepatocytes from CCl4-Treated Fibrotic Mice. Int. J. Mol. Sci. 2020, 21, 2305. [Google Scholar] [CrossRef] [Green Version]
- Ganz, M.; Szabo, G. Immune and inflammatory pathways in NASH. Hepatol. Int. 2013, 7 (Suppl. S2), 771–781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moschen, A.R.; Molnar, C.; Geiger, S.; Graziadei, I.; Ebenbichler, C.F.; Weiss, H.; Kaser, S.; Kaser, A.; Tilg, H. Anti-inflammatory effects of excessive weight loss: Potent suppression of adipose interleukin 6 and tumour necrosis factor alpha expression. Gut 2010, 59, 1259–1264. [Google Scholar] [CrossRef]
- Cusi, K. Role of obesity and lipotoxicity in the development of nonalcoholic steatohepatitis: Pathophysiology and clinical implications. Gastroenterology 2012, 142, 711–725. [Google Scholar] [CrossRef] [PubMed]
- Parthasarathy, G.; Revelo, X.; Malhi, H. Pathogenesis of Nonalcoholic Steatohepatitis: An Overview. Hepatol. Commun. 2020, 4, 478–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Marsh, S.; Hu, J.; Feng, W.; Wu, C. The Pathogenesis of Nonalcoholic Fatty Liver Disease: Interplay between Diet, Gut Microbiota, and Genetic Background. Gastroenterol. Res. Pract. 2016, 2016, 2862173. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Febbraio, M.; Wada, T.; Zhai, Y.; Kuruba, R.; He, J.; Lee, J.H.; Khadem, S.; Ren, S.; Li, S.; et al. Hepatic fatty acid transporter Cd36 is a common target of LXR, PXR, and PPARgamma in promoting steatosis. Gastroenterology 2008, 134, 556–567. [Google Scholar] [CrossRef]
- Wilson, C.G.; Tran, J.L.; Erion, D.M.; Vera, N.B.; Febbraio, M.; Weiss, E.J. Hepatocyte-Specific Disruption of CD36 Attenuates Fatty Liver and Improves Insulin Sensitivity in HFD-Fed Mice. Endocrinology 2016, 157, 570–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsusue, K.; Aibara, D.; Hayafuchi, R.; Matsuo, K.; Takiguchi, S.; Gonzalez, F.J.; Yamano, S. Hepatic PPARgamma and LXRalpha independently regulate lipid accumulation in the livers of genetically obese mice. FEBS Lett. 2014, 588, 2277–2281. [Google Scholar] [CrossRef]
- Woods, A.; Williams, J.R.; Muckett, P.J.; Mayer, F.V.; Liljevald, M.; Bohlooly, Y.M.; Carling, D. Liver-Specific Activation of AMPK Prevents Steatosis on a High-Fructose Diet. Cell Rep. 2017, 18, 3043–3051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esquejo, R.M.; Salatto, C.T.; Delmore, J.; Albuquerque, B.; Reyes, A.; Shi, Y.; Moccia, R.; Cokorinos, E.; Peloquin, M.; Monetti, M.; et al. Activation of Liver AMPK with PF-06409577 Corrects NAFLD and Lowers Cholesterol in Rodent and Primate Preclinical Models. EBioMedicine 2018, 31, 122–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fullerton, M.D.; Galic, S.; Marcinko, K.; Sikkema, S.; Pulinilkunnil, T.; Chen, Z.P.; O’Neill, H.M.; Ford, R.J.; Palanivel, R.; O’Brien, M.; et al. Single phosphorylation sites in Acc1 and Acc2 regulate lipid homeostasis and the insulin-sensitizing effects of metformin. Nat. Med. 2013, 19, 1649–1654. [Google Scholar] [CrossRef] [Green Version]
- Foretz, M.; Hebrard, S.; Leclerc, J.; Zarrinpashneh, E.; Soty, M.; Mithieux, G.; Sakamoto, K.; Andreelli, F.; Viollet, B. Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J. Clin. Investig. 2010, 120, 2355–2369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, M.S.; Kim, J.H.; Kim, H.J.; Chang, K.C.; Park, S.W. Honokiol activates the LKB1-AMPK signaling pathway and attenuates the lipid accumulation in hepatocytes. Toxicol. Appl. Pharmacol. 2015, 284, 113–124. [Google Scholar] [CrossRef]
- Lin, J.; Handschin, C.; Spiegelman, B.M. Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab. 2005, 1, 361–370. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.W.; Sheng, H.; Bai, Y.F.; Weng, Y.Y.; Fan, X.Y.; Lou, L.J.; Zhang, F. Neohesperidin enhances PGC-1alpha-mediated mitochondrial biogenesis and alleviates hepatic steatosis in high fat diet fed mice. Nutr. Diabetes 2020, 10, 27. [Google Scholar] [CrossRef]
- Kleiner, D.E.; Brunt, E.M.; Van Natta, M.; Behling, C.; Contos, M.J.; Cummings, O.W.; Ferrell, L.D.; Liu, Y.C.; Torbenson, M.S.; Unalp-Arida, A.; et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005, 41, 1313–1321. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primers (5′–3′) | Reverse Primers (5′–3′) |
|---|---|---|
| Atgl | GGCCAACGCCACTCACATCTAC | CACGGATGGTCTTCACCAGGTTG |
| Cd36 | CTGGGACCATTGGTGATGAAA | CACCACTCCAATCCCAAGTAAG |
| Fasn | AGTGGACGCACCTTAGAGGCAG | ACTTGCTGCACTTCTTGGACACG |
| Scd1 | CAACTTCACCACGTTCTTCATC | CCCGTCTCCAGTTCTCTTAATC |
| Pparγ | CGGTGTGTATGAAGCCATCT | TAAGGAACTCGCGTGTGATAAA |
| Fabp1 | GTACCAATTGCAGAGCCAGGAGA | GGTCCATAGGTGATGGTGAGTTTG |
| Pgc-1α | AGCCGTGACCACTGACAACGAG | GCTGCATGGTTCTGAGTGCTAAG |
| Cpt1a | GAAGTGTCGGCAGACCTATT | GTCCTCCTCTCTATATCCCTGTT |
| Acox-1 | CGCACATCTTGGATGGTAGT | GGCTTCGAGTGAGGAAGTTATAG |
| Nrf1 | GAGCACGGAGTGACCCAAAC | TGTACGTGGCTACATGGACCT |
| Mfn1 | TACTGGACTCAGTAAACGTGGC | CTCCGTGACCTCCTTGATCT |
| Mfn2 | ACCGTCAAGAAGGATAAGCGACAC | GTGTTCCTGTGGGTGTCTTCAAGG |
| Opa1 | CCAAGAACGAGTTGGAGAAGATGC | CACGTCATTGCATTCCAGCTCAGA |
| P2y2r | GTGCTCTACTTCGTCACCACCAG | CCATAAGCACGTAACAGACCAGGA |
| Drp1 | ACCAAAGTACCTGTAGGCGATC | CATGGCATCAGTACCCGCAT |
| Cox IV | CGCTGAGCCTGATTGGCAAGAGA | TGGCAGACAGCATCGTGACATGG |
| Atp5a1 | CCATGCCTCTAACACTCGACTTCA | CGGGTTCCAAGTTCAGGGACATAC |
| Sod1 | CAGGGAACCATCCACTTCGAGC | CTGCACTGGTACAGCCTTGTGTA |
| Sod2 | CCACCGAGGAGAAGTACCACGAG | CTCCTTATTGAAGCCAAGCCAGCC |
| Gapdh | ATGACATCACAGGTGAGCTGAAGG | CTCAAACCAGCCTTTCAGAATGGC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dusabimana, T.; Park, E.J.; Je, J.; Jeong, K.; Yun, S.P.; Kim, H.J.; Kim, H.; Park, S.W. P2Y2R Deficiency Ameliorates Hepatic Steatosis by Reducing Lipogenesis and Enhancing Fatty Acid β-Oxidation through AMPK and PGC-1α Induction in High-Fat Diet-Fed Mice. Int. J. Mol. Sci. 2021, 22, 5528. https://doi.org/10.3390/ijms22115528
Dusabimana T, Park EJ, Je J, Jeong K, Yun SP, Kim HJ, Kim H, Park SW. P2Y2R Deficiency Ameliorates Hepatic Steatosis by Reducing Lipogenesis and Enhancing Fatty Acid β-Oxidation through AMPK and PGC-1α Induction in High-Fat Diet-Fed Mice. International Journal of Molecular Sciences. 2021; 22(11):5528. https://doi.org/10.3390/ijms22115528
Chicago/Turabian StyleDusabimana, Theodomir, Eun Jung Park, Jihyun Je, Kyuho Jeong, Seung Pil Yun, Hye Jung Kim, Hwajin Kim, and Sang Won Park. 2021. "P2Y2R Deficiency Ameliorates Hepatic Steatosis by Reducing Lipogenesis and Enhancing Fatty Acid β-Oxidation through AMPK and PGC-1α Induction in High-Fat Diet-Fed Mice" International Journal of Molecular Sciences 22, no. 11: 5528. https://doi.org/10.3390/ijms22115528